Androgen Receptor Antagonists and Anti-Prostate Cancer Activities of Some Newly Synthesized Substituted Fused Pyrazolo-, Triazolo- and Thiazolo-Pyrimidine Derivatives

Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
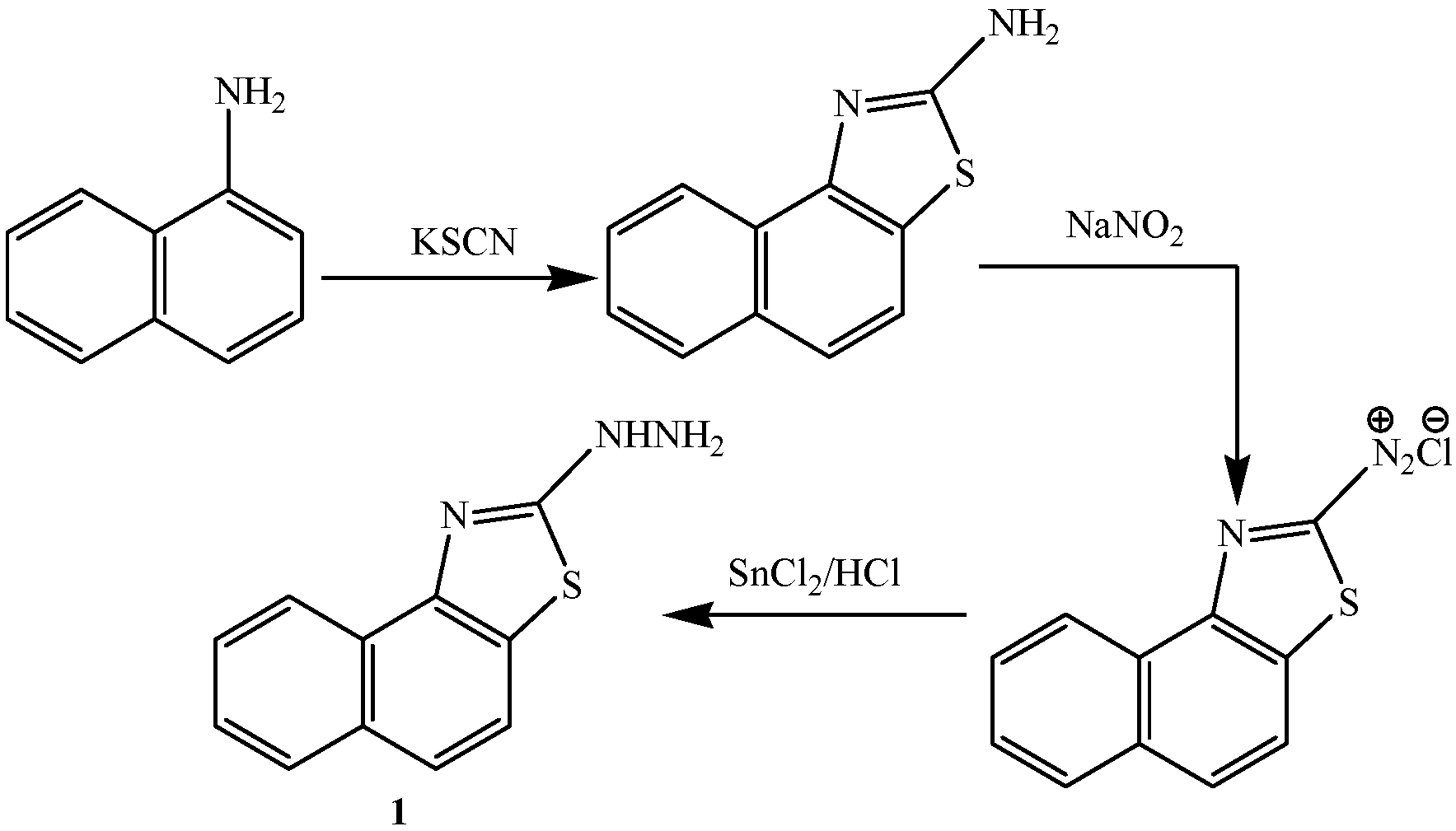
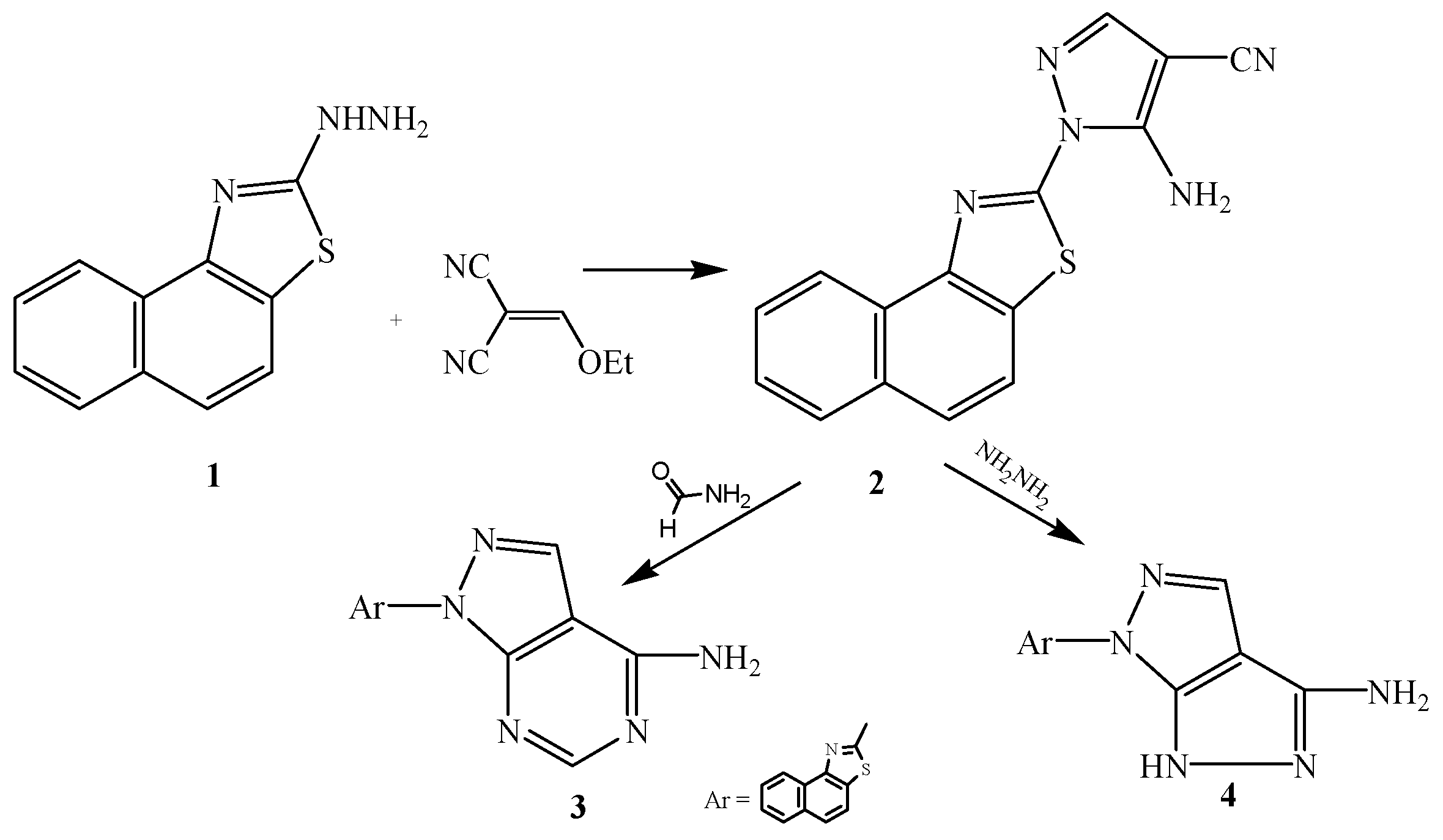
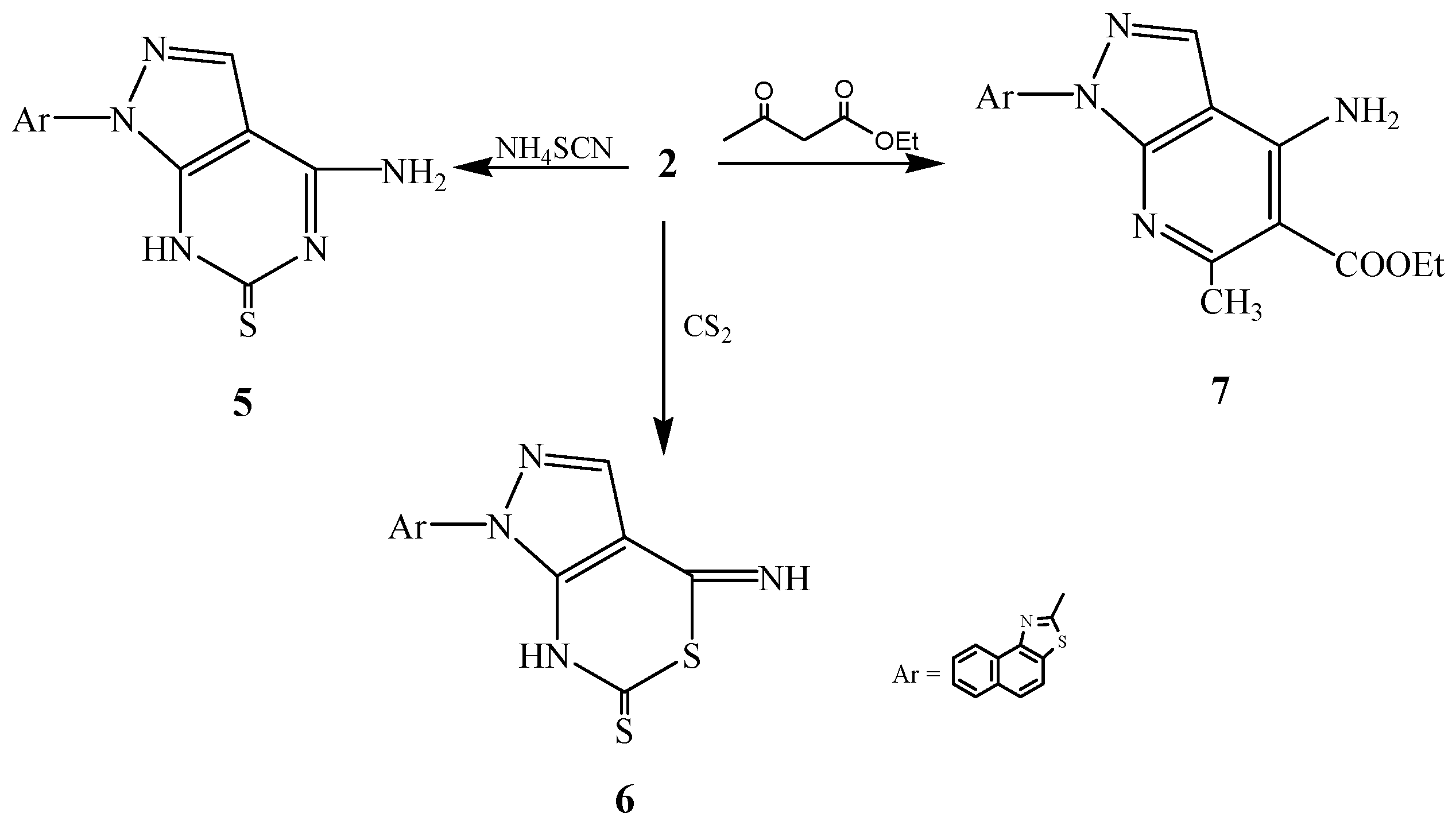
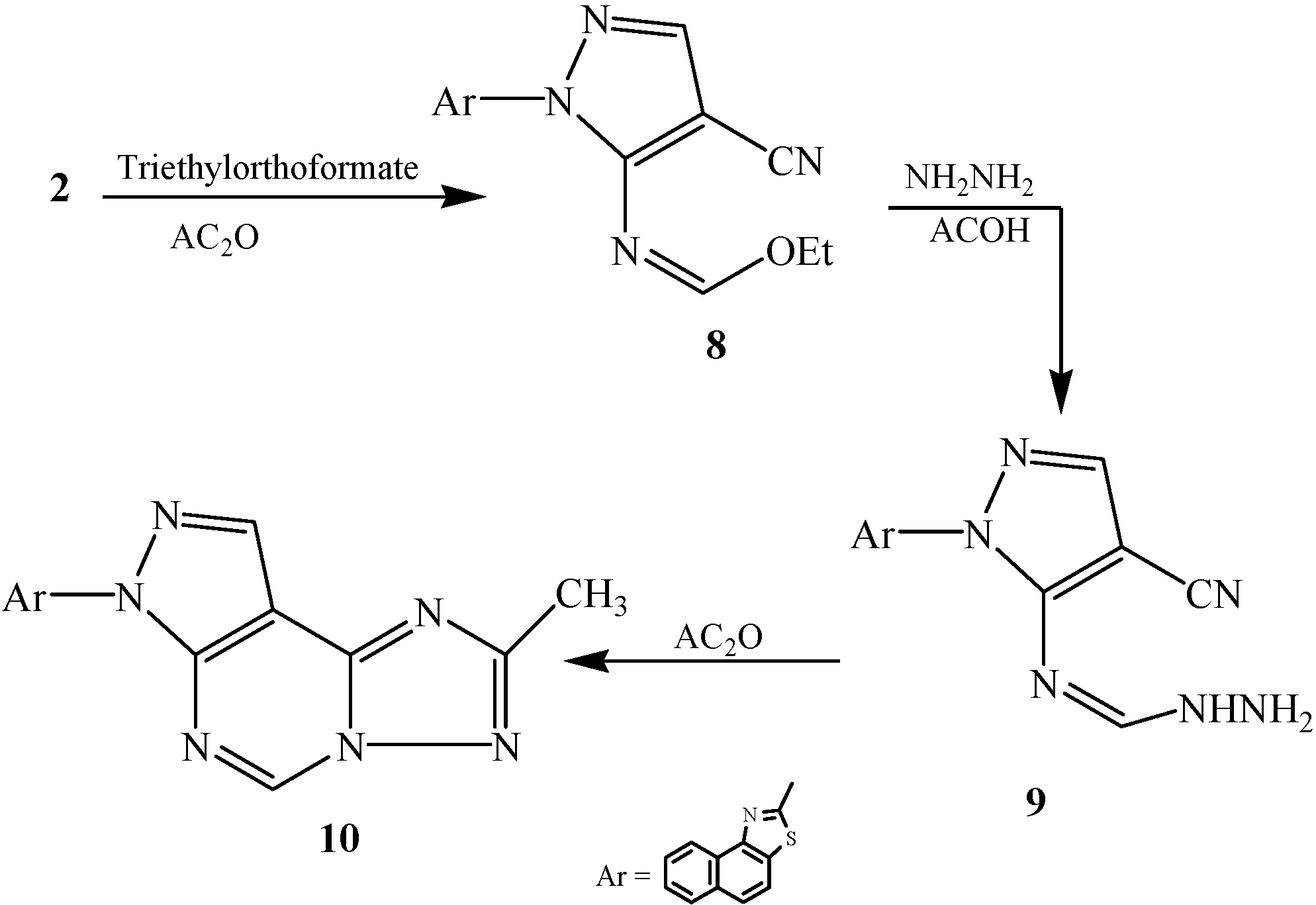
2.2. Pharmacological Evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 (µM) |
|---|---|
| Bicalutamide | 0.89 |
| 1 | Inactive |
| 2 | 0.031 |
| 3 | 0.037 |
| 4 | 0.034 |
| 5 | 0.041 |
| 6 | 0.044 |
| 7 | 0.057 |
| 8 | 0.077 |
| 9 | 0.075 |
| 10 | 0.023 |
| 11 | 0.039 |
| 12 | 0.091 |
| 13 | 0.029 |
| Compound No. | % (Inhibition) | ED50 (mg/kg) |
|---|---|---|
| Bicalutamide | 75 | 1.6 |
| 1 | Inactive | Inactive |
| 2 | 95.49 | 0.049 |
| 3 | 91 | 0.063 |
| 4 | 93.98 | 0.051 |
| 5 | 89.09 | 0.089 |
| 6 | 88.76 | 0.092 |
| 7 | 87.56 | 0.094 |
| 8 | 85.43 | 0.097 |
| 9 | 86.56 | 0.095 |
| 10 | 98.87 | 0.071 |
| 11 | 90.01 | 0.045 |
| 12 | 84.65 | 0.098 |
| 13 | 97.56 | 0.047 |
| Compound No. | IC50 (nM) PC-3 | IC50 (nM) LNCaP |
|---|---|---|
| Bicalutamide | 822 | 619.00 |
| 1 | Inactive | Inactive |
| 2 | 156.78 | 51.47 |
| 3 | 177.45 | 58.58 |
| 4 | 178.6 | 56.59 |
| 5 | 190.1 | 64.28 |
| 6 | 201.23 | 72.39 |
| 7 | 256.78 | 74.56 |
| 8 | 278.67 | 78.54 |
| 9 | 268.77 | 76.65 |
| 10 | 109.29 | 43.54 |
| 11 | 180.01 | 61.58 |
| 12 | 288.78 | 79.98 |
| 13 | 145.67 | 49.65 |
2.3. Determination of Acute Toxicity (Lethal Dose 50 (LD50))
