Advanced Glycation End Product-Induced Astrocytic Differentiation of Cultured Neurospheres through Inhibition of Notch-Hes1 Pathway-Mediated Neurogenesis

Abstract
:1. Introduction
2. Results and Discussion
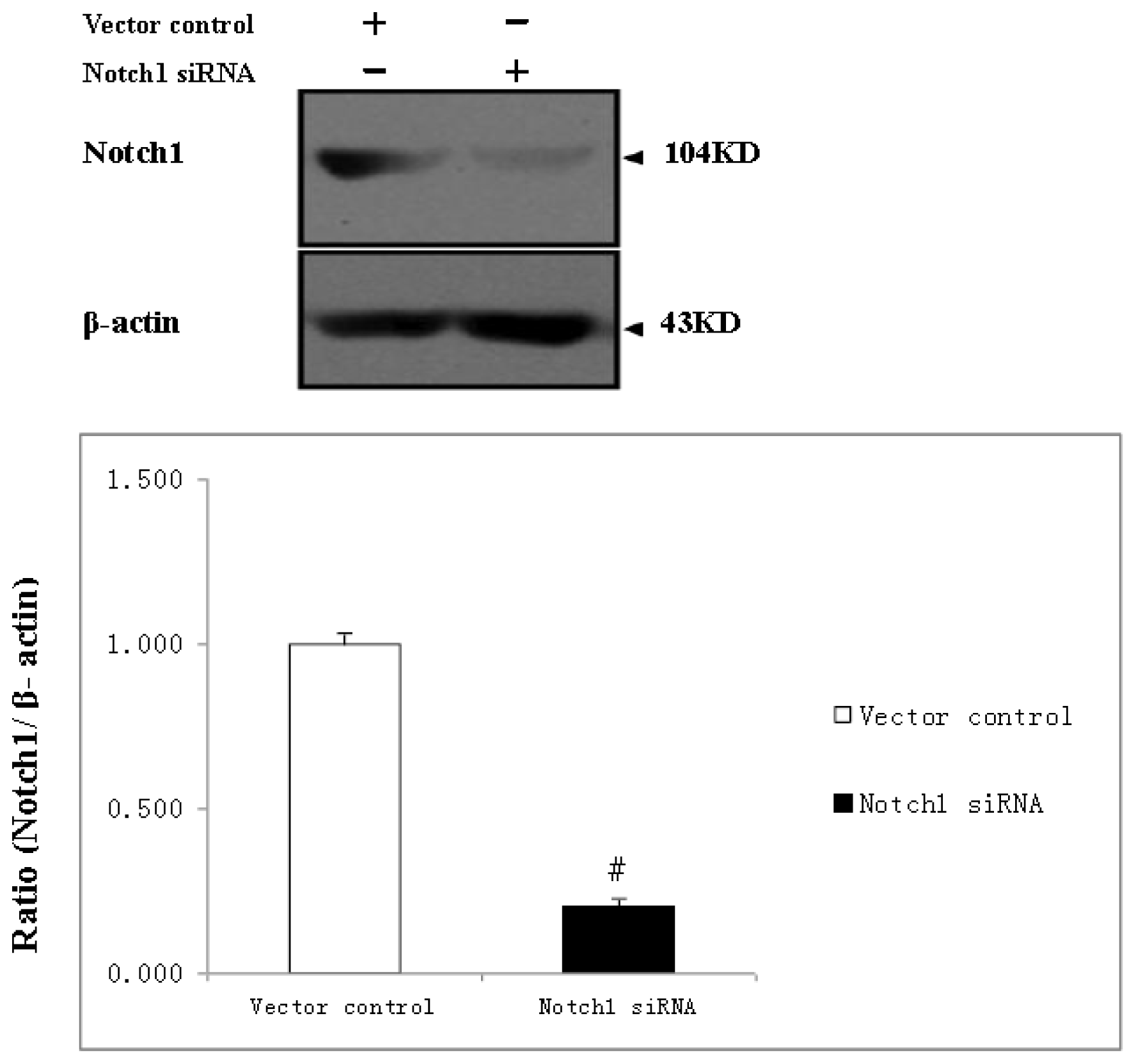
2.1. Reduced Efficacy of shRNAs
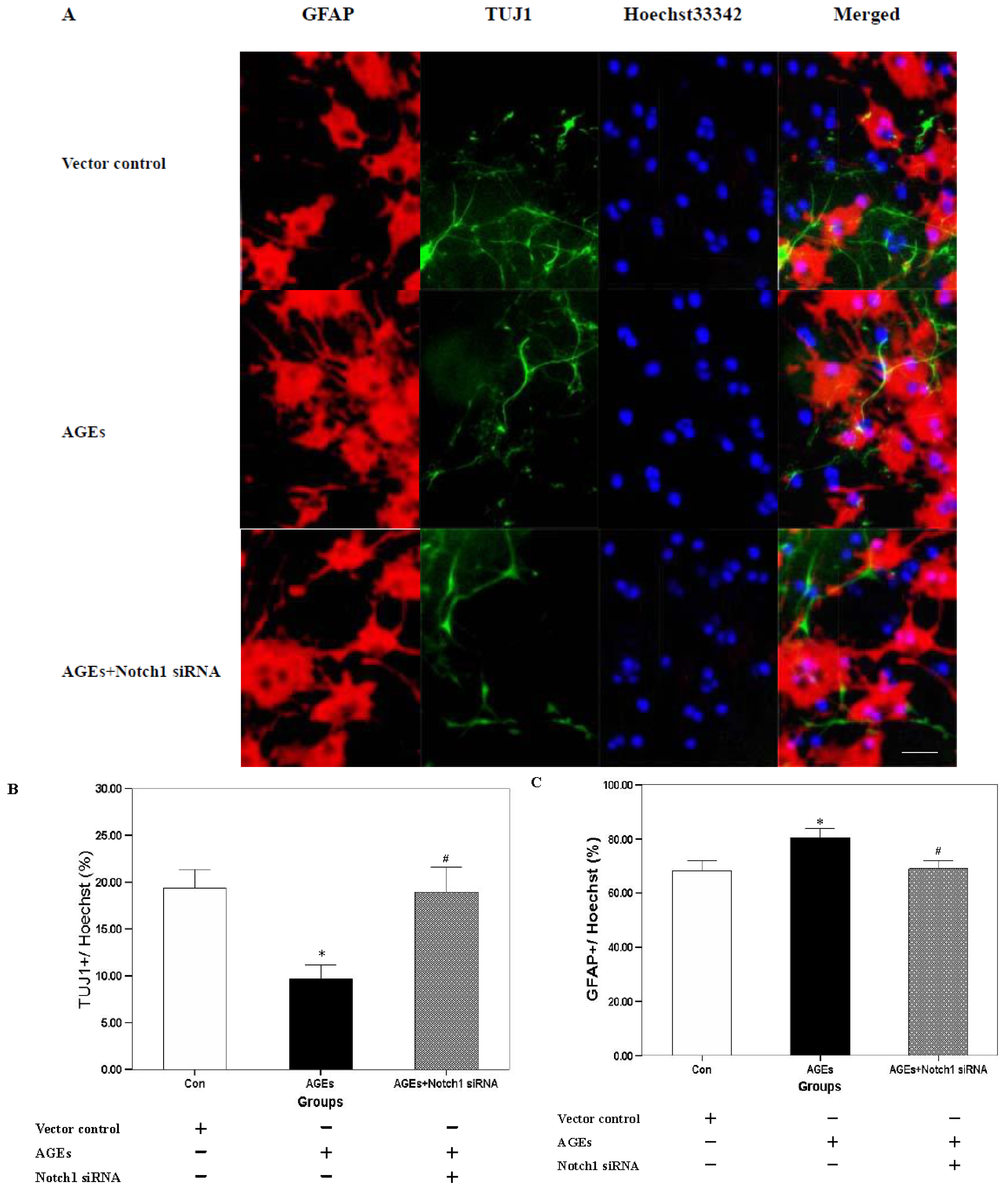
2.2. Effect of Notch-Hes1 Pathway Interference on the Differentiation of NSCs Incubated with AGE-BSA
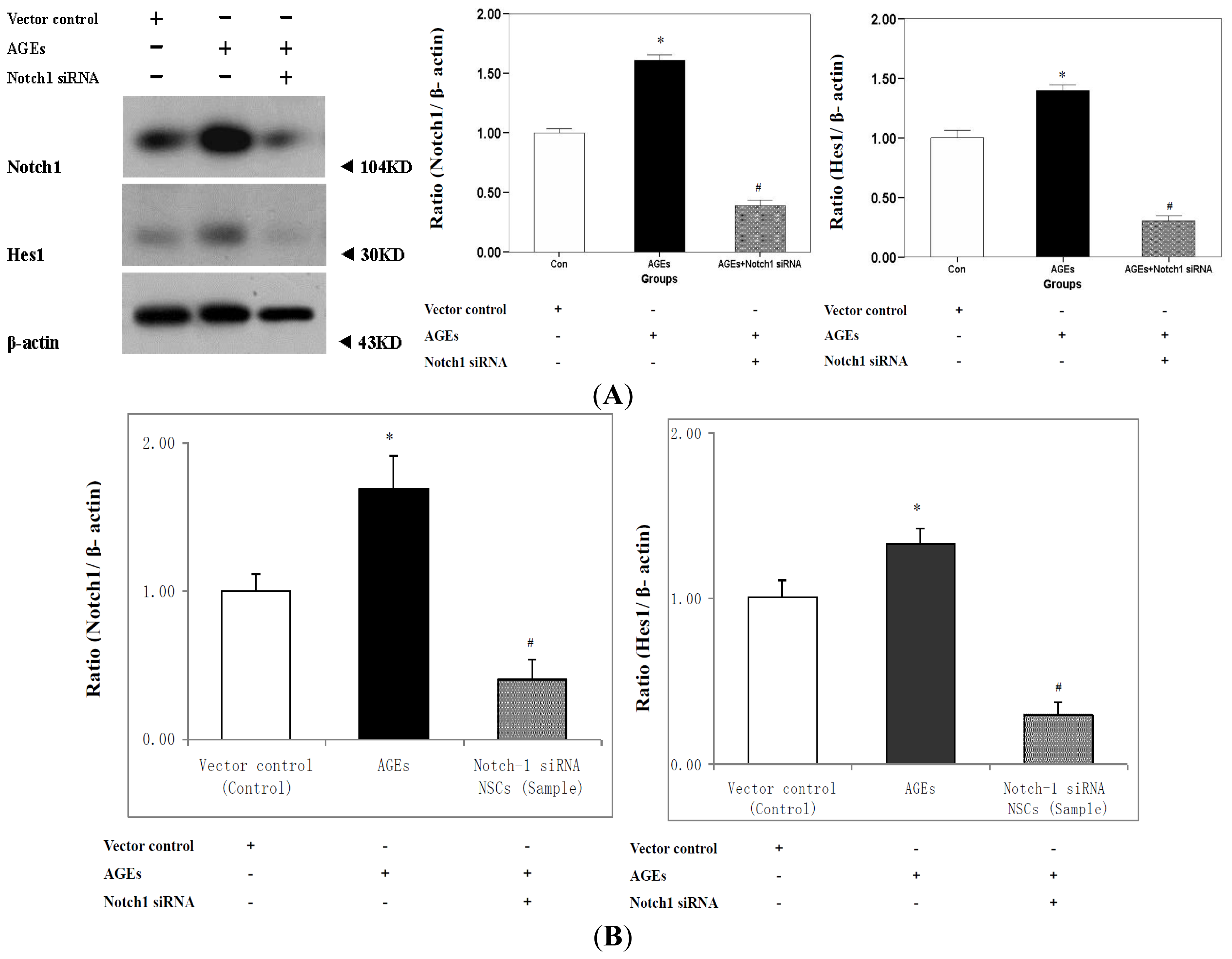
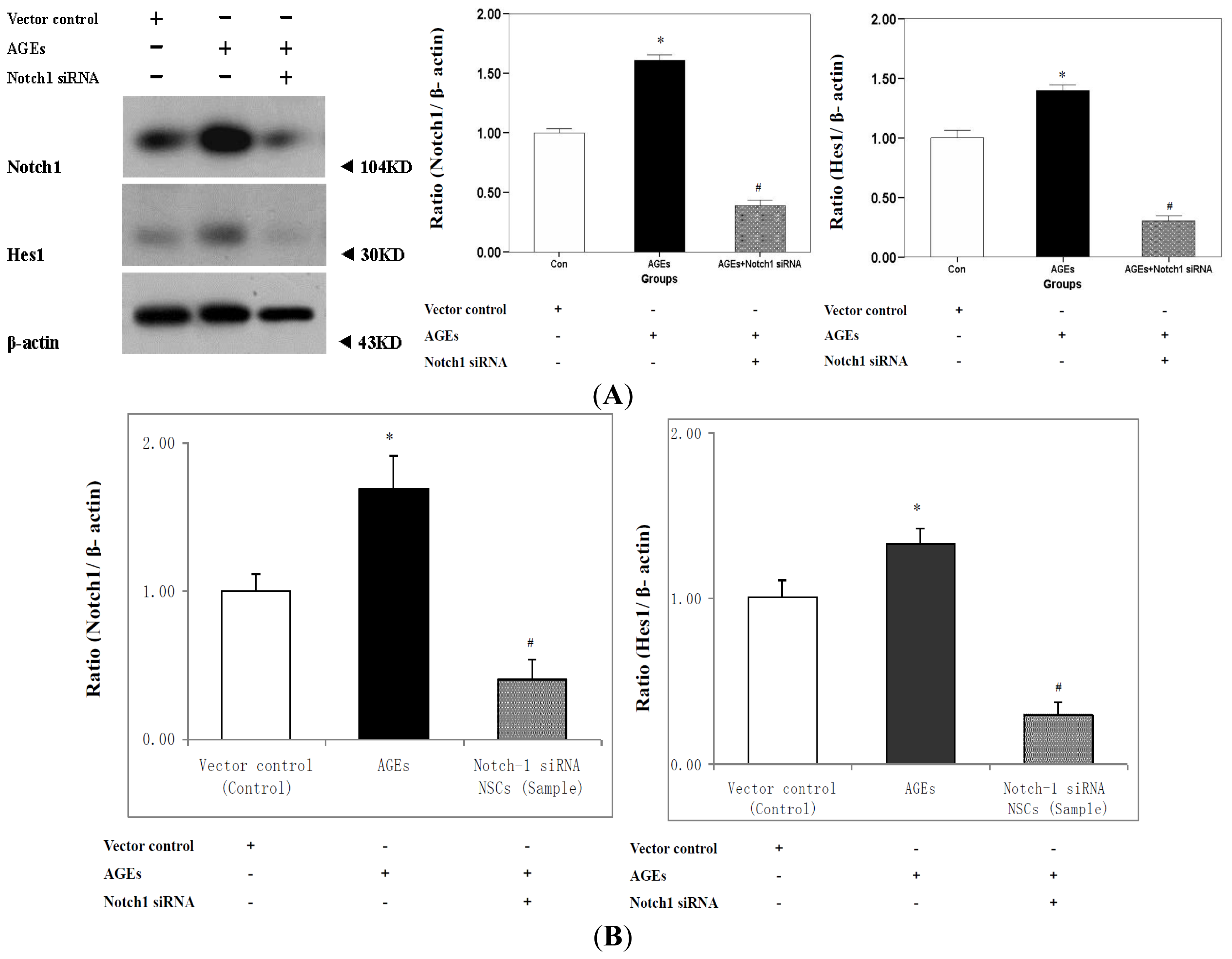
2.3. Expression of Notch-Hes1 Components of the NSCs Incubated with AGE-BSA
2.4. Discussion
3. Experimental Section
3.2. Cell Culture and Transfection
3.3. AGE-BSA Preparations
3.4. Evaluation of Differentiation
3.5. Protein and Gene Expression of Notch-Hes1 Pathway Components
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol 2004, 61, 661. [Google Scholar]
- Elias, M.F.; Elias, P.K.; Sullivan, L.M.; Wolf, P.A.; D’Agostino, R.B. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiol. Aging 2005, 26, 11–16. [Google Scholar]
- Elias, P.K.; Elias, M.F.; D’Agostino, R.B.; Cupples, L.A.; Wilson, P.W.; Silbershatz, H.; Wolf, P.A. NIDDM and blood pressure as risk factors for poor cognitive performance: The Framingham Study. Diabetes Care 1997, 20, 1388–1395. [Google Scholar]
- Yaffe, K.; Blackwell, T.; Kanaya, A.; Davidowitz, N.; Barrett-Connor, E.; Krueger, K. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology 2004, 63, 658–663. [Google Scholar]
