Stem Cells: The Pursuit of Genomic Stability

Abstract
:1. Introduction
2. DNA Damage and Repair Status during Reprogramming
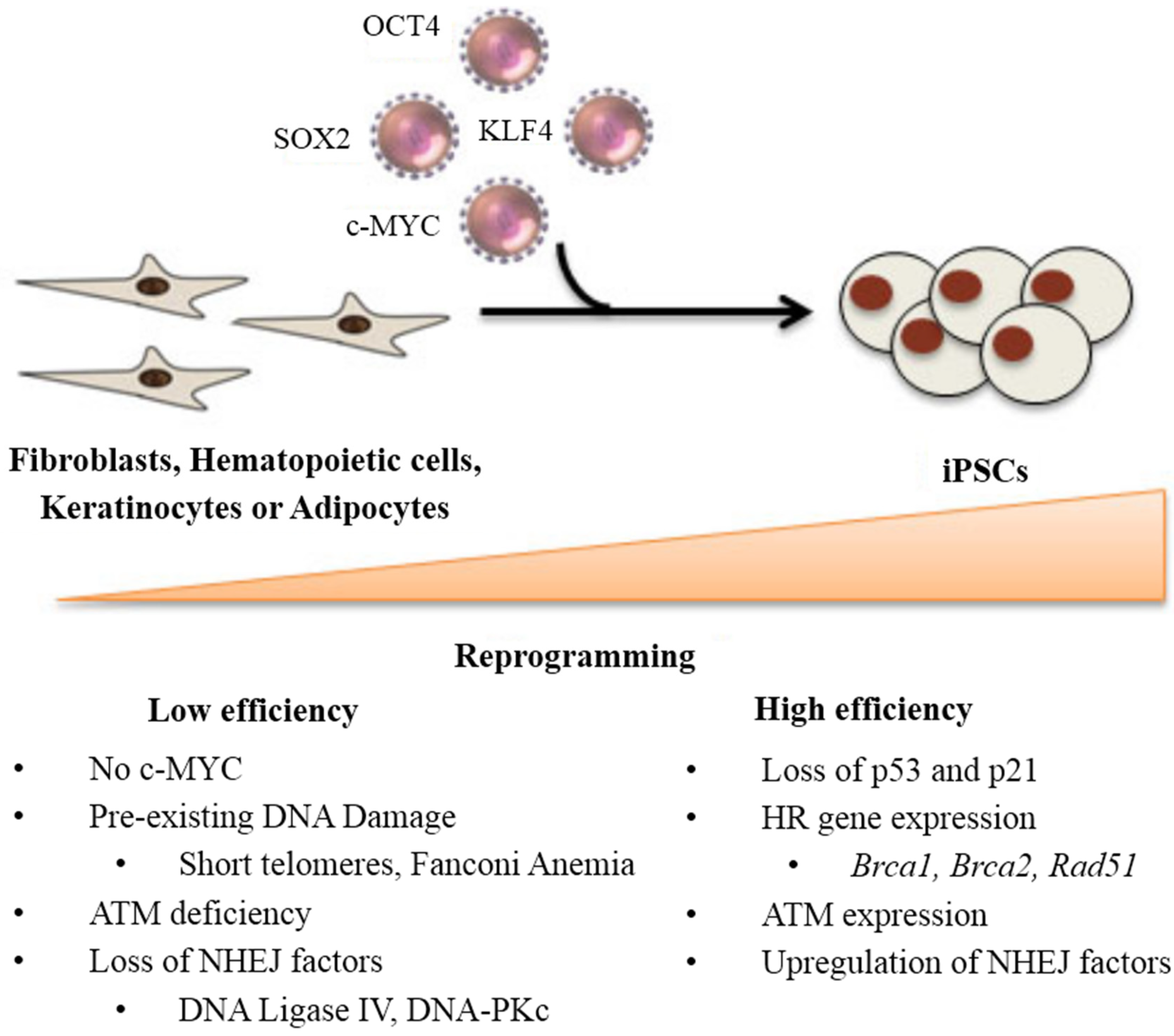
3. Stem Cell Response to DNA Damage

{kind=link}
{kind=link}
| Stem Cell Type | Single-Strand Breaks | Double-Strand Breaks | Apoptosis Sensitivity | References | |
|---|---|---|---|---|---|
| HR | NHEJ | ||||
| Pluripotent | |||||
| Human ESCs, iPSCs | ++ | +++ | ++ | +++ | [46,47,48,49,50,51,52,53,54,55,56,57,58] |
| Mouse ESCs, iPSCs | * | +++ | − | +++ | [59,60] |
| Multipotent | |||||
| Neural stem cells | * | +++ | ++ | ++ | [57,61,62,63,64,65] |
| Mesenchymal stem cells | * | +++ | +++ | − | [63,66,67] |
| Hematopoietic stem cells | ++ | * | ++ | +++ | [68,69,70] |
3.1. Pluripotent Stem Cells
3.2. Multipotent Stem Cells
3.3. Summary of Single-Strand (ss-) and Double-Strand (ds-) Breaks Repair Strategies
4. Apoptotic Susceptibility in iPSC Therapeutic Development
5. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martins-Taylor, K.; Xu, R.H. Concise review: Genomic stability of human induced pluripotent stem cells. Stem Cells 2012, 30, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Nagaria, P.; Robert, C.; Rassool, F.V. DNA double-strand break response in stem cells: Mechanisms to maintain genomic integrity. Biochim. Biophys. Acta 2013, 1830, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Eminli, S.; Foudi, A.; Stadtfeld, M.; Maherali, N.; Ahfeldt, T.; Mostoslavsky, G.; Hock, H.; Hochedlinger, K. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 2009, 41, 968–976. [Google Scholar] [CrossRef]
- Wang, J.; Gu, Q.; Hao, J.; Bai, D.; Liu, L.; Zhao, X.; Liu, Z.; Wang, L.; Zhou, Q. Generation of induced pluripotent stem cells with high efficiency from human umbilical cord blood mononuclear cells. Genomics Proteomics Bioinform. 2013, 11, 304–311. [Google Scholar] [CrossRef]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Sun, N.; Panetta, N.J.; Gupta, D.M.; Wilson, K.D.; Lee, A.; Jia, F.; Hu, S.; Cherry, A.M.; Robbins, R.C.; Longaker, M.T.; et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad Sci. USA 2009, 106, 15720–15725. [Google Scholar] [CrossRef]
- Jaenisch, R.; Young, R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008, 132, 567–582. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar] [PubMed]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; de Pinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar] [CrossRef]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisua Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef]
- Kinoshita, T.; Nagamatsu, G.; Kosaka, T.; Takubo, K.; Hotta, A.; Ellis, J.; Suda, T. Ataxia-telangiectasia mutated (ATM) deficiency decreases reprogramming efficiency and leads to genomic instability in iPS cells. Biochem. Biophys. Res. Commun. 2011, 407, 321–326. [Google Scholar] [CrossRef]
