Palmitic Acid-Induced Neuron Cell Cycle G2/M Arrest and Endoplasmic Reticular Stress through Protein Palmitoylation in SH-SY5Y Human Neuroblastoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
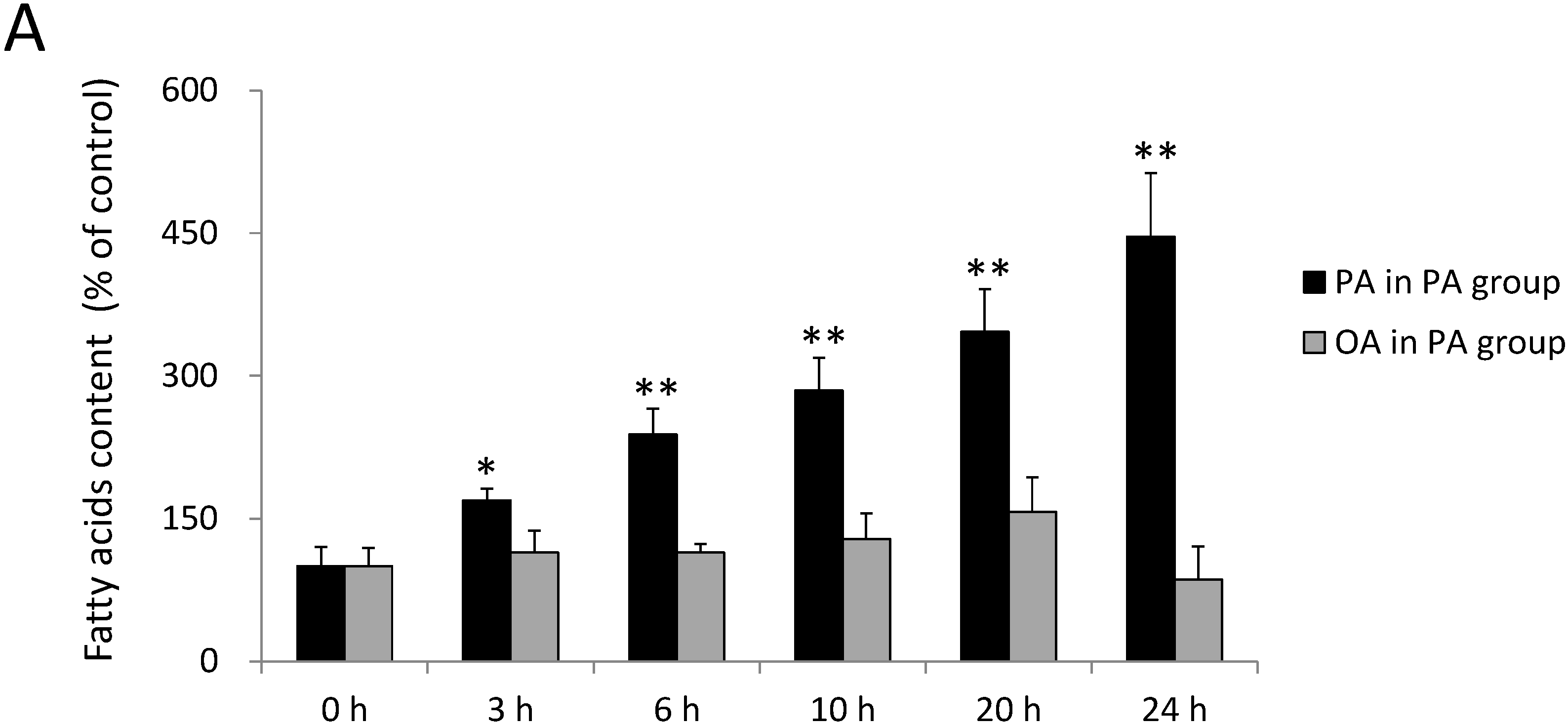
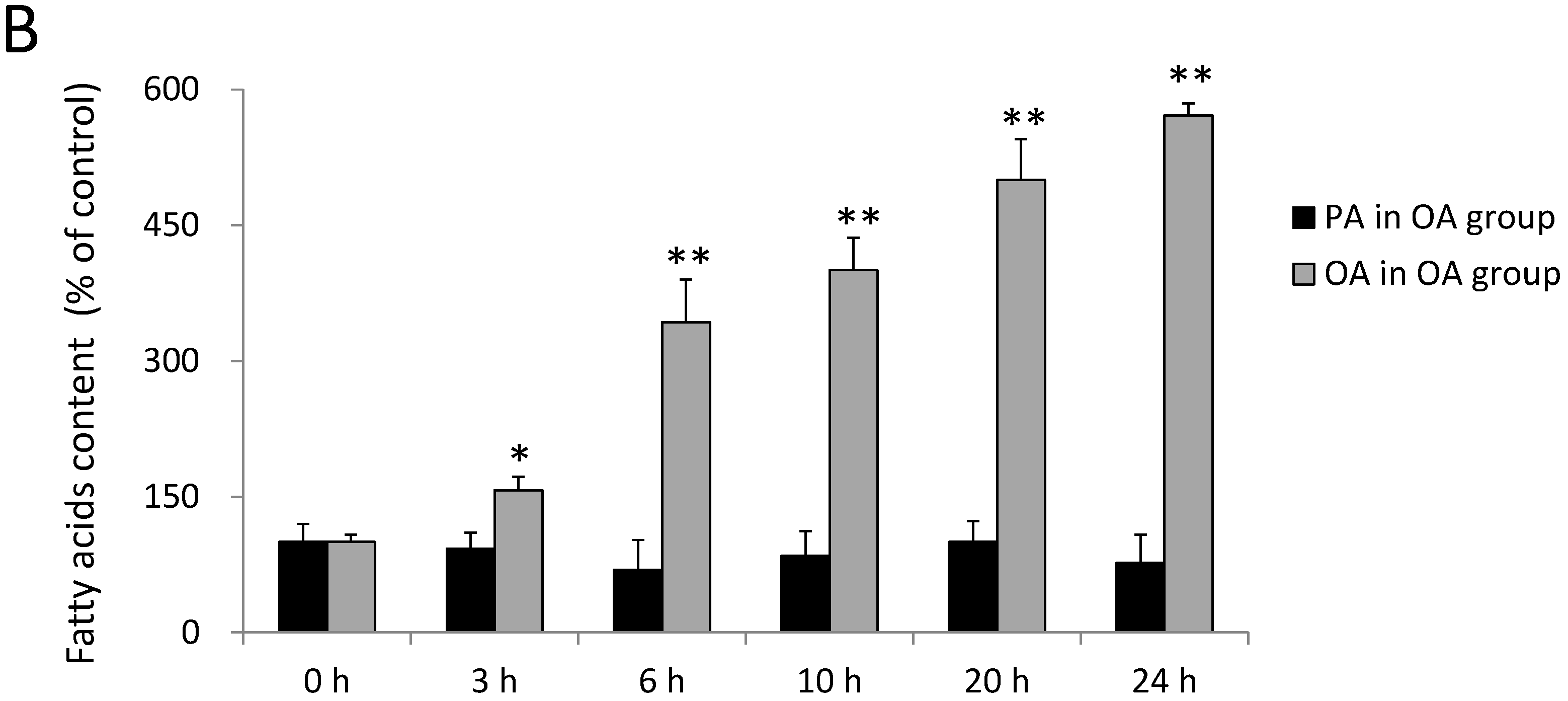
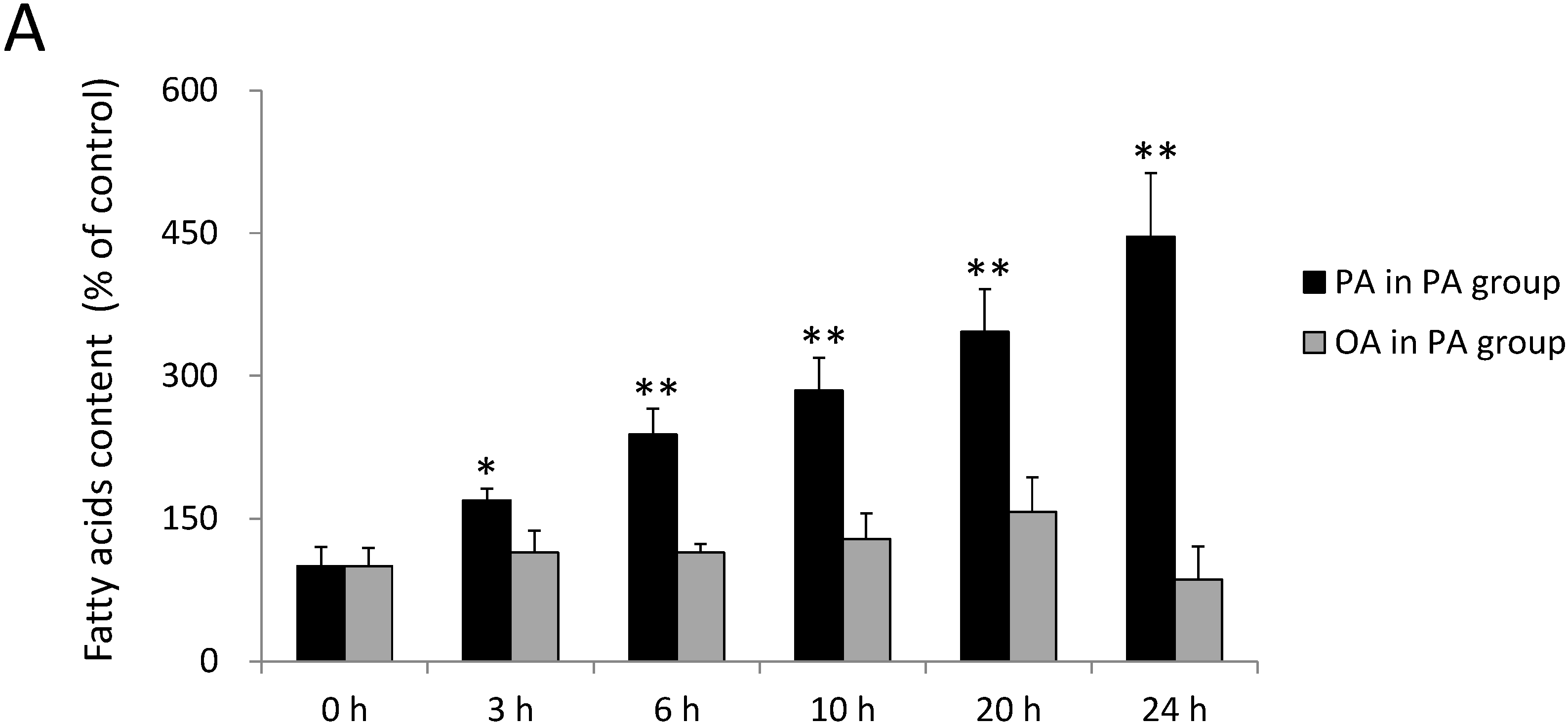
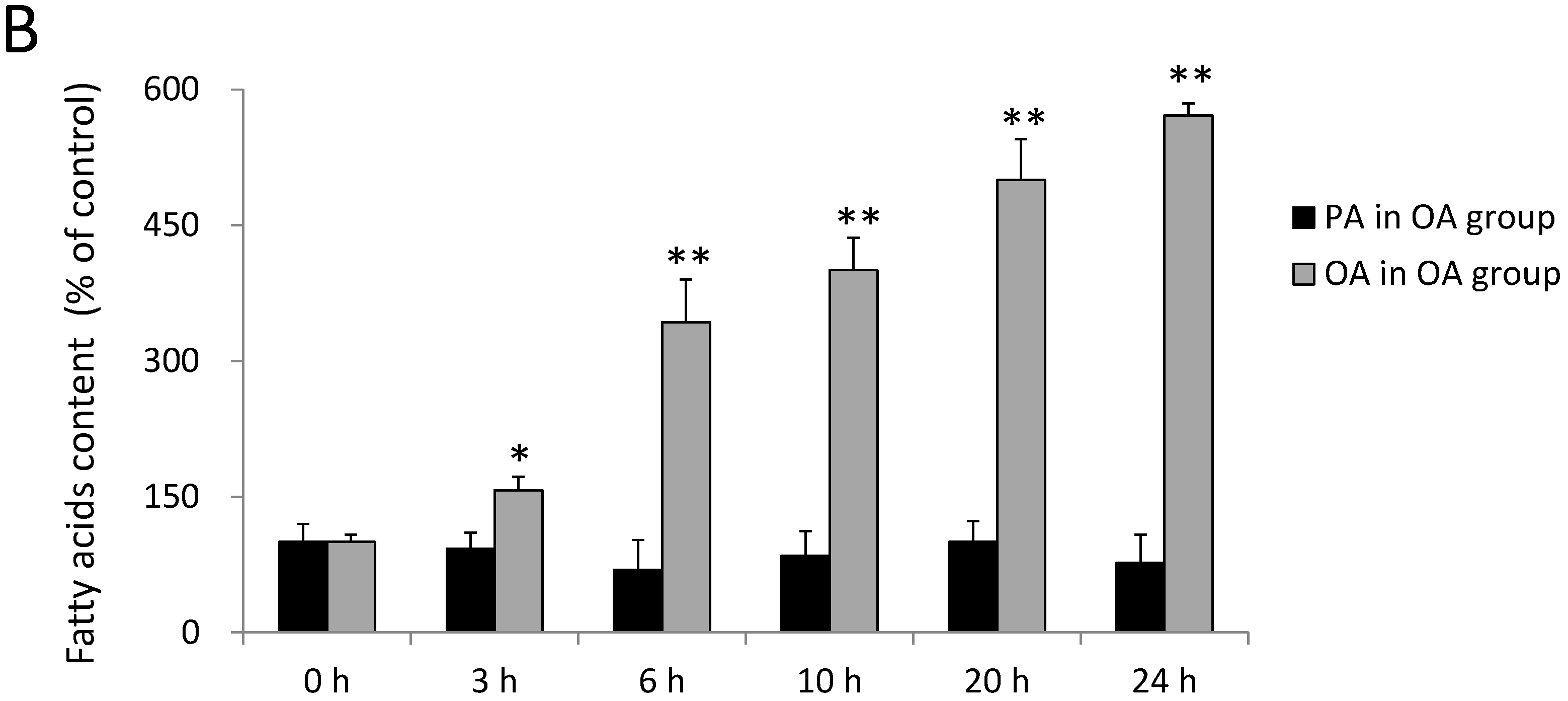
2.1. Incorporation of FAs into SH-SY5Y Cells
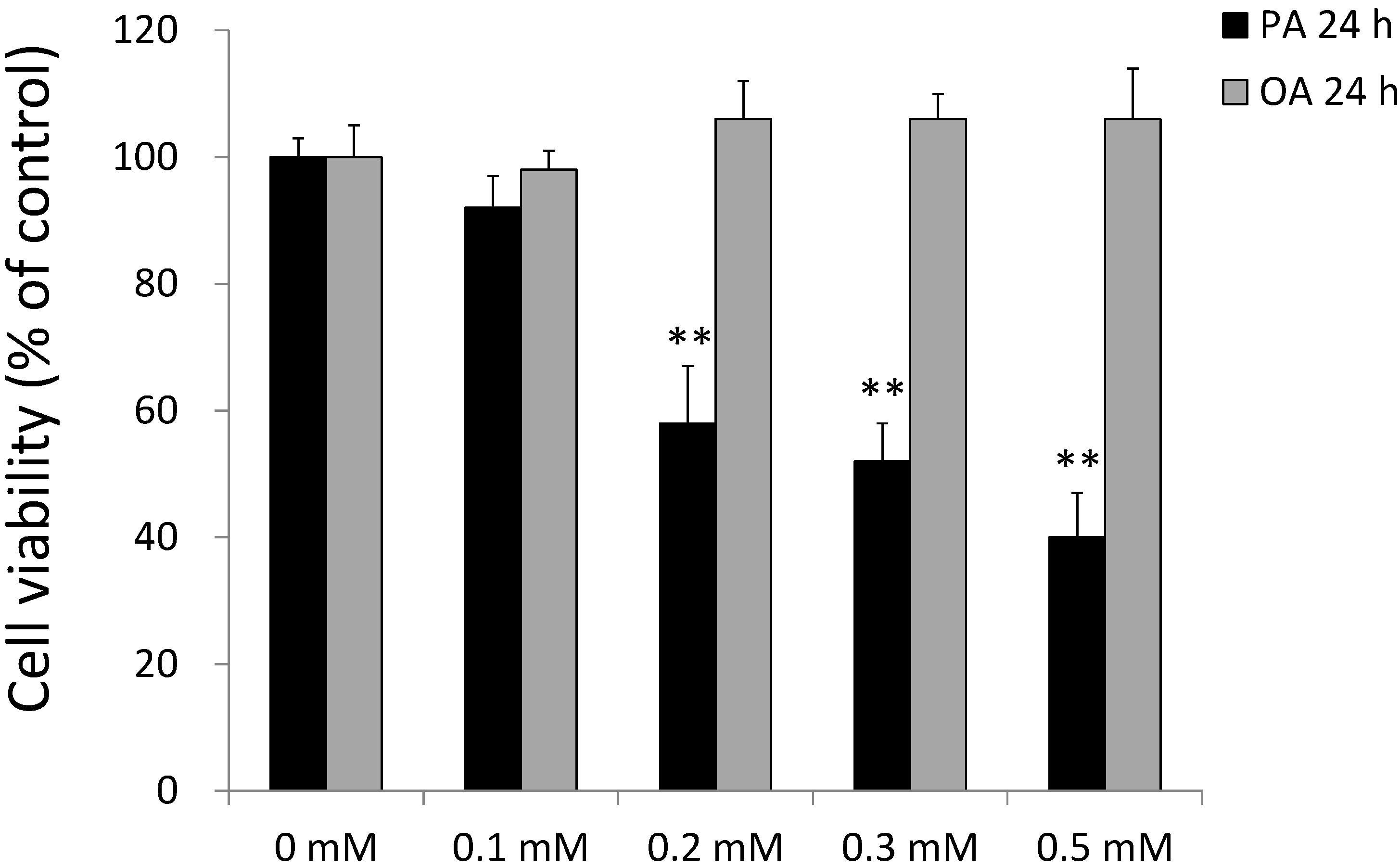
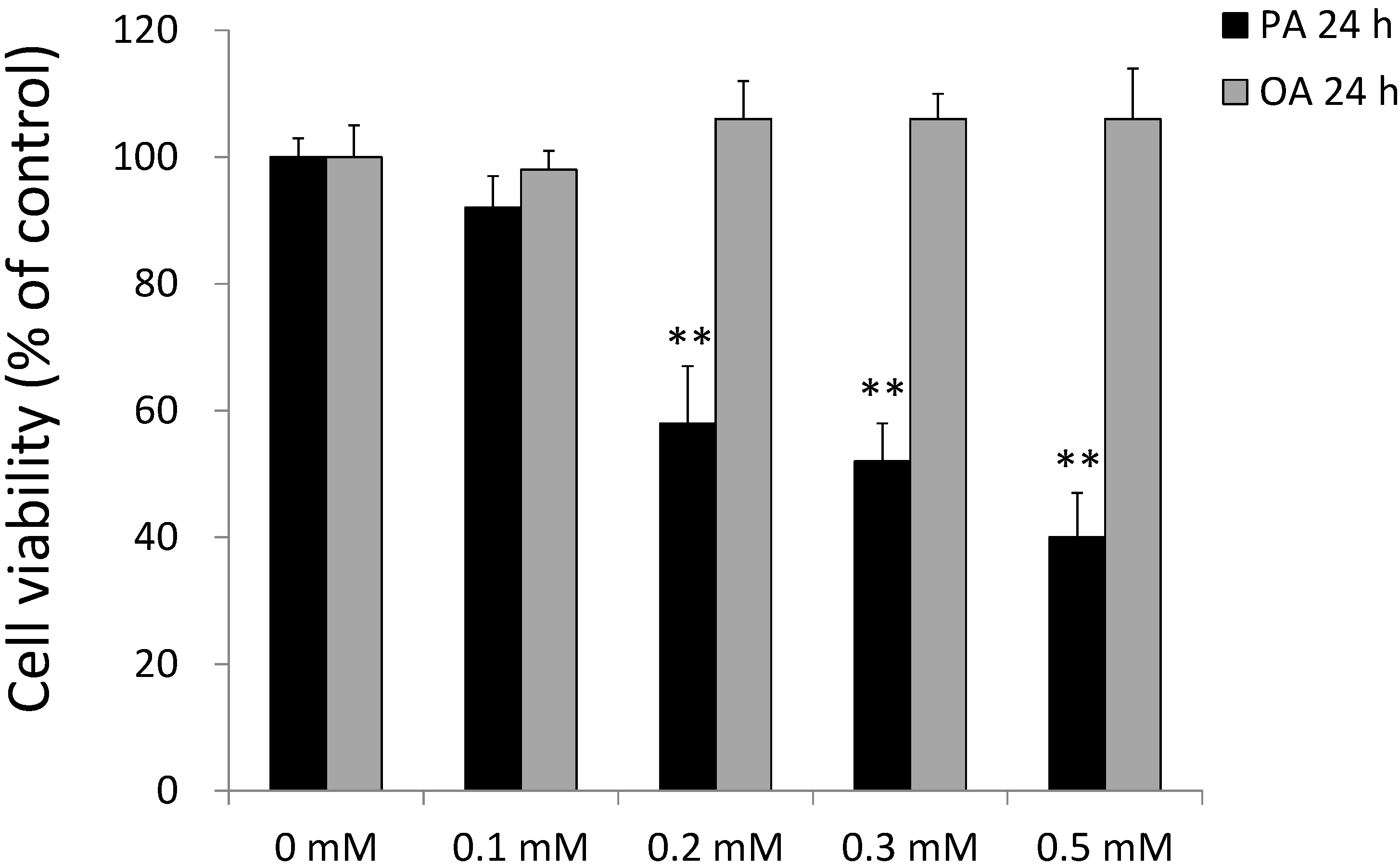
2.2. Effects of PA and OA on SH-SY5Y Cell Viability
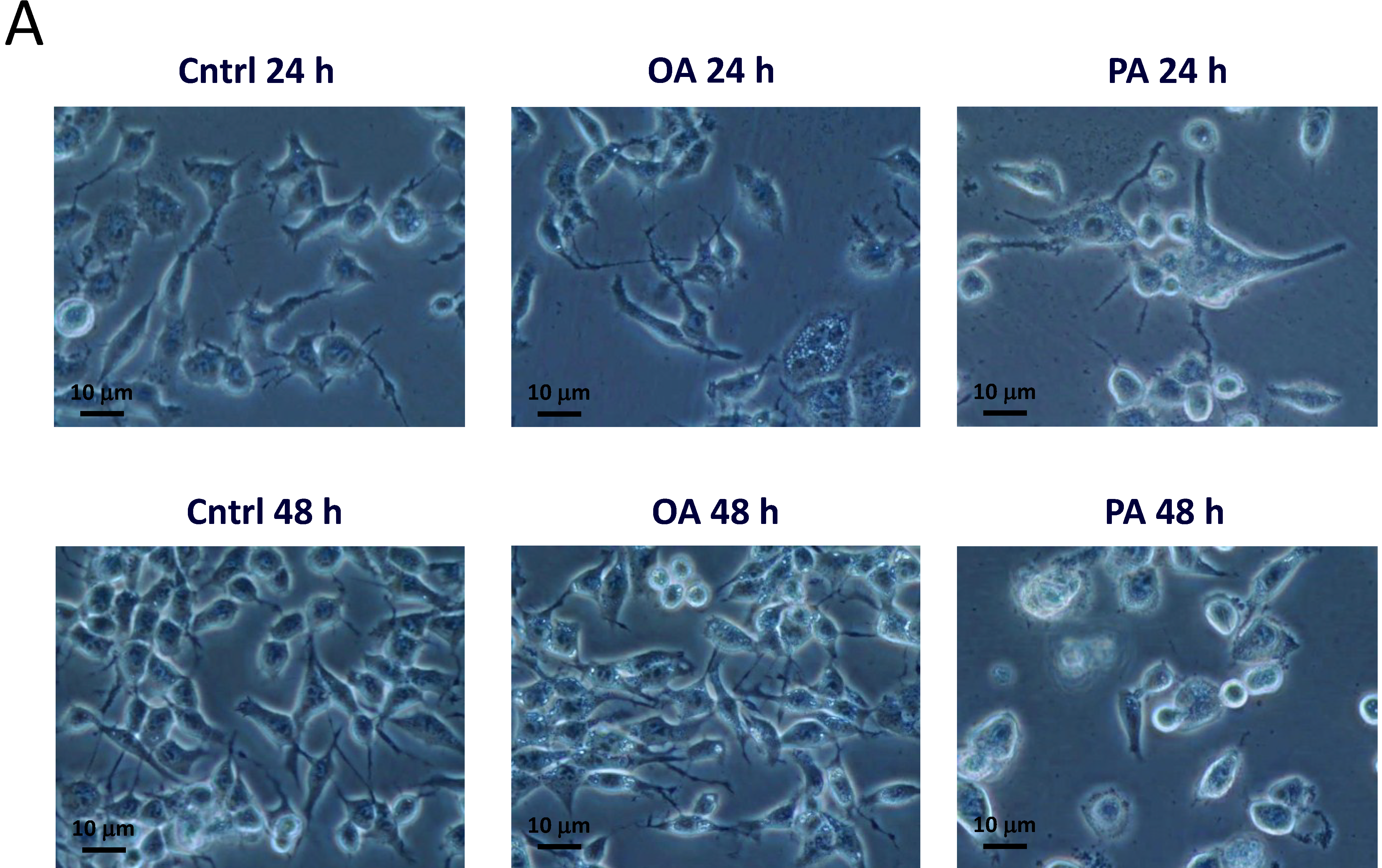
2.3. Effects of PA on SH-SY5Y Cell Morphology

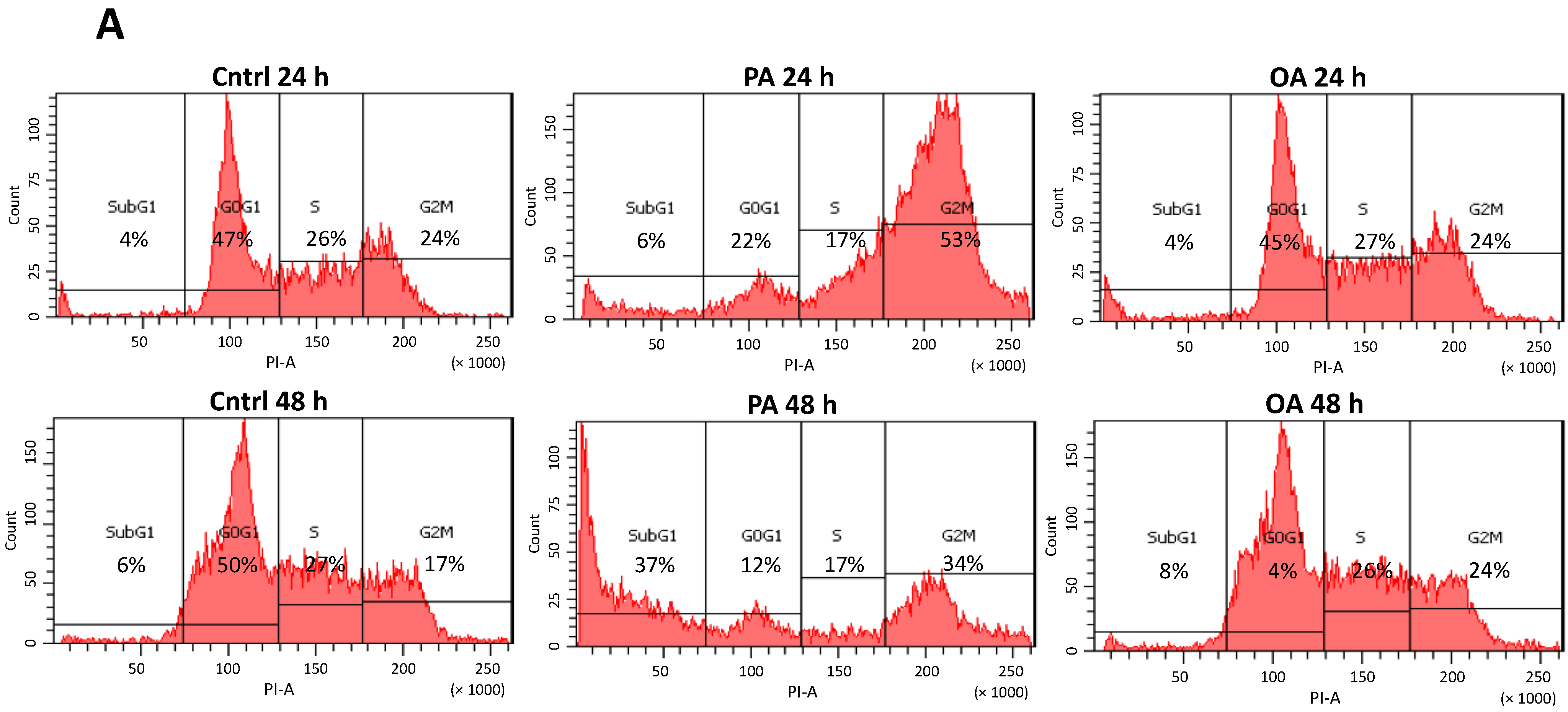
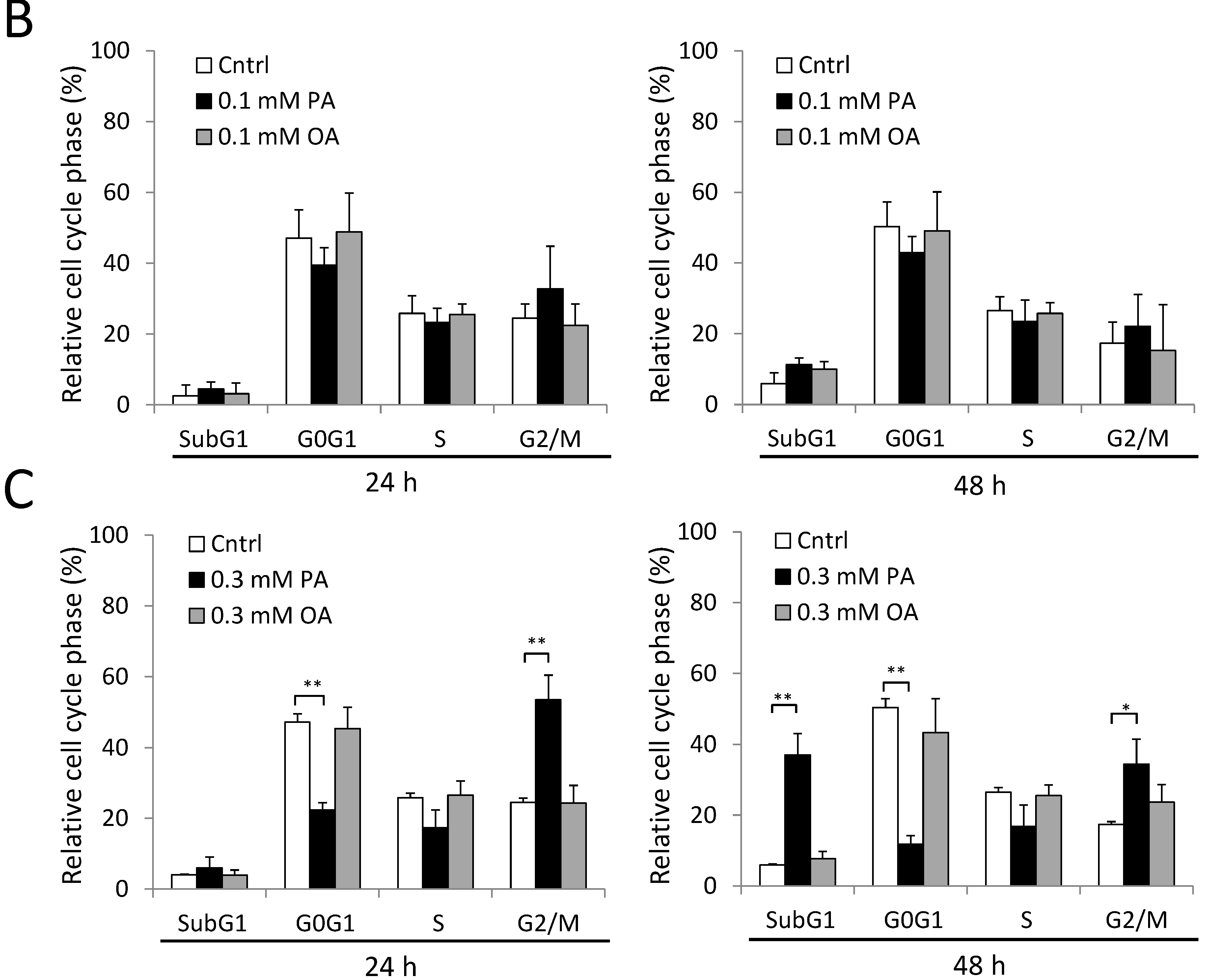
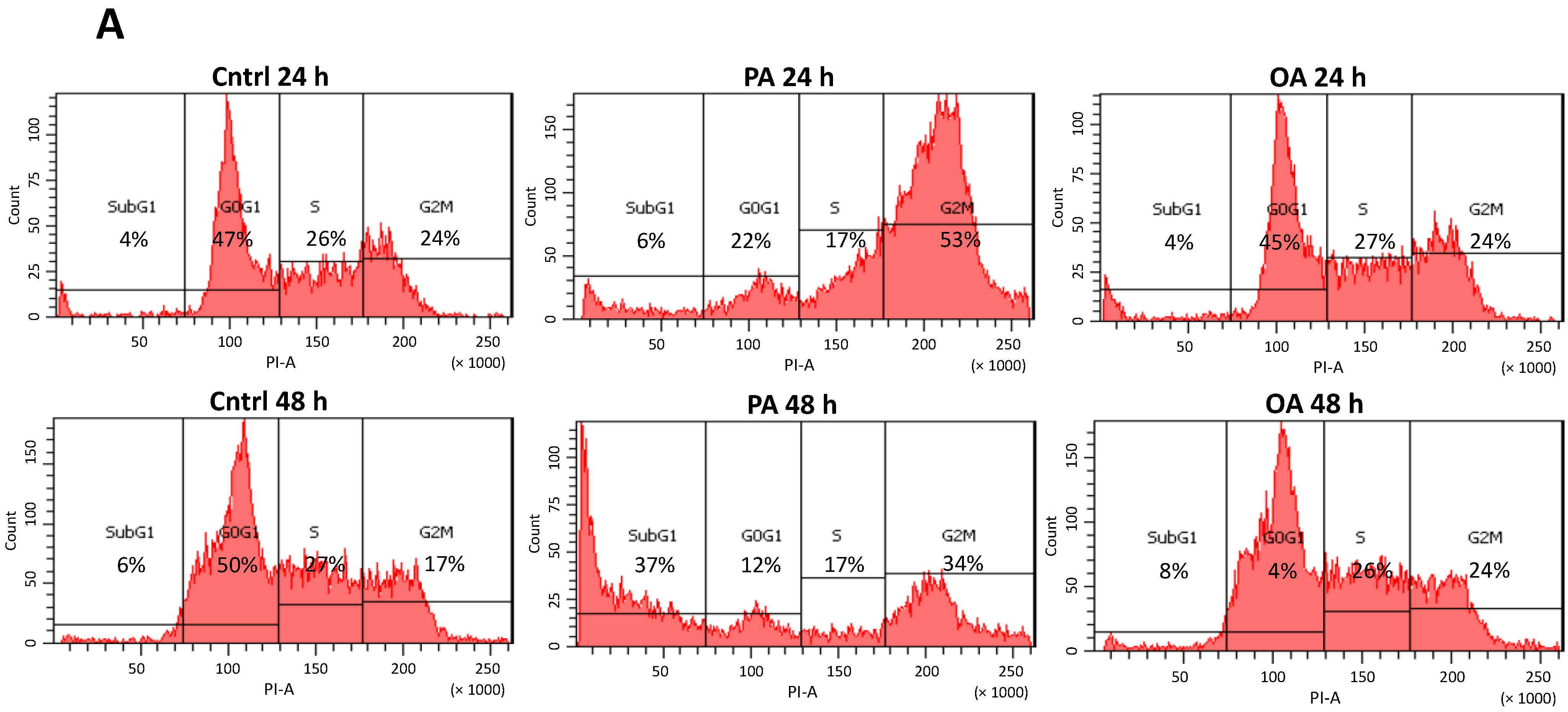
2.4. Cell Cycle G2/M Arrest Induced by PA in SH-SY5Y Cells

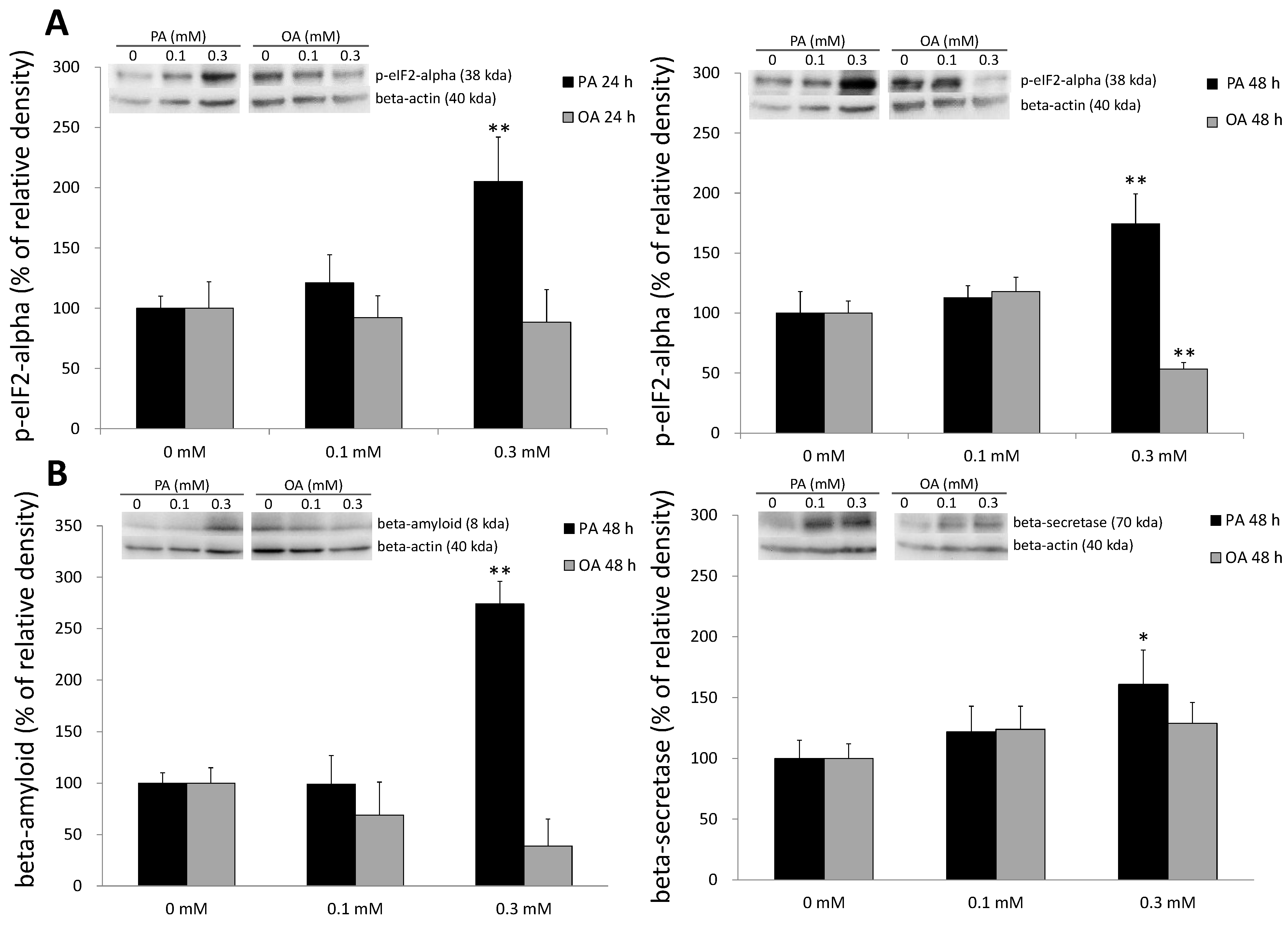
2.5. ER Stress and Beta-Amyloid Accumulation Induced by PA

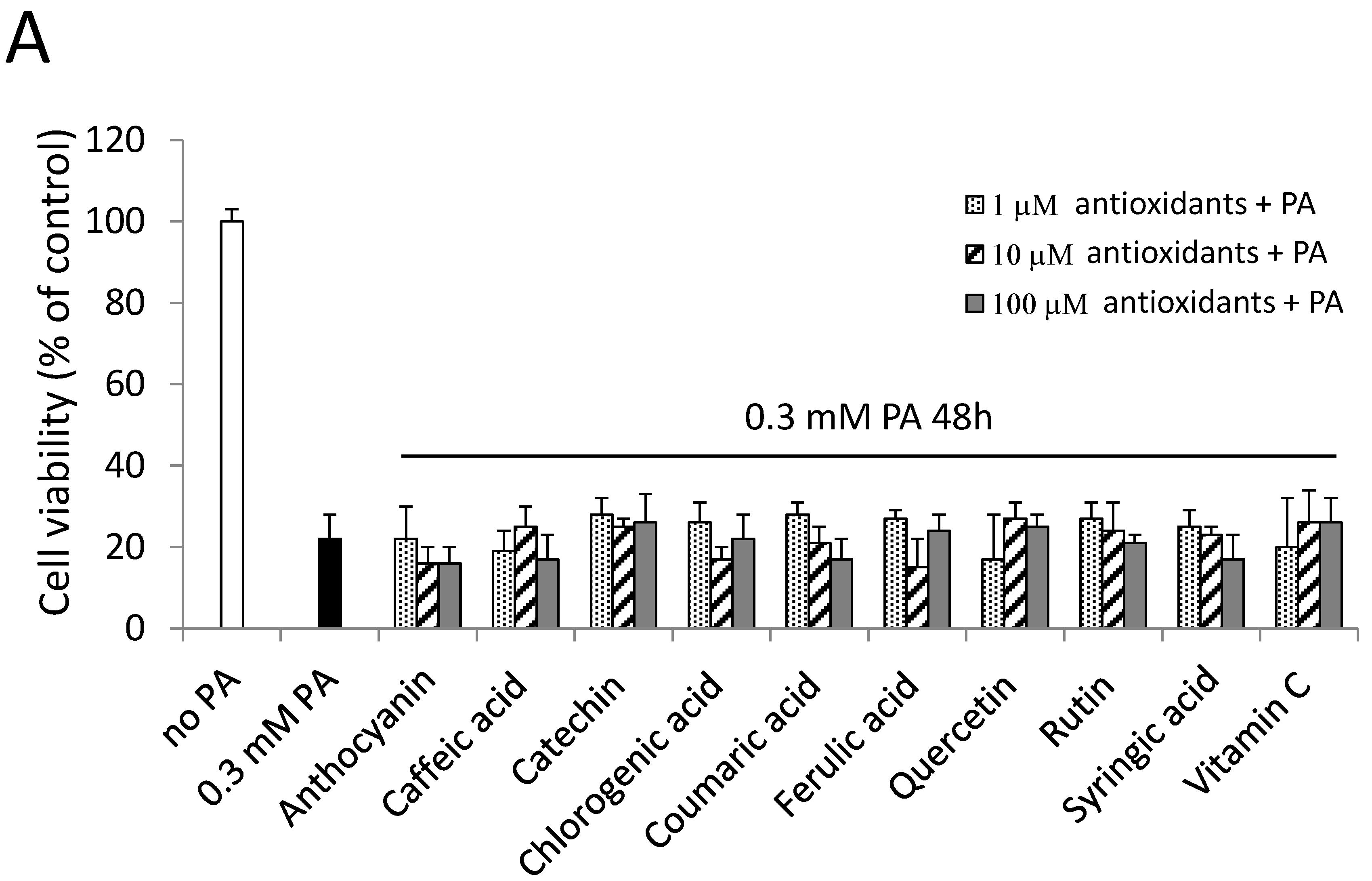
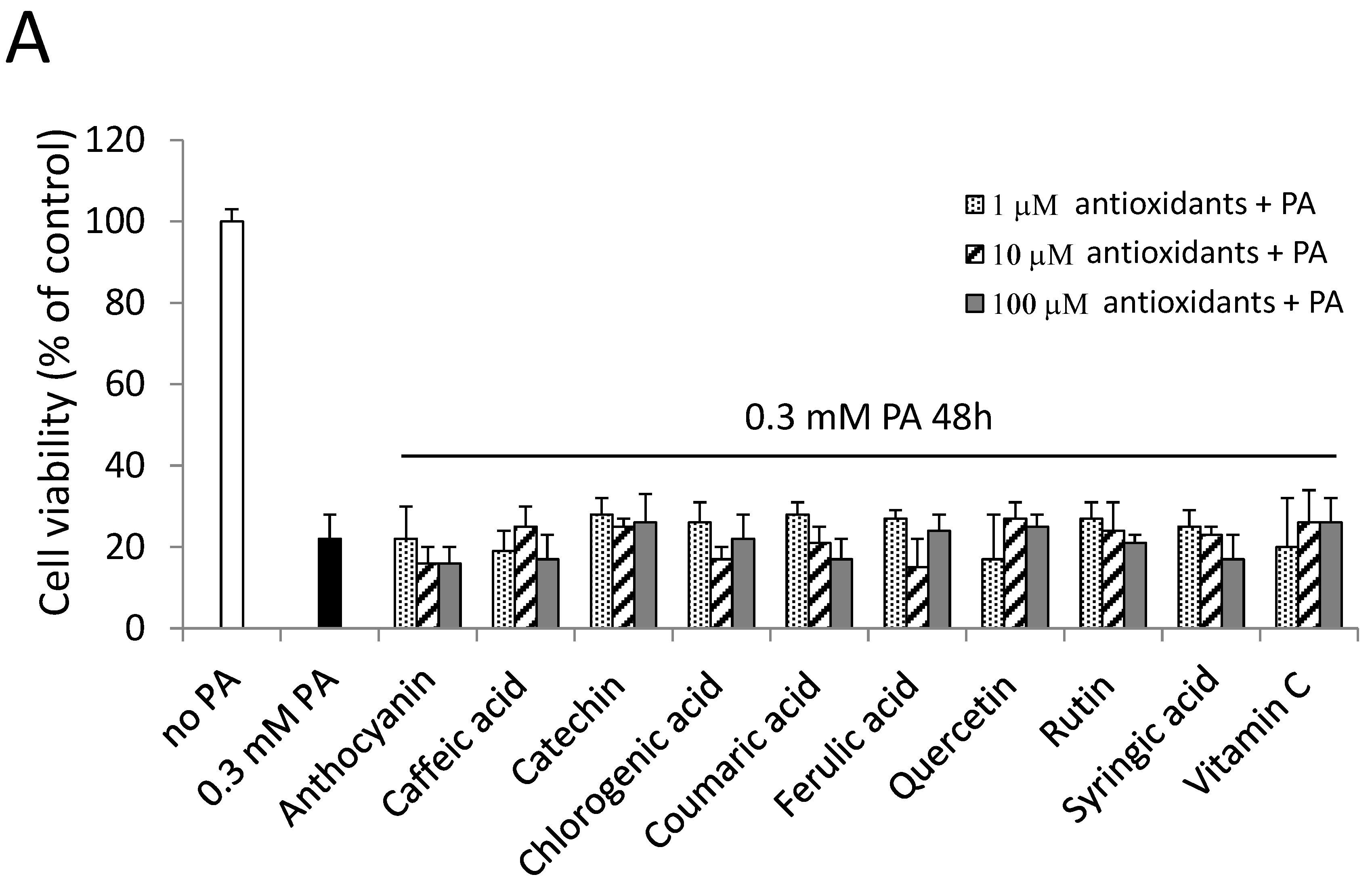
2.6. Antioxidants and a PKC Inhibitor could not Alleviate PA-Induced Cell Death

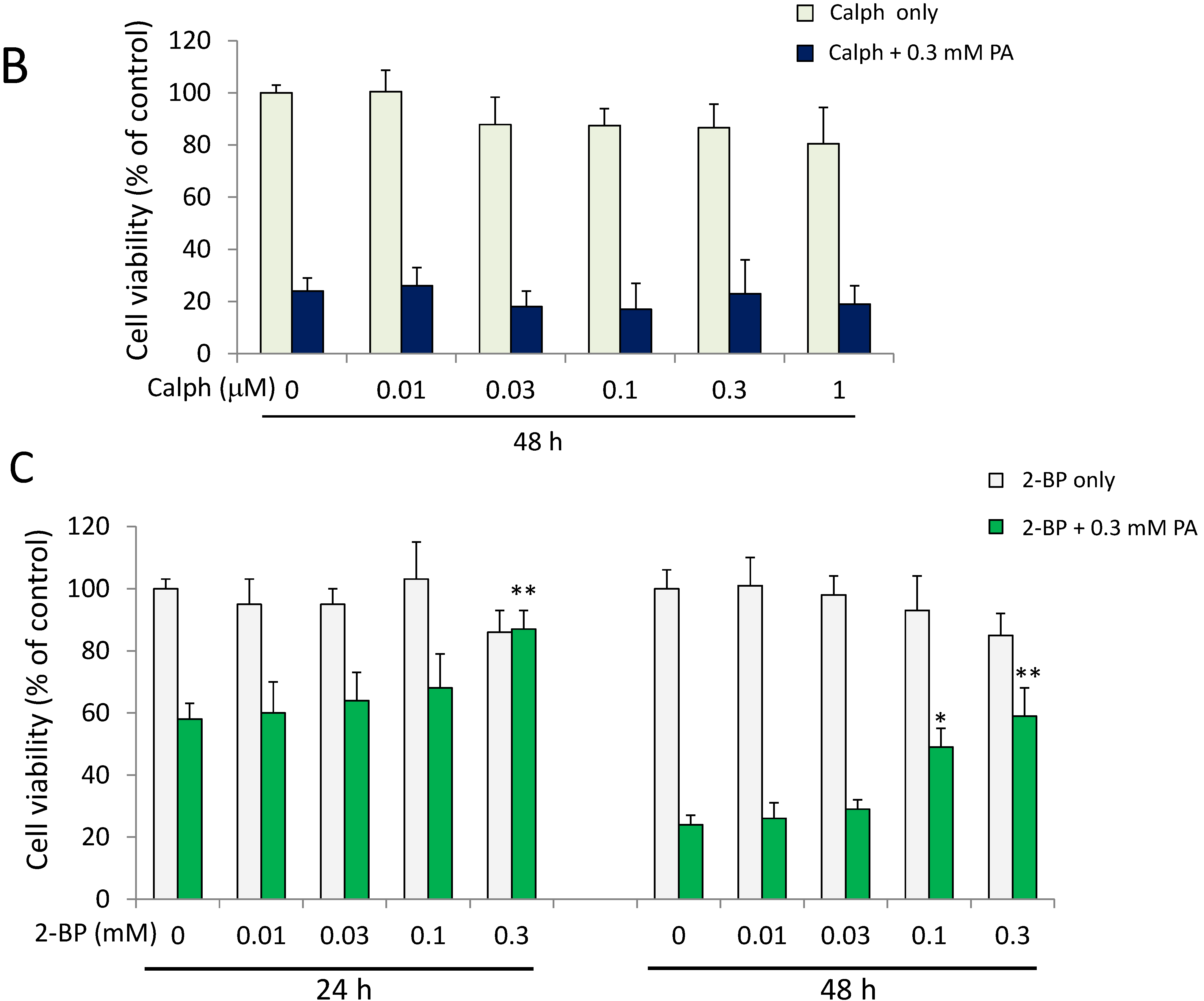
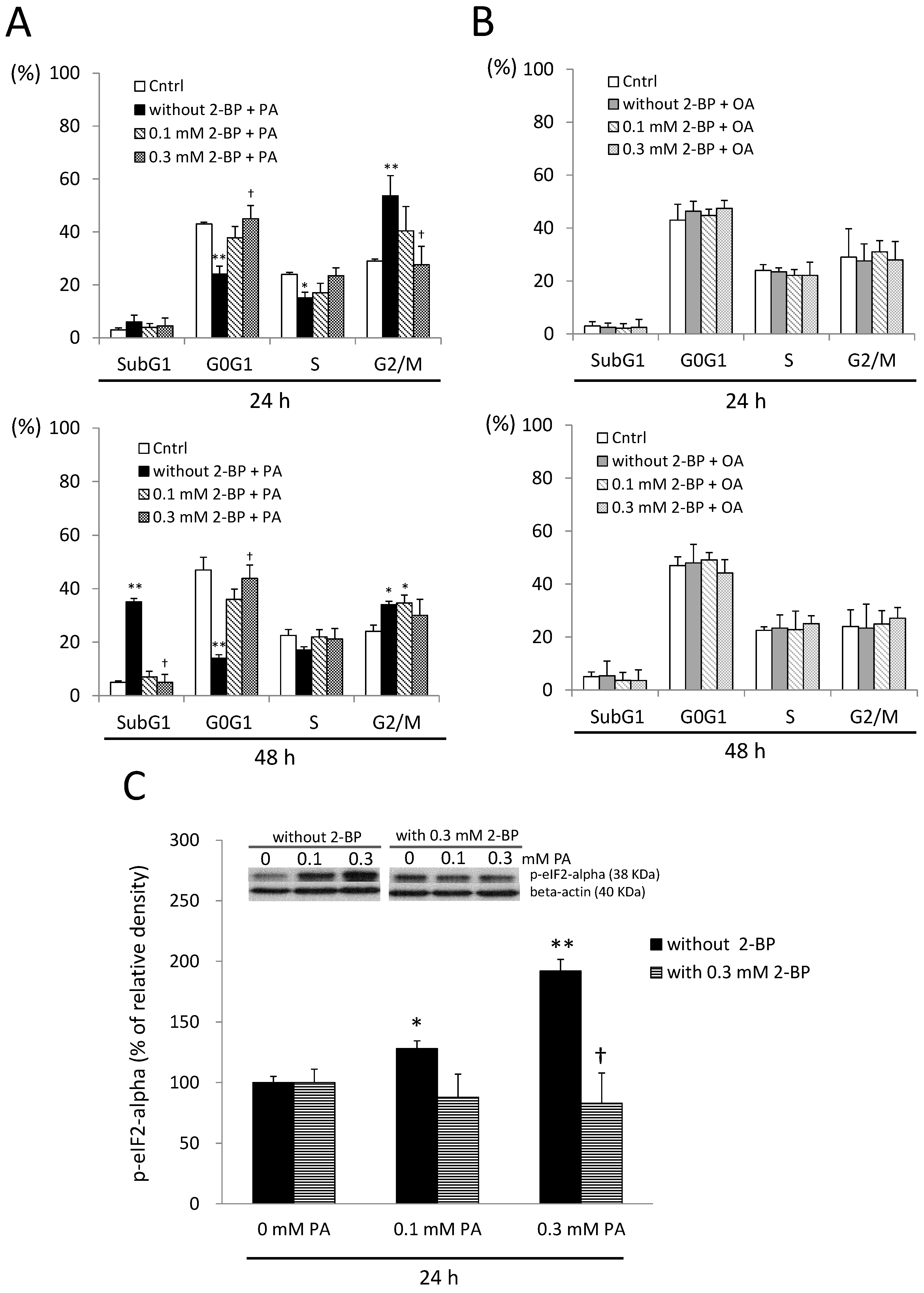
2.7. A Protein Palmitoylation Inhibitor Attenuated PA-Induced G2/M Arrest and ER Stress

2.8. Discussion
3. Experimental Section
3.1. Cell Culture Preparation
3.2. FA Treatment
3.3. Cell Viability Assay
3.4. Cell Size Measurement
3.5. FA Composition Analysis
3.6. Cell Cycle Analysis
3.7. Immunoblotting
3.8. Pretreatment with Antioxidants
3.9. PKC Inhibitor Treatment
3.10. Palmitoylation Inhibitor Treatment
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; D’Introno, A.; Colacicco, A.M.; Capurso, C.; Del Parigi, A.; Capurso, S.; Gadaleta, A.; Capurso, A.; Panza, F. Dietary fatty acids intake: Possible role in cognitive decline and dementia. Exp. Gerontol. 2005, 40, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Kalmijn, S. Fatty acid intake and the risk of dementia and cognitive decline: A review of clinical and epidemiological studies. J. Nutr. Health Aging 1999, 4, 202–207. [Google Scholar]
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.; Hofman, A.; Breteler, M. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann. Neurol. 1997, 42, 776–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Schneider, J.; Wilson, R.S. Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 194. [Google Scholar] [CrossRef] [PubMed]
