Genetic Variants of GPER/GPR30, a Novel Estrogen-Related G Protein Receptor, Are Associated with Human Seminoma

Abstract
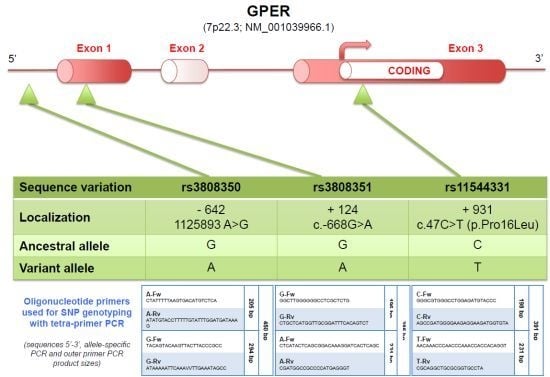
:
1. Introduction
2. Results and Discussion
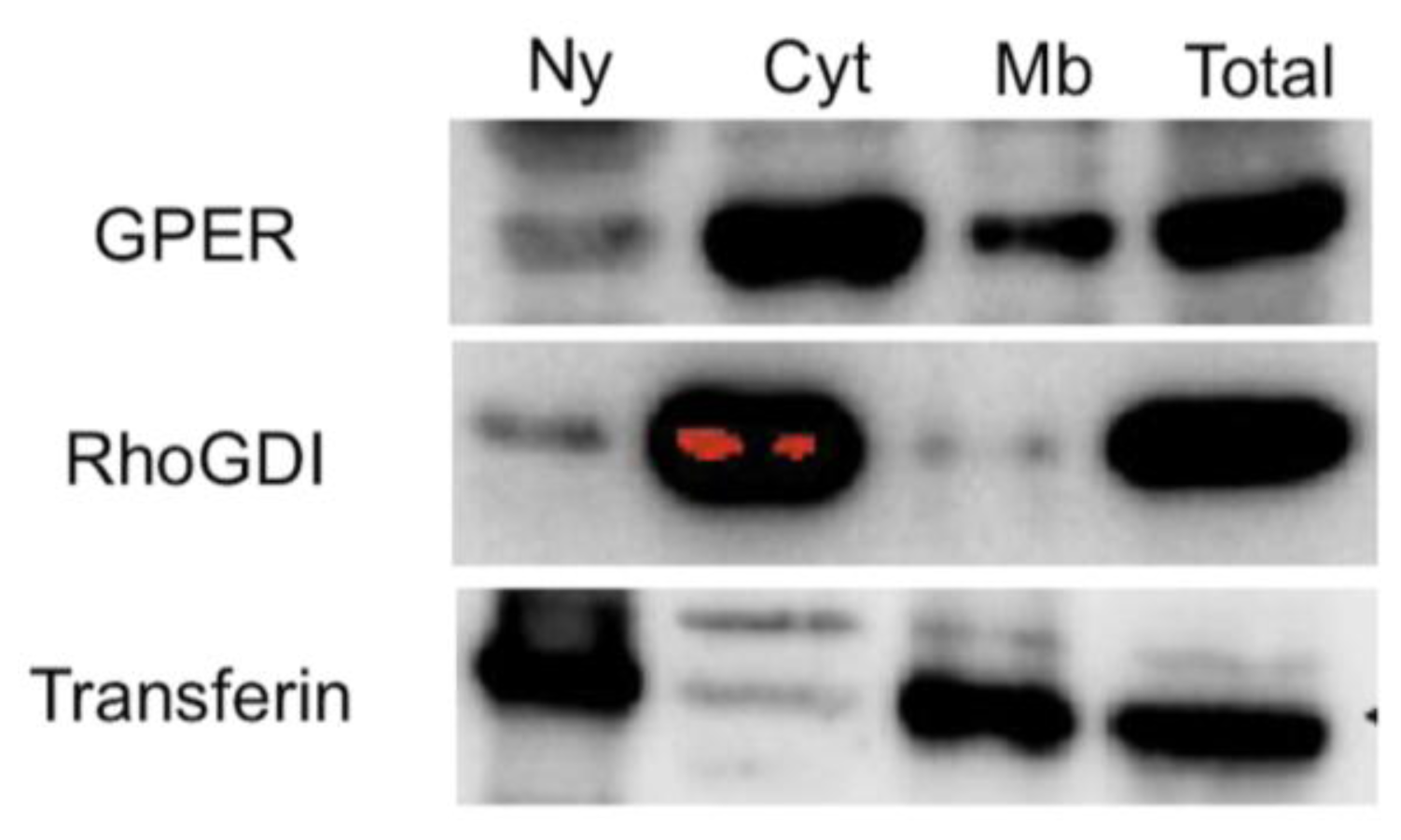
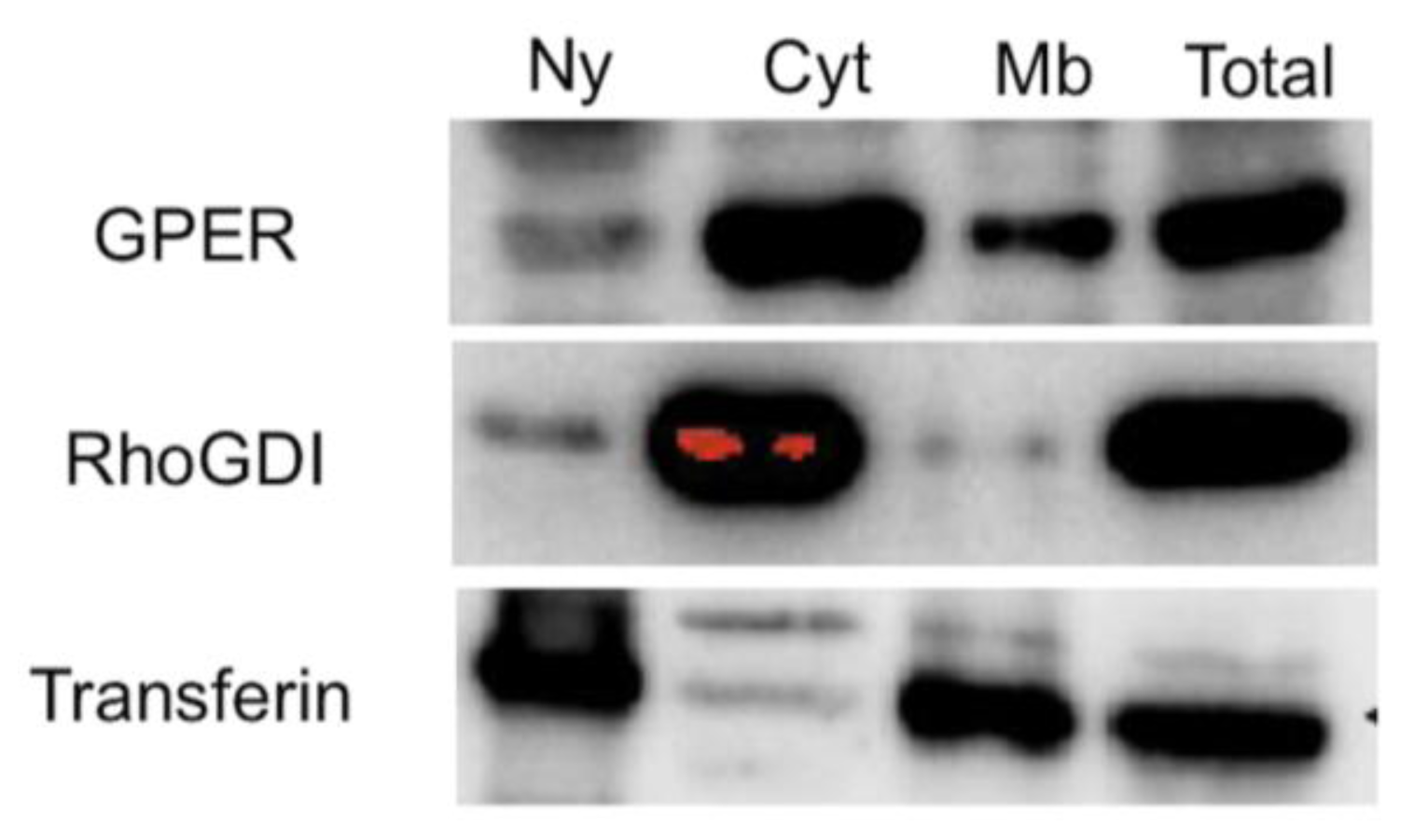
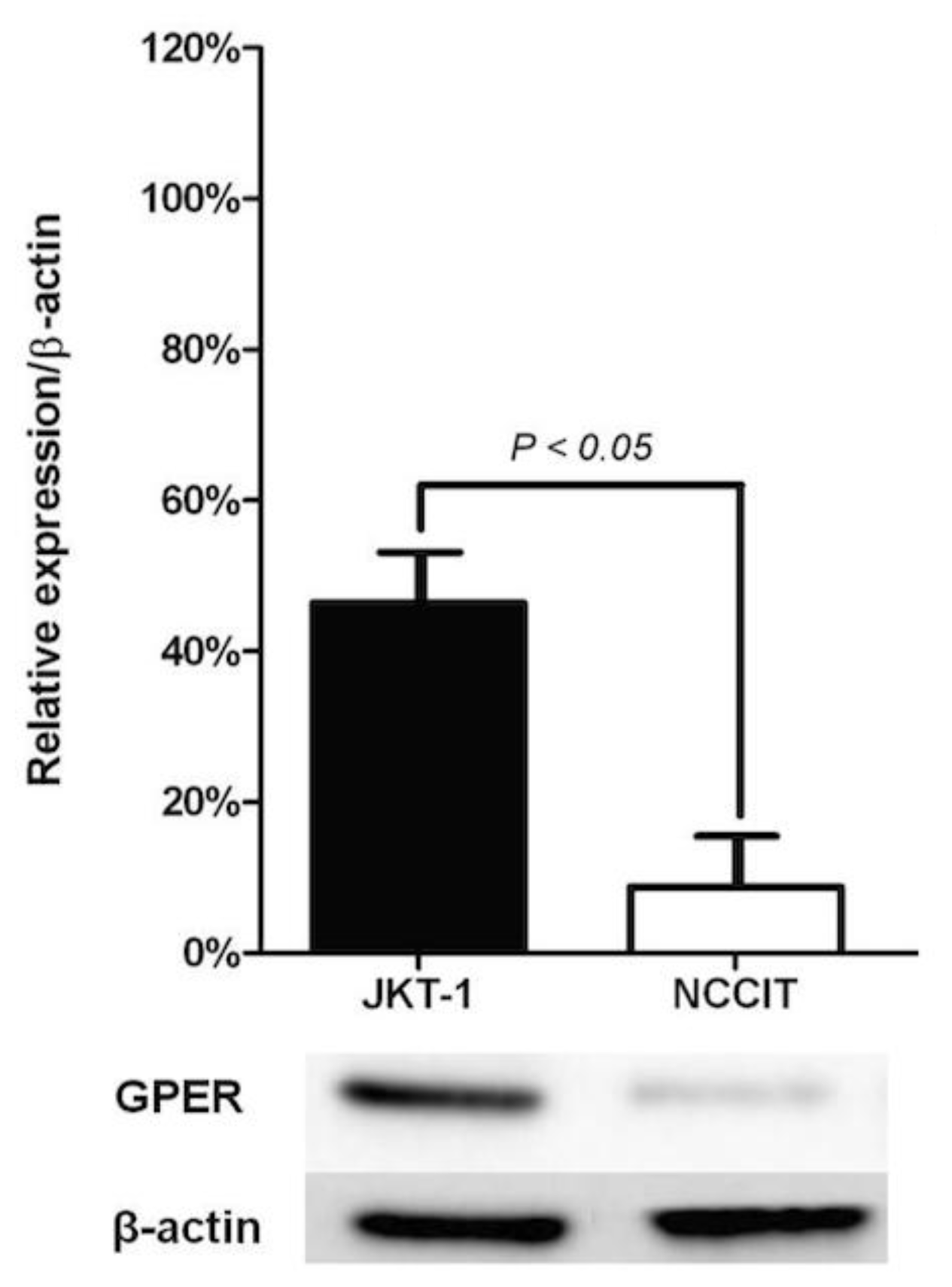