- Little, A.A.; Edwards, J.L.; Feldman, E.L. Diabetic neuropathies. Pract. Neurol 2007, 7, 82–92. [Google Scholar]
- Gispen, W.H.; Biessels, G.-J. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci 2000, 23, 542–549. [Google Scholar]
- Bucala, R.; Cerami, A. Advanced glycosylation: Chemistry, biology, and implications for diabetes and aging. Adv. Pharmacol 1992, 23, 1–34. [Google Scholar]
- John, W.G.; Lamb, E.J. The Maillard or browning reaction in diabetes. Eye 1993, 7, 230–237. [Google Scholar]
- Henle, T.; Miyata, T. Advanced glycation end products in uremia. Adv. Renal Replace. Ther 2003, 10, 321–331. [Google Scholar]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. Ser. A 2010, 65, 963–975. [Google Scholar]
- Garay-Sevilla, M.; Regalado, J.; Malacara, J.; Nava, L.; Wrobel-Zasada, K.; Castro-Rivas, A.; Wrobel, K. Advanced glycosylation end products in skin, serum, saliva and urine and its association with complications of patients with type 2 diabetes mellitus. J. Endocrinol. Invest 2005, 28, 223–230. [Google Scholar]
- Schmidt, A.M.; Yan, S.D.; Stern, D.M. The dark side of glucose. Nat. Med 1995, 1, 1002–1004. [Google Scholar]
- Rojas, A.; Morales, M.A. Advanced glycation and endothelial functions: A link towards vascular complications in diabetes. Life Sci 2004, 76, 715–730. [Google Scholar]
- Wada, R.; Nishizawa, Y.; Yagihashi, N.; Takeuchi, M.; Ishikawa, Y.; Yasumura, K.; Nakano, M.; Yagihashi, S. Effects of OPB-9195, anti-glycation agent, on experimental diabetic neuropathy. Eur. J. Clin. Invest 2001, 31, 513–520. [Google Scholar]