- Fraser, T.; Tayler, H.; Love, S. Fatty acid composition of frontal, temporal and parietal neocortex in the normal human brain and in Alzheimer’s disease. Neurochem. Res. 2010, 35, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Cipolla, M.; Chiang, J.; Arakaki, X.; Harrington, M.G. Human Cerebrospinal Fluid Fatty Acid Levels Differ between Supernatant Fluid and Brain-Derived Nanoparticle Fractions, and Are Altered in Alzheimer’s Disease. PLoS One 2014, 9, e100519. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-[beta]-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.C.; Smith, S.R.; Champagne, C.M.; Most, M.M.; Lefevre, M.; DeLany, J.P.; Denkins, Y.M.; Rood, J.C.; Veldhuis, J.; Bray, G.A. Effects of diets enriched in saturated (palmitic), monounsaturated (oleic), or trans (elaidic) fatty acids on insulin sensitivity and substrate oxidation in healthy adults. Diabetes Care 2002, 25, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Gunstone, F.D.; Harwood, J.L.; Dijkstra, A.J. The lipid handbook with CD-ROM; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Morrison, C.D.; Pistell, P.J.; Ingram, D.K.; Johnson, W.D.; Liu, Y.; Fernandez-Kim, S.O.; White, C.L.; Purpera, M.N.; Uranga, R.M.; Bruce-Keller, A.J. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: Implications for decreased Nrf2 signaling. J. Neurochem. 2010, 114, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Heude, B.; Ducimetière, P.; Berr, C. Cognitive decline and fatty acid composition of erythrocyte membranes—The EVA Study. Am. J. Clin. Nutr. 2003, 77, 803–808. [Google Scholar] [PubMed]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.M.; Belsham, D.D. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: Rescue of resistance and apoptosis through adenosine 5' monophosphate-activated protein kinase activation. Endocrinology 2010, 151, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Ulloth, J.E.; Casiano, C.A.; De Leon, M. Palmitic and stearic fatty acids induce caspase-dependent and-independent cell death in nerve growth factor differentiated PC12 cells. J. Neurochem. 2003, 84, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Kaufman, J.S.; Satia, J.A.; Rosamond, W.; Folsom, A.R. Plasma n−3 fatty acids and the risk of cognitive decline in older adults: The Atherosclerosis Risk in Communities Study. Am. J. Clin. Nutr. 2007, 85, 1103–1111. [Google Scholar] [PubMed]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, M.; Ngandu, T.; Rovio, S.; Helkala, E.-L.; Uusitalo, U.; Viitanen, M.; Nissinen, A.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Fat intake at midlife and risk of dementia and Alzheimer’s disease: A population-based study. Dement. Geriatr. Cogn. Disord. 2006, 22, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Baquer, N.Z.; Hothersall, J.S.; McLean, P. Function and regulation of the pentose phosphate pathway in brain. Curr. Top. Cell. Regul. 1988, 29, 265–289. [Google Scholar] [PubMed]