- Lee, P.; Martin, N.T.; Nakamura, K.; Azghadi, S.; Amiri, M.; Ben-David, U.; Perlman, S.; Gatti, R.A.; Hu, H.; Lowry, W.E. SMRT compounds abrogate cellular phenotypes of ataxia telangiectasia in neural derivatives of patient-specific hiPSCs. Nat. Commun. 2013, 4, 1824. [Google Scholar] [CrossRef]
- Nayler, S.; Gatei, M.; Kozlov, S.; Gatti, R.; Mar, J.C.; Wells, C.A.; Lavin, M.; Wolvetang, E. Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl. Med. 2012, 1, 523–535. [Google Scholar] [CrossRef]
- Fukawatase, Y.; Toyoda, M.; Okamura, K.; Nakamura, K.; Nakabayashi, K.; Takada, S.; Yamazaki-Inoue, M.; Masuda, A.; Nasu, M.; Hata, K.; et al. Ataxia telangiectasia derived iPS cells show preserved X-ray sensitivity and decreased chromosomal instability. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- Wyles, S.P.; Faustino, R.S.; Li, X.; Terzic, A.; Nelson, T.J. Systems-based technologies in profiling the stem cell molecular framework for cardioregenerative medicine. Stem Cell Rev. 2014, 1–10. [Google Scholar]
- Gonzalez, F.; Georgieva, D.; Vanoli, F.; Shi, Z.D.; Stadtfeld, M.; Ludwig, T.; Jasin, M.; Huangfu, D. Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep. 2013, 3, 651–660. [Google Scholar] [CrossRef]
- Soyombo, A.A.; Wu, Y.; Kolski, L.; Rios, J.J.; Rakheja, D.; Chen, A.; Kehler, J.; Hampel, H.; Coughran, A.; Ross, T.S. Analysis of induced pluripotent stem cells from a BRCA1 mutant family. Stem Cell Rep. 2013, 1, 336–349. [Google Scholar] [CrossRef]
- Tilgner, K.; Neganova, I.; Moreno-Gimeno, I.; Al-Aama, J.Y.; Burks, D.; Yung, S.; Singhapol, C.; Saretzki, G.; Evans, J.; Gorbunova, V.; et al. A human iPSC model of ligase IV deficiency reveals an important role for NHEJ-mediated-DSB repair in the survival and genomic stability of induced pluripotent stem cells and emerging haematopoietic progenitors. Cell Death Differ. 2013, 20, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef]
- Felgentreff, K.; Du, L.; Weinacht, K.G.; Dobbs, K.; Bartish, M.; Giliani, S.; Schlaeger, T.; DeVine, A.; Schambach, A.; Woodbine, L.J.; et al. Differential role of nonhomologous end joining factors in the generation, DNA damage response, and myeloid differentiation of human induced pluripotent stem cells. Proc. Natl. Acad Sci. USA 2014, 111, 8889–8894. [Google Scholar] [CrossRef]
- Salomoni, P. Reprogramming and genome integrity: Role of non-homologous end joining. Cell Death Differ. 2013, 20, 1285–1286. [Google Scholar] [CrossRef]
- Molina-Estevez, F.J.; Lozano, M.L.; Navarro, S.; Torres, Y.; Grabundzija, I.; Ivics, Z.; Samper, E.; Bueren, J.A.; Guenechea, G. Brief report: Impaired cell reprogramming in nonhomologous end joining deficient cells. Stem Cells 2013, 31, 1726–1730. [Google Scholar] [CrossRef]
- Muller, L.U.; Milsom, M.D.; Harris, C.E.; Vyas, R.; Brumme, K.M.; Parmar, K.; Moreau, L.A.; Schambach, A.; Park, I.H.; London, W.B.; et al. Overcoming reprogramming resistance of Fanconi anemia cells. Blood 2012, 119, 5449–5457. [Google Scholar] [CrossRef]