| Compound No. | LD50 (mg/kg) |
|---|---|
| 1 | 1164.45 |
| 2 | 163.46 |
| 3 | 174.56 |
| 4 | 265.67 |
| 5 | 266.58 |
| 6 | 257.79 |
| 7 | 146.80 |
| 8 | 235.99 |
| 9 | 124.89 |
| 10 | 315.78 |
| 11 | 324.57 |
| 12 | 234.46 |
| 13 | 443.35 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Synthetic Procedures
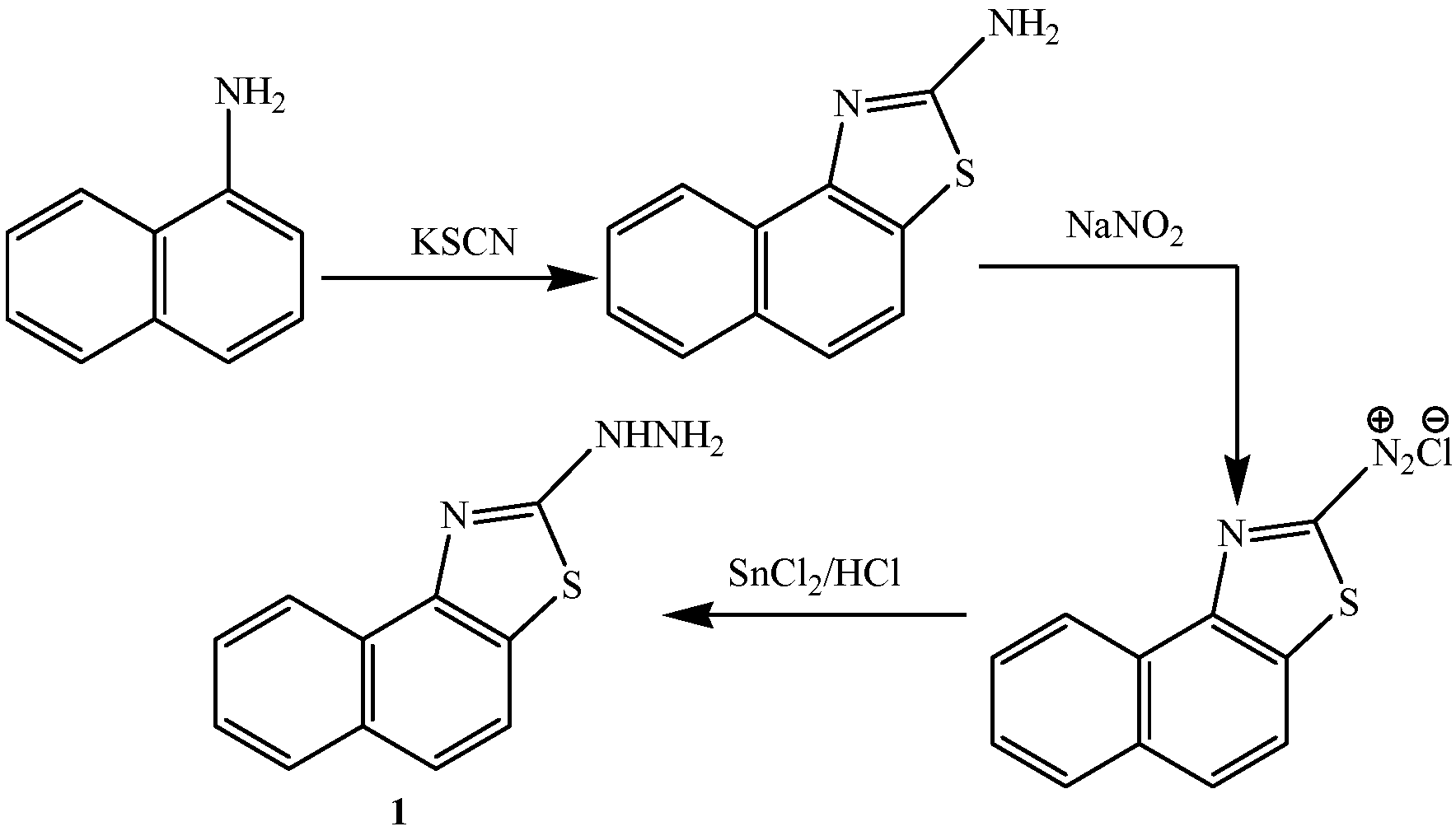
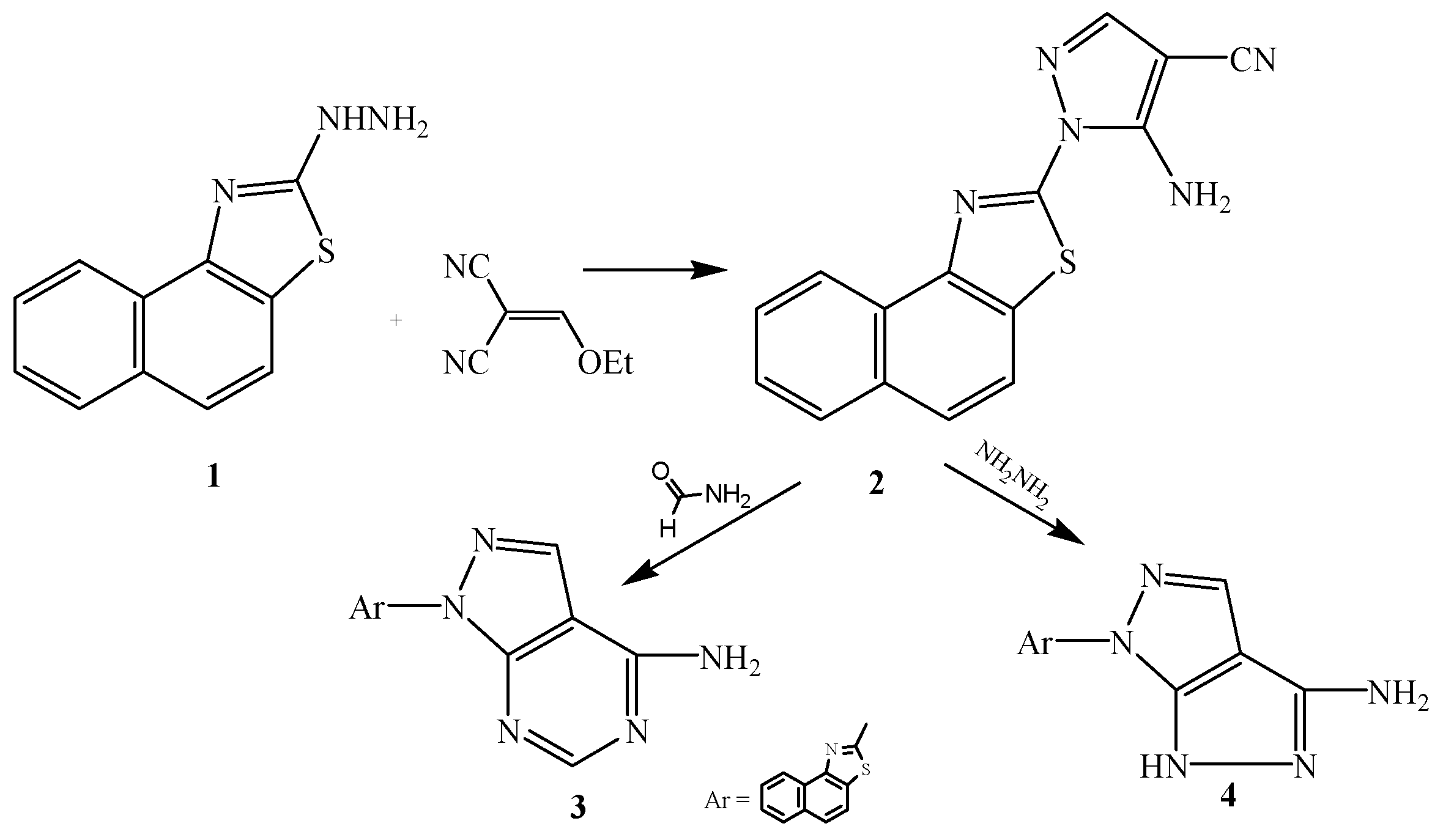
3.2.1. Synthesis of 2-[3-Amino-4-cyanopyrazol-2-yl]naphthalino[1,2-d]thiazole (2)
3.2.2. Synthesis of 2-[6-Aminopyrimidino[4,5-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (3)
3.2.3. Synthesis of 2-[5-Amino-3H-pyrazolo[3,4-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (4)
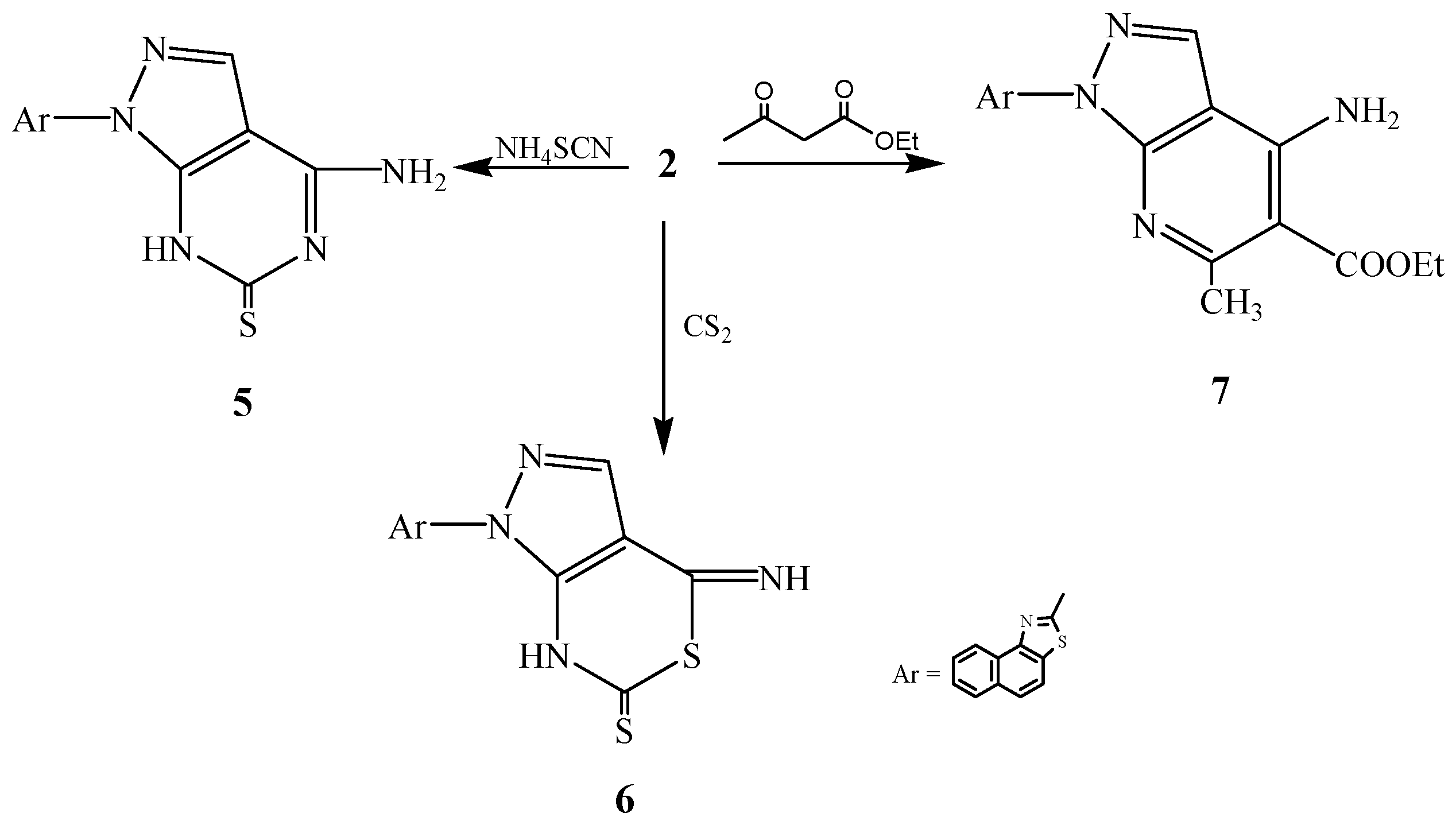
3.2.4. Synthesis of 2-[6-Amino-3H-4-thiopyrimidino[4,5-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (5)
3.2.5. Synthesis of 2-[6-Imino-3H-4-thiothiazino[4,5-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (6)
3.2.6. Synthesis of 2-[6-Amino-5-ethoxycarbonyl-4-methylpyridino[2,3-c]pyrazol-2-yl]naphthalino-[1,2-d]thiazole (7)
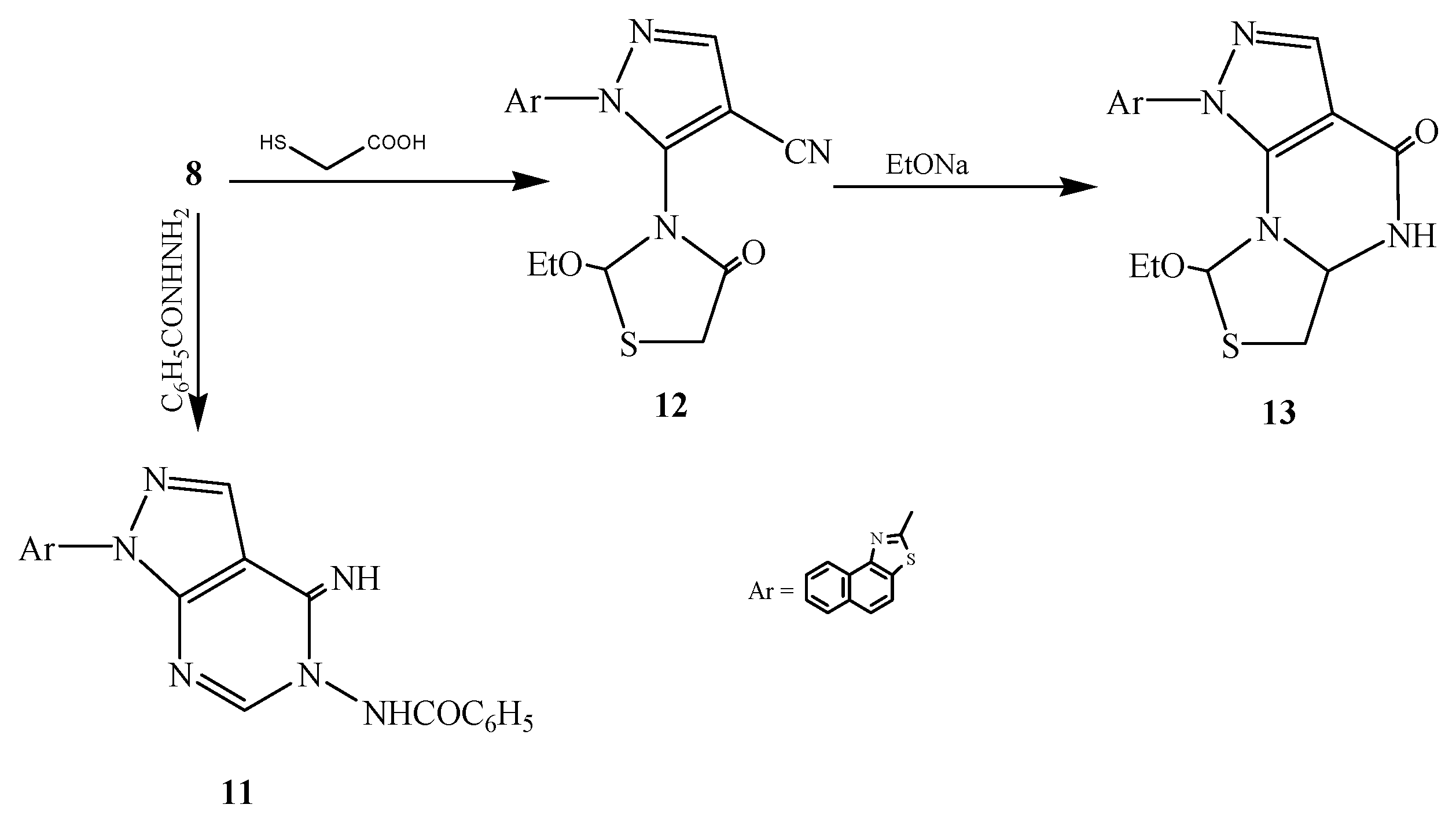
3.2.7. Synthesis of 2-[3-Ethylimidoformat-4-cyanopyrazol-2-yl]naphthalino[1,2-d]thiazole (8)
3.2.8. Synthesis of 2-[3-Imidoformic hydrazido-4-cyanopyrazol-2-yl]naphthalino[1,2-d]thiazole (9)
3.2.9. Synthesis of 2-[6-Methyl[1,2,4]triazolo[2,3-c]pyrimidino[6,5-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (10)
3.2.10. Synthesis of 2-[7-Benzamido-6-iminopyrimidino[4,5-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (11)