- Takeuchi, M.; Yamagishi, S.-i. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimers disease. Curr. Pharm. Des 2008, 14, 973–978. [Google Scholar]
- Takeuchi, M.; Kikuchi, S.; Sasaki, N.; Suzuki, T.; Watai, T.; Iwaki, M.; Bucala, R.; Si, Y. Involvement of advanced glycation end-products (AGEs) in Alzheimers disease. Curr. Alzheimer Res 2004, 1, 39–46. [Google Scholar]
- Sato, T.; Iwaki, M.; Shimogaito, N.; Wu, X.; Yamagishi, S.-i.; Takeuchi, M. TAGE (toxic AGEs) theory in diabetic complications. Curr. Mol. Med 2006, 6, 351–358. [Google Scholar]
- Yamagishi, S.-i.; Imaizumi, T. Diabetic vascular complications: Pathophysiology, biochemical basis and potential therapeutic strategy. Curr. Pharm. Des 2005, 11, 2279–2299. [Google Scholar]
- Wang, S.-H.; Sun, Z.-L.; Guo, Y.-J.; Yuan, Y.; Li, L. PPARγ-mediated advanced glycation end products regulation of neural stem cells. Mol. Cell. Endocrinol 2009, 307, 176–184. [Google Scholar]
- Alenzi, F.Q.; Bahkali, A.H. Stem cells: Biology and clinical potential. Afr. J. Biotechnol 2011, 10, 19929–19940. [Google Scholar]
- Paspala, S.; Murthy, T.; Mahaboob, V.; Habeeb, M. Pluripotent stem cells—A review of the current status in neural regeneration. Neurol. India 2011, 59, 558. [Google Scholar]
- Abrous, D.N.; Koehl, M.; le Moal, M. Adult neurogenesis: From precursors to network and physiology. Physiol. Rev 2005, 85, 523–569. [Google Scholar]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci 2008, 11, 309–317. [Google Scholar]