- Hamilton, J.A.; Brunaldi, K. A model for fatty acid transport into the brain. J. Mol. Neurosci. 2007, 33, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Chan, C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci. Lett. 2005, 384, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Sheng, L.; Masserang, A.; Chan, C. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci. Lett. 2006, 406, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Spinas, G.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on β-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Umegaki, H. Type 2 diabetes as a risk factor for cognitive impairment: Current insights. Clin. Interv. Aging 2014, 9, 1011. [Google Scholar] [CrossRef] [PubMed]
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Desai, G.; Zheng, C.; Geetha, T.; Mathews, S.T.; White, B.D.; Huggins, K.W.; Zizza, C.A.; Broderick, T.L.; Babu, J.R. The Pancreas-Brain Axis: Insight into Disrupted Mechanisms Associating Type 2 Diabetes and Alzheimer’s Disease. J. Alzheimers Dis. 2014, 42, 347–356. [Google Scholar] [PubMed]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Vidoni, E.D.; Honea, R.A.; Burns, J.M. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 2014, 35, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.T.; Sheen, Y.J.; Kao, C.D.; Chang, C.A.; Hu, Y.C.; Lin, J.L. Association between the characteristics of metabolic syndrome and Alzheimer’s disease. Metab. Brain Dis. 2013, 28, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Stephan, B.; Wells, J.; Brayne, C.; Albanese, E.; Siervo, M. Increased fructose intake as a risk factor for dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A. The emerging role of dietary fructose in obesity and cognitive decline. Nutr. J. 2013, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Clore, J.N.; Allred, J.; White, D.; Li, J.; Stillman, J. The role of plasma fatty acid composition in endogenous glucose production in patients with type 2 diabetes mellitus. Metabolism 2002, 51, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, C.E.; Winocur, G. High-fat diets, insulin resistance and declining cognitive function. Neurobiol. Aging 2005, 26, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Dagogo-Jack, S.E.; Wiethop, B.V.; Murphy, C.; Nevins, R.T.; Fleischman, S.; Rice, V.; Newcomer, J.W.; Cryer, P.E. Effects of hyperglycemia on memory and hormone levels in dementia of the Alzheimer type: A longitudinal study. Behav. Neurosci. 1993, 107, 926. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.-X.; Manly, J.J.; Mayeux, R. Relation of diabetes to mild cognitive impairment. Arch. Neurol. 2007, 64, 570. [Google Scholar] [CrossRef] [PubMed]