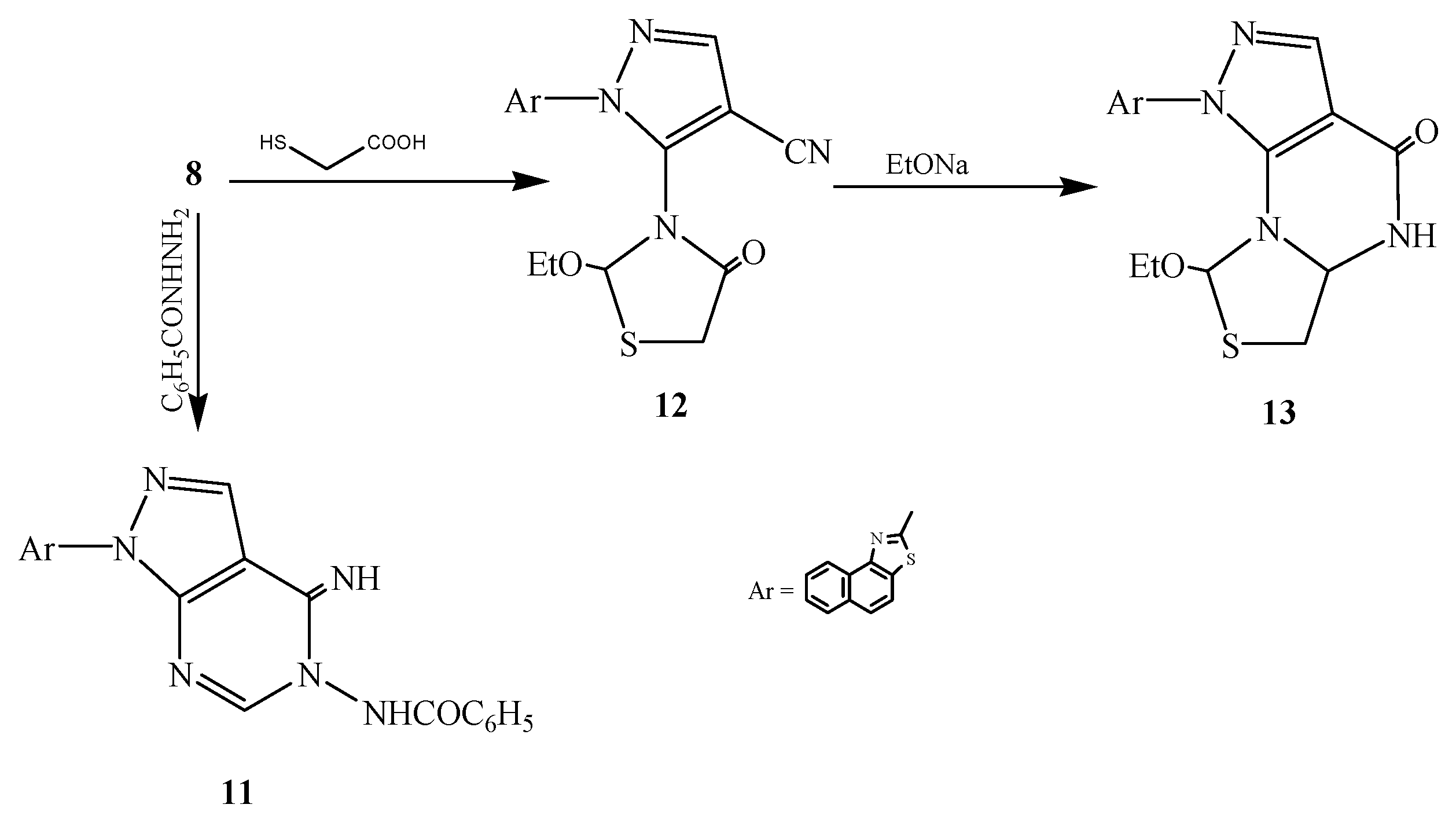
3.2.11. Synthesis of 2-[3-(2-Ethoxy-4-oxo-5-dihydro-1,3-thiazol-3-yl)-4-cyanopyrazol-2-yl]-naphthalino[1,2-d]thiazole (12)
3.2.12. Synthesis of 2-[3-Ethoxy-5-dihydro-6H-7-oxo-[1,3]thiazolo[3,4-b]pyrimidino[4,5-c]pyrazol-2-yl]naphthalino[1,2-d]thiazole (13)
3.3. Pharmacological Evaluation
3.3.1. Animals
3.3.2. Evaluation of Transcriptional Activity for Human Androgen Receptor
Establishment of Chinese Hamster Cell Model
Activities of the Tested Compounds to Inhibit Androgen Receptor Mediated Transcription Induced by Dihydrotestosterone (DHT) (AR Antagonistic Activity)
3.3.3. In Vivo Evaluation of Anti-Androgenic Activities in Castrated Immature Rats
3.3.4. Anti-Prostate Cancer Screening Anti-Androgenic Bioassay in Human Prostate Cancer Cells
4. Structure Activity Relationship (SAR)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Akbari, J.B.; Mehta, K.B.; Pathak, S.J.; Joshi, H.S. Synthesis and antimicrobial activity of some new pyrazolo[3,4-d]pyrimidines and thiazolo[4,5-d]pyrimidines. Indian J. Chem. 2008, 47, 477–480. [Google Scholar]
- Habib, N.S.; Soliman, R.; El-Tombary, A.A.; El-Hawash, S.A.; Shaaban, O.G. Synthesis of thiazolo[4,5-d]pyrimidine derivatives as potential antimicrobial agents. Arch. Pharm. Res. 2007, 30, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Walters, I.; Austin, C.; Austin, R.; Bonnert, R.; Cage, P.; Christie, M.; Ebden, M.; Gardiner, S.; Grahames, C.; Hill, S.; et al. Evaluation of a series of bicyclic CXCR2 antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Revankar, G.R.; Ojwang, J.O.; Mustain, S.D.; Rando, R.F.; de Clercq, E.; Huffman, J.H.; Drach, J.C.; Sommadossi, J.P.; Lewis, A.F. Thiazolo[4,5-d]pyrimidines. Part II. Synthesis and anti-human cytomegalovirus activity in vitro of certain acyclonucleosides and acyclonucleotides derived from the guanine analogue 5-aminothiazolo [4,5-d] pyrimidine-2,7(3H,6H)-dione. Antivir. Chem. Chemother. 1998, 9, 53–63. [Google Scholar] [PubMed]
- Beck, J.P.; Curry, M.A.; Chorvat, R.J.; Fitzgerald, L.W.; Gilligan, P.J.; Zaczek, R.; Trainor, G.L. Thiazolo[4,5-d]pyrimidine thiones and -ones as corticotropin-releasing hormone (CRH-R1) receptor antagonists. Bioorg. Med. Chem. Lett. 1999, 9, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Rida, S.M.; Habib, N.S.; Badawey, E.A.; Fahmy, H.T.; Ghozlan, H.A. Synthesis of novel thiazolo[4,5-d]pyrimidine derivatives for antimicrobial, anti-HIV and anticancer investigation. Pharmazie 1996, 51, 927–931. [Google Scholar] [PubMed]
- Fahmy, H.T.; Rostom, S.A.; Bekhit, A.A. Synthesis and antitumor evaluation of new polysubstituted thiazole and derived thiazolo[4,5-d]pyrimidine systems. Arch. Pharm. 2002, 335, 213–222. [Google Scholar] [CrossRef]
- Hammam, A.G.; Fahmy, A.F.M.; Amr, A.E.; Mohamed, A.M. Synthesis of novel tricyclicheterocyclic compounds as potential anticancer agents using chromanone and thiochromanoneas synthons. Indian J. Chem. 2003, 42, 1985–1993. [Google Scholar]