- Zhang, W.J.; Tan, Y.F.; Yue, J.; Vranic, M.; Wojtowicz, J. Impairment of hippocampal neurogenesis in streptozotocin-treated diabetic rats. Acta Neurol. Scand 2008, 117, 205–210. [Google Scholar]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.-K.; Kittappa, R.; McKay, R.D. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar]
- Kawai, T.; Takagi, N.; Nakahara, M.; Takeo, S. Changes in the expression of Hes5 and Mash1 mRNA in the adult rat dentate gyrus after transient forebrain ischemia. Neurosci. Lett 2005, 380, 17–20. [Google Scholar]
- Lindsell, C.E.; Boulter, J.; diSibio, G.; Gossler, A.; Weinmaster, G. Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol. Cell. Neurosci 1996, 8, 14–27. [Google Scholar]
- Le Borgne, R.; Bardin, A.; Schweisguth, F. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development 2005, 132, 1751–1762. [Google Scholar]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res 2005, 306, 343–348. [Google Scholar]
- Lasky, J.L.; Wu, H. Notch signaling, brain development, and human disease. Pediatr. Res 2005, 57, 104R–109R. [Google Scholar]
- Chiba, S. Concise review: Notch signaling in stem cell systems. Stem Cells 2006, 24, 2437–2447. [Google Scholar]
- Grandbarbe, L.; Bouissac, J.; Rand, M.; de Angelis, M.H.; Artavanis-Tsakonas, S.; Mohier, E. δ-Notch signaling controls the generation of neurons/glia from neural stem cells in a stepwise process. Development 2003, 130, 1391–1402. [Google Scholar]