- Sakurai, Y.; Komatsu, K.; Agematsu, K.; Matsuoka, M. DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirology 2009, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kim, D.; Kim, C.H.; Mills, R.E.; Chang, M.Y.; Iskow, R.C.; Ko, S.; Moon, J.I.; Choi, H.W.; Yoo, P.S.M.; et al. Increased genomic integrity of an improved protein-based mouse induced pluripotent stem cell method compared with current viral-induced strategies. Stem Cells Transl. Med. 2014, 3, 599–609. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef]
- Bru, T.; Clarke, C.; McGrew, M.J.; Sang, H.M.; Wilmut, I.; Blow, J.J. Rapid induction of pluripotency genes after exposure of human somatic cells to mouse ES cell extracts. Exp. Cell Res. 2008, 314, 2634–2642. [Google Scholar] [CrossRef]
- Yakubov, E.; Rechavi, G.; Rozenblatt, S.; Givol, D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 2010, 394, 189–193. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef]
- Lee, M.R.; Prasain, N.; Chae, H.D.; Kim, Y.J.; Mantel, C.; Yoder, M.C.; Broxmeyer, H.E. Epigenetic regulation of NANOG by miR-302 cluster-MBD2 completes induced pluripotent stem cell reprogramming. Stem Cells 2013, 31, 666–681. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Fong, Y.W.; Inouye, C.; Yamaguchi, T.; Cattoglio, C.; Grubisic, I.; Tjian, R. A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell 2011, 147, 120–131. [Google Scholar] [CrossRef]
- Jiang, J.; Lv, W.; Ye, X.; Wang, L.; Zhang, M.; Yang, H.; Okuka, M.; Zhou, C.; Zhang, X.; Liu, L.; et al. Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of iPS cells as demonstrated by tetraploid complementation. Cell Res. 2013, 23, 92–106. [Google Scholar] [CrossRef]
- Hirata, T.; Amano, T.; Nakatake, Y.; Amano, M.; Piao, Y.; Hoang, H.G.; Ko, M.S. Zscan4 transiently reactivates early embryonic genes during the generation of induced pluripotent stem cells. Sci. Rep. 2012, 2, 208. [Google Scholar]
- Tapia, N.; Scholer, H.R. p53 connects tumorigenesis and reprogramming to pluripotency. J. Exp. Med. 2010, 207, 2045–2048. [Google Scholar] [CrossRef]
- Maynard, S.; Swistowska, A.M.; Lee, J.W.; Liu, Y.; Liu, S.T.; Da Cruz, A.B.; Rao, M.; de Souza-Pinto, N.C.; Zeng, X.; Bohr, V.A. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells 2008, 26, 2266–2274. [Google Scholar] [CrossRef]
- Filion, T.M.; Qiao, M.; Ghule, P.N.; Mandeville, M.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Altieri, D.C.; Stein, G.S. Survival responses of human embryonic stem cells to DNA damage. J. Cell Physiol. 2009, 220, 586–592. [Google Scholar] [CrossRef]
- Bogomazova, A.N.; Lagarkova, M.A.; Tskhovrebova, L.V.; Shutova, M.V.; Kiselev, S.L. Error-prone nonhomologous end joining repair operates in human pluripotent stem cells during late G2. Aging (Albany NY) 2011, 3, 584–596. [Google Scholar]
- Adams, B.R.; Golding, S.E.; Rao, R.R.; Valerie, K. Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One 2010, 5, e10001. [Google Scholar] [CrossRef]
- Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. ATM-independent, high-fidelity nonhomologous end joining predominates in human embryonic stem cells. Aging (Albany NY) 2010, 2, 582–596. [Google Scholar]
- Serrano, L.; Liang, L.; Chang, Y.; Deng, L.; Maulion, C.; Nguyen, S.; Tischfield, J.A. Homologous recombination conserves DNA sequence integrity throughout the cell cycle in embryonic stem cells. Stem Cells Dev. 2011, 20, 363–374. [Google Scholar] [CrossRef]
- Fan, J.; Robert, C.; Jang, Y.Y.; Liu, H.; Sharkis, S.; Baylin, S.B.; Rassool, F.V. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat. Res. 2011, 713, 8–17. [Google Scholar] [CrossRef]
- Fung, H.; Weinstock, D.M. Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS One 2011, 6, e20514. [Google Scholar] [CrossRef]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human embryonic stem cells fail to activate CHK1 and commit to apoptosis in response to DNA replication stress. Stem Cells 2012, 30, 1385–1393. [Google Scholar] [CrossRef]
- Luo, L.Z.; Gopalakrishna-Pillai, S.; Nay, S.L.; Park, S.W.; Bates, S.E.; Zeng, X.; Iverson, L.E.; O’Connor, T.R. DNA repair in human pluripotent stem cells is distinct from that in non-pluripotent human cells. PLoS One 2012, 7, e30541. [Google Scholar] [CrossRef]
- Hyka-Nouspikel, N.; Desmarais, J.; Gokhale, P.J.; Jones, M.; Meuth, M.; Andrews, P.W.; Nouspikel, T. Deficient DNA damage response and cell cycle checkpoints lead to accumulation of point mutations in human embryonic stem cells. Stem Cells 2012, 30, 1901–1910. [Google Scholar] [CrossRef]
- Lan, M.L.; Acharya, M.M.; Tran, K.K.; Bahari-Kashani, J.; Patel, N.H.; Strnadel, J.; Giedzinski, E.; Limoli, C.L. Characterizing the radioresponse of pluripotent and multipotent human stem cells. PLoS One 2012, 7, e50048. [Google Scholar] [CrossRef]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; von Zglinicki, T.; Lako, M. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef]
- Tichy, E.D.; Pillai, R.; Deng, L.; Liang, L.; Tischfield, J.; Schwemberger, S.J.; Babcock, G.F.; Stambrook, P.J. Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem Cells Dev. 2010, 19, 1699–1711. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Armstrong, L.; Leake, A.; Lako, M.; von Zglinicki, T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004, 22, 962–971. [Google Scholar] [CrossRef]
- Schneider, L.; Fumagalli, M.; d’Adda di Fagagna, F. Terminally differentiated astrocytes lack DNA damage response signaling and are radioresistant but retain DNA repair proficiency. Cell Death Differ. 2012, 426, 582–591. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, N.; Ellerby, L.M.; Davalos, A.R.; Zeng, X.; Campisi, J.; Desprez, P.Y. Responses of human embryonic stem cells and their differentiated progeny to ionizing radiation. Biochem. Biophys. Res. Commun. 2012, 426, 100–105. [Google Scholar] [CrossRef]