- Amr, A.G.; Mohamed, A.M.; Mohamed, S.F.; Abdel-Hafez, N.A.; Hammam, A.G. Anticancer activities of some newly synthesized pyridine, pyrane, and pyrimidine derivatives. Bioorg. Med. Chem. 2006, 14, 5481–5488. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ghalia, M.H.; Amr, A.E. Synthesis and investigation of a new cyclo-(Nα-dipicolinoyl)pentapeptide of a breast and CNS cytotoxic activity and an ionophoric specifity. Amino Acids 2004, 26, 283–289. [Google Scholar] [PubMed]
- Abd El-Salam, O.I.; Fahmy, A.F.M.; Mohamed, A.M.; Elnaggar, D.H.; Hammam, A.G. Synthesis, anticancer and anti-inflammatory activities of 3,4-dihydro-7-nitrobenzo[b]oxepin-5(2H)-one and its related derivatives. World J. Chem. 2010, 5, 7–17. [Google Scholar]
- Borgio, J.F.; Bency, B.J.; Thorat, P.K.; Lonkar, A.D. Gynandropsis pentaphylla DC extracts on the production of microbial proteins. Am. J. Drug Discov. Dev. 2011, 1, 129–136. [Google Scholar] [CrossRef]
- Matloobi, M.; Kappe, C.O. Microwave-assisted solution- and solid-phase synthesis of 2-amino-4-arylpyrimidine derivatives. J. Comb. Chem. 2007, 9, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Chengguo, L.V.; Lei, M.; Yan, S. Study on the diurnal changes of net photosynthetic rate and the impact factors of Stevia rebaudiana bertoni in autumn. Am. J. Plant Physiol. 2009, 4, 18–23. [Google Scholar] [CrossRef]
- Gadhachanda, V.R.; Wu, B.; Wang, Z.; Kuhen, K.L.; Caldwell, J.; Zondler, H.; Walter, H.; Havenhand, M.; He, Y. 4-Aminopyrimidines as novel HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Masroor, M.; Khan, A.; Morris, J.B. Agrobotanical attributes, nitrogen-fixation, enzyme activities and nutraceuticals of hyacinth bean (Lablab purpureus L.): A bio-functional medicinal legume. Am. J. Plant Physiol. 2009, 4, 58–69. [Google Scholar]