- Van Praag, H.; Schinder, A.F.; Christie, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034. [Google Scholar]
- Paton, J.A.; Nottebohm, F.N. Neurons generated in the adult brain are recruited into functional circuits. Science 1984, 225, 1046–1048. [Google Scholar]
- Cameron, H.A.; Mckay, R.D. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comparat. Neurol 2001, 435, 406–417. [Google Scholar]
- Gould, E.; Tanapat, P.; McEwen, B.S.; Flügge, G.; Fuchs, E. Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc. Natl. Acad. Sci. USA 1998, 95, 3168–3171. [Google Scholar]
- Jin, K.; Minami, M.; Lan, J.Q.; Mao, X.O.; Batteur, S.; Simon, R.P.; Greenberg, D.A. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. USA 2001, 98, 4710–4715. [Google Scholar]
- Drapeau, E.; Mayo, W.; Aurousseau, C.; Le Moal, M.; Piazza, P.-V.; Abrous, D.N. Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 14385–14390. [Google Scholar]
- Haughey, N.J.; Liu, D.; Nath, A.; Borchard, A.C.; Mattson, M.P. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid β-peptide. Neuromol. Med 2002, 1, 125–135. [Google Scholar]
- Shors, T.J.; Miesegaes, G.; Beylin, A.; Zhao, M.; Rydel, T.; Gould, E. Neurogenesis in the adult is involved in the formation of trace memories. Nature 2001, 410, 372–376. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol 2010, 119, 7–35. [Google Scholar]