- Sokolov, M.; Neumann, R. Lessons learned about human stem cell responses to ionizing radiation exposures: A long road still ahead of us. Int. J. Mol. Sci. 2013, 14, 15695–15723. [Google Scholar] [CrossRef]
- Rousseau, L.; Etienne, O.; Roque, T.; Desmaze, C.; Haton, C.; Mouthon, M.A.; Bernardino-Sgherri, J.; Essers, J.; Kanaar, R.; Boussin, F.D. In vivo importance of homologous recombination DNA repair for mouse neural stem and progenitor cells. PLoS One 2012, 7, e37194. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L. Survival of neural stem cells undergoing DNA damage-induced astrocytic differentiation in self-renewal-promoting conditions in vitro. PLoS One 2014, 9, e87228. [Google Scholar] [CrossRef]
- Prendergast, A.M.; Cruet-Hennequart, S.; Shaw, G.; Barry, F.P.; Carty, M.P. Activation of DNA damage response pathways in human mesenchymal stem cells exposed to cisplatin or γ-irradiation. Cell Cycle 2011, 10, 3768–3777. [Google Scholar] [CrossRef]
- Oliver, L.; Hue, E.; Sery, Q.; Lafargue, A.; Pecqueur, C.; Paris, F.; Vallette, F.M. Differentiation-related response to DNA breaks in human mesenchymal stem cells. Stem Cells 2013, 31, 800–807. [Google Scholar] [CrossRef]
- Wang, J.; Sun, Q.; Morita, Y.; Jiang, H.; Gross, A.; Lechel, A.; Hildner, K.; Guachalla, L.M.; Gompf, A.; Hartmann, D.; et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012, 148, 1001–1014. [Google Scholar] [CrossRef]
- De Laval, B.; Pawlikowska, P.; Petit-Cocault, L.; Bilhou-Nabera, C.; Aubin-Houzelstein, G.; Souyri, M.; Pouzoulet, F.; Gaudry, M.; Porteu, F. Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell 2013, 12, 37–48. [Google Scholar]
- Cho, J.S.; Kook, S.H.; Robinson, A.R.; Niedernhofer, L.J.; Lee, B.C. Cell autonomous and nonautonomous mechanisms drive hematopoietic stem/progenitor cell loss in the absence of DNA repair. Stem Cells 2013, 31, 511–525. [Google Scholar]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef]
- Barta, T.; Vinarsky, V.; Holubcova, Z.; Dolezalova, D.; Verner, J.; Pospisilova, S.; Dvorak, P.; Hampl, A. Human embryonic stem cells are capable of executing G1/S checkpoint activation. Stem Cells 2010, 28, 1143–1152. [Google Scholar]
- Nouspikel, T. Genetic instability in human embryonic stem cells: Prospects and caveats. Future Oncol. 2013, 9, 867–877. [Google Scholar] [CrossRef]
- Lin, B.; Gupta, D.; Heinen, C.D. Human Pluripotent stem cells have a novel mismatch repair-dependent damage response. J. Biol. Chem. 2014, 289, 24314–24324. [Google Scholar] [CrossRef]
- Abbas, T.; Keaton, M.A.; Dutta, A. Genomic instability in cancer. Cold Spring Harb Perspect. Biol. 2013, 5, a012914. [Google Scholar]
- Grandela, C.; Pera, M.F.; Grimmond, S.M.; Kolle, G.; Wolvetang, E.J. p53 is required for etoposide-induced apoptosis of human embryonic stem cells. Stem Cell Res. 2007, 1, 116–128. [Google Scholar]