- Negaoui, H.; Hanane, K.; Omar, K.; Djamel, S. A model of intestinal anaphylaxis in whey sensitized Balb/c mice. Am. J. Immunol. 2009, 5, 56–60. [Google Scholar] [CrossRef]
- Khan, M.S.H.; Wagatsuma, T.; Akhter, A.; Tawaraya, K. Sterol biosynthesis inhibition by paclobutrazol induces greater aluminum (Al) sensitivity in Al-tolerant rice. Am. J. Plant Physiol. 2009, 4, 89–99. [Google Scholar] [CrossRef]
- Chang, L.C.; Spanjersberg, R.F.; von Frijtag Drabbe Künzel, J.K.; Mulder-Krieger, T.; van den Hout, G.; Beukers, M.W.; Brussee, J.; Ijzerman, A.P. 2,4,6-Trisubstituted pyrimidines as a new class of selective adenosine A1 receptor antagonists. J. Med. Chem. 2004, 47, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, S.; Shariati, S. Effect of trehalose Arabidopsis thaliana L. on Huntington’s disease. Am. J. Plant Physiol. 2010, 5, 1–6. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Hossain, M.A.; Fujita, M. Physiological and biochemical mechanisms of nitric oxide induced abiotic stress tolerance in plants. Am. J. Plant Physiol. 2010, 5, 295–324. [Google Scholar] [CrossRef]
- Ozeki, K.; Ichikawa, T.; Takehara, H.; Tanimura, K.; Sato, M.; Yaginuma, H. Studies on antiallergy agents. III. Synthesis of 2-anilino-1,6-dihydro-6-oxo-5-pyrimidinecarboxylic acids and related compounds. Chem. Pharm. Bull. 1989, 37, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Dahmardeh, M. Effect of plant density and nitrogen rate on PAR absorption and maize yield. Am. J. Plant. Physiol. 2011, 6, 44–49. [Google Scholar] [CrossRef]