- Rosendorff, C.; Beeri, M.S.; Silverman, J.M. Cardiovascular risk factors for Alzheimer’s disease. Am. J. Geriatr. Cardiol. 2007, 16, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373. [Google Scholar]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, T.; Gerstein, H.; Williamson, J. Cognitive decline and dementia in diabetes—systematic overview of prospective observational studies. Diabetologia 2005, 48, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- MacKnight, C.; Rockwood, K.; Awalt, E.; McDowell, I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement. Geriatr. Cogn. Dis. 2002, 14, 77–83. [Google Scholar] [CrossRef]
- Becker, E.B.; Bonni, A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 2004, 72, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bu, B.; Xie, M.; Zhang, M.; Yu, Z.; Tao, D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009, 89, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.K.; Cordery, D.; Denyer, G.S.; Biden, T.J. Expression profiling of palmitate-and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic β-cell function. Diabetes 2002, 51, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Gentile, C.L.; Nivala, A.M.; Wang, D.; Wei, Y.; Pagliassotti, M.J. Linking endoplasmic reticulum stress to cell death in hepatocytes: Roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1027–E1035. [Google Scholar] [CrossRef] [PubMed]
- Alencar, R.C.; Cobas, R.A.; Gomes, M.B. Assessment of cognitive status in patients with type 2 diabetes through the Mini-Mental Status Examination: A cross-sectional study. Diabetol. Metab. Syndr. 2010, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Wan, J.; Arstikaitis, P.; Takahashi, H.; Huang, K.; Bailey, A.O.; Thompson, J.X.; Roth, A.F.; Drisdel, R.C.; Mastro, R. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 2008, 456, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Dai, D.-L.; Yao, L.; Yu, H.-H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.-H.; Shen, W.; Yang, Z.-X. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Nivala, A.M.; Reese, L.; Frye, M.; Gentile, C.L.; Pagliassotti, M.J. Fatty acid-mediated endoplasmic reticulum stress in vivo: Differential response to the infusion of Soybean and Lard Oil in rats. Metabolism 2013, 62, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Lee, M.-S.; Wu, C.-R.; Cho, H.-J.; Lin, K.-Y.; Lai, G.-H.; Wang, S.-Y.; Kuo, Y.