- Liu, J.C.; Guan, X.; Ryan, J.A.; Rivera, A.G.; Mock, C.; Agrawal, V.; Letai, A.; Lerou, P.H.; Lahav, G. High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 2013, 13, 483–491. [Google Scholar] [CrossRef]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef]
- D’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar]
- Latella, L.; Lukas, J.; Simone, C.; Puri, P.L.; Bartek, J. Differentiation-induced radioresistance in muscle cells. Mol. Cell Biol. 2004, 24, 6350–6361. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Acharya, M.M.; Lan, M.L.; Kan, V.H.; Patel, N.H.; Giedzinski, E.; Tseng, B.P.; Limoli, C.L. Consequences of ionizing radiation-induced damage in human neural stem cells. Free Radic. Biol. Med. 2010, 49, 1846–1855. [Google Scholar] [CrossRef]
- Nowak, E.; Etienne, O.; Millet, P.; Lages, C.S.; Mathieu, C.; Mouthon, M.A.; Boussin, F.D. Radiation-induced H2AX phosphorylation and neural precursor apoptosis in the developing brain of mice. Radiat. Res. 2006, 165, 155–164. [Google Scholar] [CrossRef]
- Herzog, K.H.; Chong, M.J.; Kapsetaki, M.; Morgan, J.I.; McKinnon, P.J. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 1998, 280, 1089–1091. [Google Scholar] [CrossRef]
- D’Sa-Eipper, C.; Leonard, J.R.; Putcha, G.; Zheng, T.S.; Flavell, R.A.; Rakic, P.; Kuida, K.; Roth, K.A. DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development 2001, 128, 137–146. [Google Scholar]
- Roque, T.; Haton, C.; Etienne, O.; Chicheportiche, A.; Rousseau, L.; Martin, L.; Mouthon, M.A.; Boussin, F.D. Lack of a p21waf1/cip-dependent G1/S checkpoint in neural stem and progenitor cells after DNA damage in vivo. Stem Cells 2012, 30, 537–547. [Google Scholar] [CrossRef]
- Aladjem, M.I.; Spike, B.T.; Rodewald, L.W.; Hope, T.J.; Klemm, M.; Jaenisch, R.; Wahl, G.M. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol. 1998, 8, 145–155. [Google Scholar] [CrossRef]
- Lee, Y.; McKinnon, P.J. Responding to DNA double strand breaks in the nervous system. Neuroscience 2007, 145, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Dahir, G.A.; Cui, Q.; Anderson, P.; Simon, C.; Joyner, C.; Triffitt, J.; Balian, G. Pluripotential mesenchymal cells repopulate bone marrow and retain osteogenic properties. Clin. Orthop. Relat. Res. 2000, 379, S134–S145. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Lin, C.T.; Chen, W.C.; Yang, C.T.; Chen, C.C.; Liao, S.K.; Liu, J.M.; Lu, C.H.; Lee, K.D. The sensitivity of human mesenchymal stem cells to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 244–253. [Google Scholar] [CrossRef]
- Mueller, L.P.; Luetzkendorf, J.; Mueller, T.; Reichelt, K.; Simon, H.; Schmoll, H. J. Presence of mesenchymal stem cells in human bone marrow after exposure to chemotherapy: Evidence of resistance to apoptosis induction. Stem Cells 2006, 24, 2753–2765. [Google Scholar] [CrossRef]
- Rando, T.A. Stem cells, ageing and the quest for immortality. Nature 2006, 441, 1080–1086. [Google Scholar] [CrossRef]