- Robin, A.; Julienne, K.; Meslin, J.C.; Deniaud, D. Synthesis of pyridone and pyridine rings by [4 + 2] heterocyclocondensation. Tetrahedron Lett. 2004, 45, 9557–9559. [Google Scholar] [CrossRef]
- Li, A.H.; Moro, S.; Forsyth, N.; Melman, N.; Ji, X.D.; Jacobson, K.A. Synthesis, CoMFA analysis, and receptor docking of 3,5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J. Med. Chem. 1999, 42, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Vacher, B.; Bonnaud, B.; Funes, P.; Jubault, N.; Koek, W.; Assié, M.B.; Cosi, C.; Kleven, M. Novel derivatives of 2-pyridinemethylamine as selective, potent, and orally active agonists at 5-HT1A receptors. J. Med. Chem. 1999, 42, 1648–1660. [Google Scholar] [CrossRef] [PubMed]
- Nasratun, M.; Said, H.A.; Noraziah, A.; Alla, A.N.A. Immobilization of lipase from Candida rugosa on chitosan beads for transesterification reaction. Am. J. Appl. Sci. 2009, 6, 1653–1657. [Google Scholar] [CrossRef]
- Fayed, A.A.; Ghanem, S. Anti-microbial evaluation of new thiazolo pyrimidine derivatives. Eur. Sci. J. 2013, 9, 1857–7881. [Google Scholar]
- Austen, K.F.; Brocklehurst, W.E. Anaphylaxis in chopped guinea pig lung. I. Effect of peptidase substrates and inhibitors. J. Exp. Med. 1961, 113, 521–539. [Google Scholar] [CrossRef] [PubMed]