-H.; Senthil Kumar, K.; Yang, H.-L. The chalcone flavokawain B induces G2/M cell-cycle arrest and apoptosis in human oral carcinoma HSC-3 cells through the intracellular ROS generation and downregulation of the Akt/p38 MAPK signaling pathway. J. Agric. Food Chem. 2012, 60, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Barboule, N.; Lafon, C.; Chadebech, P.; Vidal, S.; Valette, A. Involvement of p21 in the PKC-induced regulation of the G2/M cell cycle transition. FEBS Lett. 1999, 444, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Breitkreutz, I.; Tonon, G.; Zhang, J.; Hayden, P.J.; Nguyen, T.; Fruehauf, J.H.; Lin, B.K.; Chauhan, D.; Hideshima, T. Targeting PKC: A novel role for beta-catenin in ER stress and apoptotic signaling. Blood 2009, 113, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.-C.; Mullen, A.; Abrams, K.; Mukhopadhyay, D.; Claffey, K.P. Role of protein kinase C isoforms in phorbol ester-induced vascular endothelial growth factor expression in human glioblastoma cells. J. Biol. Chem. 1999, 274, 15407–15414. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, A.; Andres-Lacueva, C.; Martin, A.; Lauretani, F.; Di Iorio, A.; Bartali, B.; Corsi, A.; Bandinelli, S.; Mattson, M.P.; Ferrucci, L. Low plasma N-3 fatty acids and dementia in older persons: The InCHIANTI study. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Schneider, J.A.; Wilson, R.S.; Scherr, P.A. Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 2006, 63, 1085. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Ngandu, T.; Helkala, E.L.; Tuomilehto, J.; Nissinen, A.; Soininen, H.; Kivipelto, M. Fat intake at midlife and cognitive impairment later in life: A population-based CAIDE study. Int. J. Geriatr. Psychiatr. 2008, 23, 741–747. [Google Scholar] [CrossRef]
- Morris, M.; Evans, D.; Bienias, J.; Tangney, C.; Wilson, R. Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 2004, 62, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Eitel, K.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Häring, H.-U.; Kellerer, M. Different role of saturated and unsaturated fatty acids in β-cell apoptosis. Biochem. Biophys. Res. Commun. 2002, 299, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Montell, E.; Turini, M.; Marotta, M.; Roberts, M.; Noé, V.; Macé, K.; Gómez-Foix, A.M. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E229–E237. [Google Scholar] [PubMed]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed]