2.1. Localization of GPER in Human Seminoma-Derived Cells
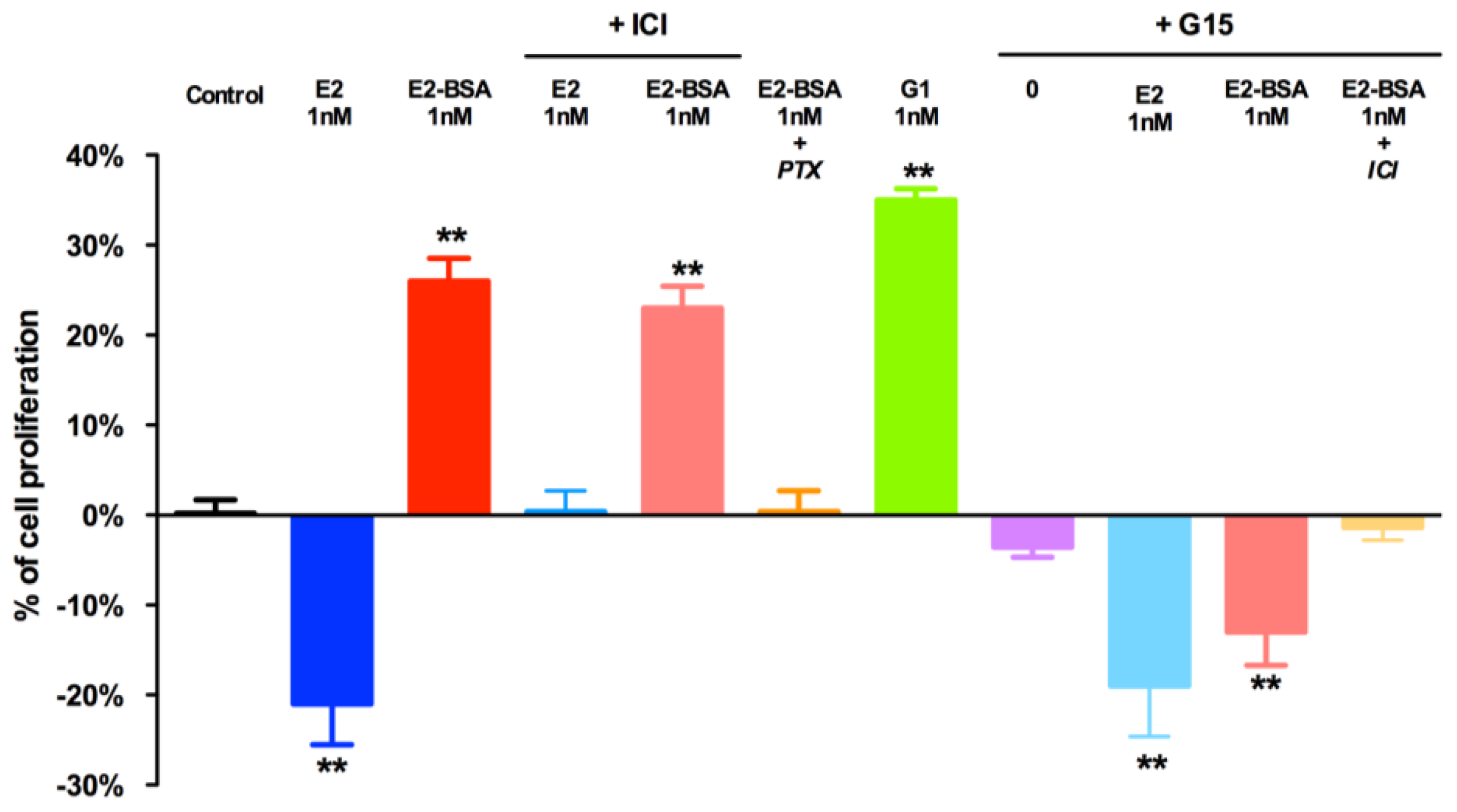