- Baydas, G.; Reiter, R.J.; Yasar, A.; Tuzcu, M.; Akdemir, I.; Nedzvetskii, V.S. Melatonin reduces glial reactivity in the hippocampus, cortex, and cerebellum of streptozotocin-induced diabetic rats. Free Radic. Biol. Med 2003, 35, 797–804. [Google Scholar]
- Lechuga-Sancho, A.M.; Arroba, A.I.; Frago, L.M.; Pañeda, C.; García-Cáceres, C.; Delgado Rubín de Célix, A.; Argente, J.; Chowen, J.A. Activation of the intrinsic cell death pathway, increased apoptosis and modulation of astrocytes in the cerebellum of diabetic rats. Neurobiol. Dis 2006, 23, 290–299. [Google Scholar]
- Saravia, F.E.; Revsin, Y.; Gonzalez Deniselle, M.C.; Gonzalez, S.L.; Roig, P.; Lima, A.; Homo-Delarche, F.; de Nicola, A.F. Increased astrocyte reactivity in the hippocampus of murine models of type 1 diabetes: The nonobese diabetic (NOD) and streptozotocin-treated mice. Brain Res 2002, 957, 345–353. [Google Scholar]
- Baydas, G.; Donder, E.; Kiliboz, M.; Sonkaya, E.; Tuzcu, M.; Yasar, A.; Nedzvetskii, V. Neuroprotection by α-lipoic acid in streptozotocin-induced diabetes. Biochemistry 2004, 69, 1001–1005. [Google Scholar]
- Pugliese, G.; Pricci, F.; Romeo, G.; Pugliese, F.; Mené, P.; Giannini, S.; Cresci, B.; Galli, G.; Rotella, C.M.; Vlassara, H. Upregulation of mesangial growth factor and extracellular matrix synthesis by advanced glycation end products via a receptor-mediated mechanism. Diabetes 1997, 46, 1881–1887. [Google Scholar]