- Staiger, K.; Staiger, H.; Weigert, C.; Haas, C.; Häring, H.-U.; Kellerer, M. Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-κB activation. Diabetes 2006, 55, 3121–3126. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.-M.; Yanase, T.; Nishi, Y.; Tanaka, A.; Saito, M.; Jin, C.-H.; Mukasa, C.; Okabe, T.; Nomura, M.; Goto, K. Saturated FFAs, palmitic acid and stearic acid, induce apoptosis in human granulosa cells. Endocrinology 2001, 142, 3590–3597. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, A.; Togliatto, G.; Rosso, A.; Dentelli, P.; Olgasi, C.; Cotogni, P.; Brizzi, M.F. Increase of palmitic acid concentration impairs endothelial progenitor cell and bone marrow-derived progenitor cell bioavailability: Role of the STAT5/PPARgamma transcriptional complex. Diabetes 2013, 62, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhao, S.; Wang, F.; Zhang, H.; Chen, Z.J.; Wang, J.; Wang, Z.; Du, Z.; Ling, E.A.; Liu, Q.; Hao, A. Palmitic acid increases apoptosis of neural stem cells via activating c-Jun N-terminal kinase. Stem Cell Res. 2013, 10, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Correia-da-Silva, G.; Valentão, P.; Teixeira, N.; Andrade, P.B. Palmitic Acid and Ergosta-7, 22-dien-3-ol Contribute to the Apoptotic Effect and Cell Cycle Arrest of an Extract from Marthasterias glacialis L. in Neuroblastoma Cells. Mar. Drugs 2013, 12, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Herman-Antosiewicz, A.; Singh, A.V.; Lew, K.L.; Srivastava, S.K.; Kamath, R.; Brown, K.D.; Zhang, L.; Baskaran, R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J. Biol. Chem. 2004, 279, 25813–25822. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.G.; Heilman, D.W.; Parker, A.E.; Green, M.R. The viral protein Apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev. 2004, 18, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fåhraeus, R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Melrose, J.; Chan, C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, S.; Rabinovitz, S.; Carasso, R.L.; Mostofsky, D.I. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiol. Aging 2002, 23, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Pagliassotti, M.J. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol. Cell. Biochem. 2007, 303, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Madaro, L.; Marrocco, V.; Carnio, S.; Sandri, M.; Bouché, M. Intracellular signaling in ER stress-induced autophagy in skeletal muscle cells. FASEB J. 2013, 27, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Larroque-Cardoso, P.; Swiader, A.; Ingueneau, C.; Nègre-Salvayre, A.; Elbaz, M.; Reyland, M.; Salvayre, R.; Vindis, C. Role of protein kinase C δ in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. 2013, 4, e520. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.C.; Kemp, C.J.; Elias, C.F.; Abplanalp, W.; Herman, J.P.; Migrenne, S.; Lefevre, A.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Yu, F.; et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J. Clin. Invest. 2009, 119, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Voss, O.H.; Kim, S.; Wewers, M.D.; Doseff, A.I. Regulation of monocyte apoptosis by the protein kinase Cdelta-dependent phosphorylation of caspase-3. J. Biol. Chem. 2005, 280, 17371–17379. [Google Scholar] [CrossRef] [PubMed]
- Rocks, O.; Gerauer, M.; Vartak, N.; Koch, S.; Huang, Z.P.; Pechlivanis, M.; Kuhlmann, J.; Brunsveld, L.; Chandra, A.; Ellinger, B.; et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 2010, 141, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Greaves, J.; Prescott, G.R.; Gorleku, O.A.; Chamberlain, L.H. The fat controller: Roles of palmitoylation in intracellular protein trafficking and targeting to membrane microdomains (Review). Mol. Membr. Biol. 2009, 26, 67–79. [Google Scholar] [PubMed]
- Salaun, C.; Greaves, J.; Chamberlain, L.H. The intracellular dynamic of protein palmitoylation. J. Cell Biol. 2010, 191, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Bijlmakers, M.-J.; Marsh, M. The on–off story of protein palmitoylation. Trends Cell Biol. 2003, 13, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflammation 2009, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.C.; Green, C.D.; Olson, L.K.; Moxley, M.A.; Corbett, J.A. A role for aberrant protein palmitoylation in FFA-induced ER stress and β-cell death. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1390–E1398. [Google Scholar] [CrossRef] [PubMed]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Use of analogs and inhibitors to study the functional significance of protein palmitoylation. Methods 2006, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Webb, Y.; Hermida-Matsumoto, L.; Resh, M.D. Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem. 2000, 275, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Smotrys, J.E.; Linder, M.E. Palmitoylation of intracellular signaling proteins: Regulation and function. Ann. Rev. Biochem. 2004, 73, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, M.; Ittrich, C.; Wiedmann, V.; Knyazev, Y.; Weninger, A.; Riemenschneider, M.; Hartmann, T. New Alzheimer amyloid beta responsive genes identified in human neuroblastoma cells by hierarchical clustering. PLoS One 2009, 4, e6779. [Google Scholar] [CrossRef] [PubMed]