- Kinoyama, I.; Taniguchi, N.; Kawaminami, E.; Nozawa, E.; Koutoku, H.; Furutani, T.; Kudoh, M.; Okada, M. N-Arylpiperazine-1-carboxamide derivatives: A novel series of orally active nonsteroidal androgen receptor antagonists. Chem. Pharm. Bull. 2005, 53, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Shi, Q.; Nyarko, A.K.; Bastow, K.F.; Wu, C.C.; Su, C.Y.; Shih, C.C.; Lee, K.H. Antitumor agents. 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents. J. Med. Chem. 2006, 49, 3963–3972. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahashwan, S.A.; Fayed, A.A.; Ramadan, M.A.; Amr, A.E.-G.E.; Al-Harbi, N.O. Androgen Receptor Antagonists and Anti-Prostate Cancer Activities of Some Newly Synthesized Substituted Fused Pyrazolo-, Triazolo- and Thiazolo-Pyrimidine Derivatives. Int. J. Mol. Sci. 2014, 15, 21587-21602. https://doi.org/10.3390/ijms151121587
Bahashwan SA, Fayed AA, Ramadan MA, Amr AE-GE, Al-Harbi NO. Androgen Receptor Antagonists and Anti-Prostate Cancer Activities of Some Newly Synthesized Substituted Fused Pyrazolo-, Triazolo- and Thiazolo-Pyrimidine Derivatives. International Journal of Molecular Sciences. 2014; 15(11):21587-21602. https://doi.org/10.3390/ijms151121587
Chicago/Turabian StyleBahashwan, Saleh A., Ahmed A. Fayed, Mohamed A. Ramadan, Abd El-Galil E. Amr, and Naif O. Al-Harbi. 2014. "Androgen Receptor Antagonists and Anti-Prostate Cancer Activities of Some Newly Synthesized Substituted Fused Pyrazolo-, Triazolo- and Thiazolo-Pyrimidine Derivatives" International Journal of Molecular Sciences 15, no. 11: 21587-21602. https://doi.org/10.3390/ijms151121587