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713. [Google Scholar] [CrossRef]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Sahin, E.; Depinho, R.A. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 2010, 464, 520–528. [Google Scholar] [CrossRef]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef]
- Brimble, S.N.; Zeng, X.; Weiler, D.A.; Luo, Y.; Liu, Y.; Lyons, I.G.; Freed, W.; Robins, A.J.; Rao, M.S.; Schulz, T.C. Karyotypic stability, genotyping, differentiation, feeder-free maintenance, and gene expression sampling in three human embryonic stem cell lines derived prior to August 9, 2001. Stem Cells Dev. 2004, 13, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Arking, D.E.; Shivapurkar, N.; Ikeda, M.; Stastny, V.; Kassauei, K.; Sui, G.; Cutler, D.J.; Liu, Y.; Brimble, S.N.; et al. Genomic alterations in cultured human embryonic stem cells. Nat. Genet. 2005, 37, 1099–1103. [Google Scholar] [CrossRef]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef]
- Lefort, N.; Feyeux, M.; Bas, C.; Feraud, O.; Bennaceur-Griscelli, A.; Tachdjian, G.; Peschanski, M.; Perrier, A.L. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat. Biotechnol. 2008, 26, 1364–1366. [Google Scholar] [CrossRef]
- Martins-Taylor, K.; Nisler, B.S.; Taapken, S.M.; Compton, T.; Crandall, L.; Montgomery, K.D.; Lalande, M.; Xu, R.H. Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 488–491. [Google Scholar] [CrossRef]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, C.; Liu, H.; Sun, Y. Induced pluripotent stem cells are sensitive to DNA damage. Genomics Proteomics Bioinform. 2013, 11, 320–326. [Google Scholar] [CrossRef]
- Van Sloun, P.P.; Jansen, J.G.; Weeda, G.; Mullenders, L.H.; van Zeeland, A.A.; Lohman, P.H.; Vrieling, H. The role of nucleotide excision repair in protecting embryonic stem cells from genotoxic effects of UV-induced DNA damage. Nucleic Acids Res. 1999, 27, 3276–3282. [Google Scholar] [CrossRef]
- Roos, W.P.; Christmann, M.; Fraser, S.T.; Kaina, B. Mouse embryonic stem cells are hypersensitive to apoptosis triggered by the DNA damage O6-methylguanine due to high E2F1 regulated mismatch repair. Cell Death Differ. 2007, 14, 1422–1432. [Google Scholar] [CrossRef]
- Corbet, S.W.; Clarke, A.R.; Gledhill, S.; Wyllie, A.H. p53-dependent and -independent links between DNA-damage, apoptosis and mutation frequency in ES cells. Oncogene 1999, 18, 1537–1544. [Google Scholar] [CrossRef]
- Momcilovic, O.; Knobloch, L.; Fornsaglio, J.; Varum, S.; Easley, C.; Schatten, G. DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS One 2010, 5, e13410. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Nelson, T.J.; Kane, G.C.; Martinez-Fernandez, A.; Crespo-Diaz, R.J.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Induced pluripotent stem cell intervention rescues ventricular wall motion disparity, achieving biological cardiac resynchronization post-infarction. J. Physiol. 2013, 591, 4335–4349. [Google Scholar]