- Hamid, R.; Rotshteyn, Y.; Rabadi, L.; Parikh, R.; Bullock, P. Comparison of alamar blue and MTT assays for high through-put screening. Toxicol In Vitro 2004, 18, 703. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane-Stanley, G. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Dobrzyń, A.; Górski, J. Ceramides and sphingomyelins in skeletal muscles of the rat: Content and composition. Effect of prolonged exercise. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E277–E285. [Google Scholar] [PubMed]
- Tserng, K.-Y.; Griffin, R. Quantitation and molecular species determination of diacylglycerols, phosphatidylcholines, ceramides, and sphingomyelins with gas chromatography. Anal. Biochem. 2003, 323, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Juan, G. DNA content measurement for DNA ploidy and cell cycle analysis. Curr. Protoc. Cytom. 2001. [Google Scholar] [CrossRef]
- Lee, K.-T.; Chen, Y.-H.; Lin, C.-I.; Chiu, W.-C.; Liao, H.; Lin, S.-H. Consumption of oriental plums improved the cognitive performance and modulated the cerebral neurodegeneration-related protein expressions in rats with nicotinamide/streptozotocin-induced diabetes. Food Nutr. Sci. 2013, 4, 1145–1154. [Google Scholar] [CrossRef]
- Westermann, P.; Knoblich, M.; Maier, O.; Lindschau, C.; Haller, H. Protein kinase C bound to the Golgi apparatus supports the formation of constitutive transport vesicles. Biochem. J. 1996, 320, 651–658. [Google Scholar] [PubMed]
- Bruns, R.F.; Miller, F.D.; Merriman, R.L.; Howbert, J.J.; Heath, W.F.; Kobayashi, E.; Takahashi, I.; Tamaoki, T.; Nakano, H. Inhibition of protein kinase C by calphostin C is light-dependent. Biochem. Biophys. Res. Commun. 1991, 176, 288–293. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, Y.-H.; Lin, C.-I.; Liao, H.; Chen, Y.-H.; Lin, S.-H. Palmitic Acid-Induced Neuron Cell Cycle G2/M Arrest and Endoplasmic Reticular Stress through Protein Palmitoylation in SH-SY5Y Human Neuroblastoma Cells. Int. J. Mol. Sci. 2014, 15, 20876-20899. https://doi.org/10.3390/ijms151120876
Hsiao Y-H, Lin C-I, Liao H, Chen Y-H, Lin S-H. Palmitic Acid-Induced Neuron Cell Cycle G2/M Arrest and Endoplasmic Reticular Stress through Protein Palmitoylation in SH-SY5Y Human Neuroblastoma Cells. International Journal of Molecular Sciences. 2014; 15(11):20876-20899. https://doi.org/10.3390/ijms151120876
Chicago/Turabian StyleHsiao, Yung-Hsuan, Ching-I Lin, Hsiang Liao, Yue-Hua Chen, and Shyh-Hsiang Lin. 2014. "Palmitic Acid-Induced Neuron Cell Cycle G2/M Arrest and Endoplasmic Reticular Stress through Protein Palmitoylation in SH-SY5Y Human Neuroblastoma Cells" International Journal of Molecular Sciences 15, no. 11: 20876-20899. https://doi.org/10.3390/ijms151120876