2.2. Role of GPER in Human Seminoma-Derived Cells Proliferation
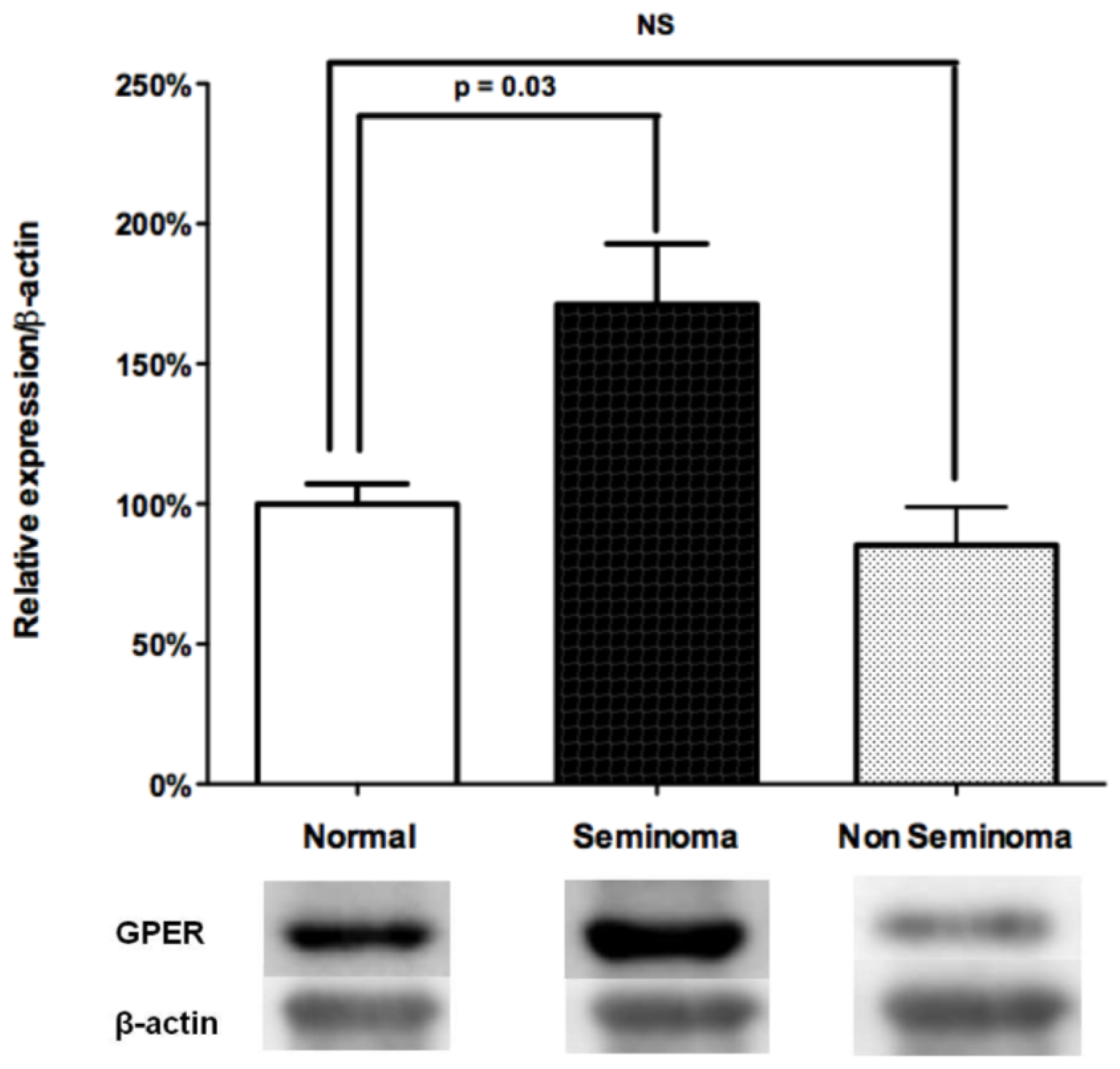