- Nelson, T.J.; Martinez-Fernandez, A.; Terzic, A. Induced pluripotent stem cells: Developmental biology to regenerative medicine. Nat. Rev. Cardiol. 2010, 7, 700–710. [Google Scholar]
- Iglesias-Garcia, O.; Pelacho, B.; Prosper, F. Induced pluripotent stem cells as a new strategy for cardiac regeneration and disease modeling. J. Mol. Cell Cardiol. 2013, 62, 43–50. [Google Scholar] [CrossRef]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H. Lessons from human teratomas to guide development of safe stem cell therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef]
- Lee, M.O.; Moon, S.H.; Jeong, H.C.; Yi, J.Y.; Lee, T.H.; Shim, S.H.; Rhee, Y.H.; Lee, S.H.; Oh, S.J.; Lee, M.Y.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad Sci. USA 2013, 110, E3281–E3290. [Google Scholar] [CrossRef]
- Ben-David, U.; Gan, Q.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef]
- Lim, T.T.; Geisen, C.; Hesse, M.; Fleischmann, B.K.; Zimmermann, K.; Pfeifer, A. Lentiviral vector mediated thymidine kinase expression in pluripotent stem cells enables removal of tumorigenic cells. PLoS One 2013, 8, e70543. [Google Scholar] [CrossRef]
- Tan, H.L.; Fong, W.J.; Lee, E.H.; Yap, M.; Choo, A. mAb 84, a cytotoxic antibody that kills undifferentiated human embryonic stem cells via oncosis. Stem Cells 2009, 27, 1792–1801. [Google Scholar] [CrossRef]
- Choo, A.B.; Tan, H.L.; Ang, S.N.; Fong, W.J.; Chin, A.; Lo, J.; Zheng, L.; Hentze, H.; Philp, R.J.; Oh, S.; et al. Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells 2008, 26, 1454–1463. [Google Scholar] [CrossRef]
- Smith, A.J.; Nelson, N.G.; Oommen, S.; Hartjes, K.A.; Folmes, C.D.; Terzic, A.; Nelson, T.J. Apoptotic susceptibility to DNA damage of pluripotent stem cells facilitates pharmacologic purging of teratoma risk. Stem Cells Transl. Med. 2012, 1, 709–718. [Google Scholar] [CrossRef]
- Ezoe, S. Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int. J. Environ. Res. Public Health 2012, 9, 2444–2453. [Google Scholar] [CrossRef]
- Wyles, S.P.; Yamada, S.; Oommen, S.; Maleszewski, J.J.; Beraldi, R.; Martinez-Fernandez, A.; Terzic, A.; Nelson, T.J. Inhibition of DNA topoisomerase II selectively reduces the threat of tumorigenicity following induced pluripotent stem cell-based myocardial therapy. Stem Cells Dev. 2014, 23, 2274–2282. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyles, S.P.; Brandt, E.B.; Nelson, T.J. Stem Cells: The Pursuit of Genomic Stability. Int. J. Mol. Sci. 2014, 15, 20948-20967. https://doi.org/10.3390/ijms151120948
Wyles SP, Brandt EB, Nelson TJ. Stem Cells: The Pursuit of Genomic Stability. International Journal of Molecular Sciences. 2014; 15(11):20948-20967. https://doi.org/10.3390/ijms151120948
Chicago/Turabian StyleWyles, Saranya P., Emma B. Brandt, and Timothy J. Nelson. 2014. "Stem Cells: The Pursuit of Genomic Stability" International Journal of Molecular Sciences 15, no. 11: 20948-20967. https://doi.org/10.3390/ijms151120948