- Wang, S.-H.; Guo, Y.-J.; Yuan, Y.; Li, L.; Li, F.-F.; Ye, K.-P.; Huang, Y. PPARγ-mediated advanced glycation end products regulate neural stem cell proliferation but not neural differentiation through the BDNF-CREB pathway. Toxicol. Lett 2011, 206, 339–346. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC t method. Methods 2001, 25, 402–408. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Genes | Primers |
|---|---|
| Notch-1-F | 5′-CCGCTGTGAGTCGGTCATTA-3′ |
| Notch-1-R | 5′-GGCACCTACAGATGAATCCA-3′ |
| Hes1-F | 5′-TTCAGCGAGTGCATGAACGA-3′ |
| Hes1-R | 5′-GTAGGTCATGGCGTTGATCT-3′ |
| β-actin-F | 5′-CCTAGGCACCAGGGTGTGAT-3′ |
| β-actin-R | 5′-TTGGTGACAATGCCGTGTTC-3′ |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, Y.; Wang, P.; Sun, H.; Cai, R.; Xia, W.; Wang, S. Advanced Glycation End Product-Induced Astrocytic Differentiation of Cultured Neurospheres through Inhibition of Notch-Hes1 Pathway-Mediated Neurogenesis. Int. J. Mol. Sci. 2014, 15, 159-170. https://doi.org/10.3390/ijms15010159
Guo Y, Wang P, Sun H, Cai R, Xia W, Wang S. Advanced Glycation End Product-Induced Astrocytic Differentiation of Cultured Neurospheres through Inhibition of Notch-Hes1 Pathway-Mediated Neurogenesis. International Journal of Molecular Sciences. 2014; 15(1):159-170. https://doi.org/10.3390/ijms15010159
Chicago/Turabian StyleGuo, Yijing, Pin Wang, Haixia Sun, Rongrong Cai, Wenqing Xia, and Shaohua Wang. 2014. "Advanced Glycation End Product-Induced Astrocytic Differentiation of Cultured Neurospheres through Inhibition of Notch-Hes1 Pathway-Mediated Neurogenesis" International Journal of Molecular Sciences 15, no. 1: 159-170. https://doi.org/10.3390/ijms15010159