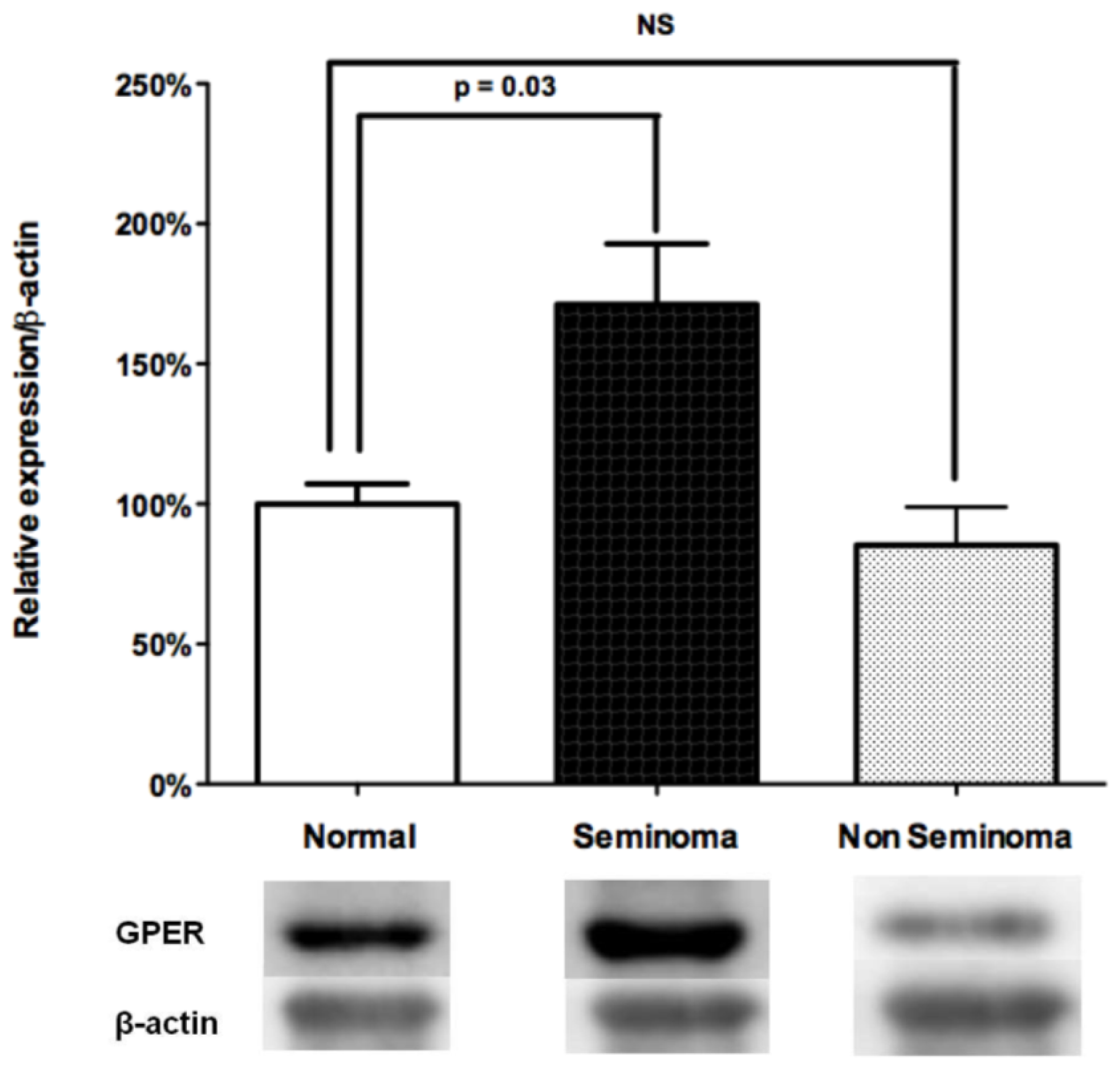
2.3. Overexpression of GPER in Human Seminomas
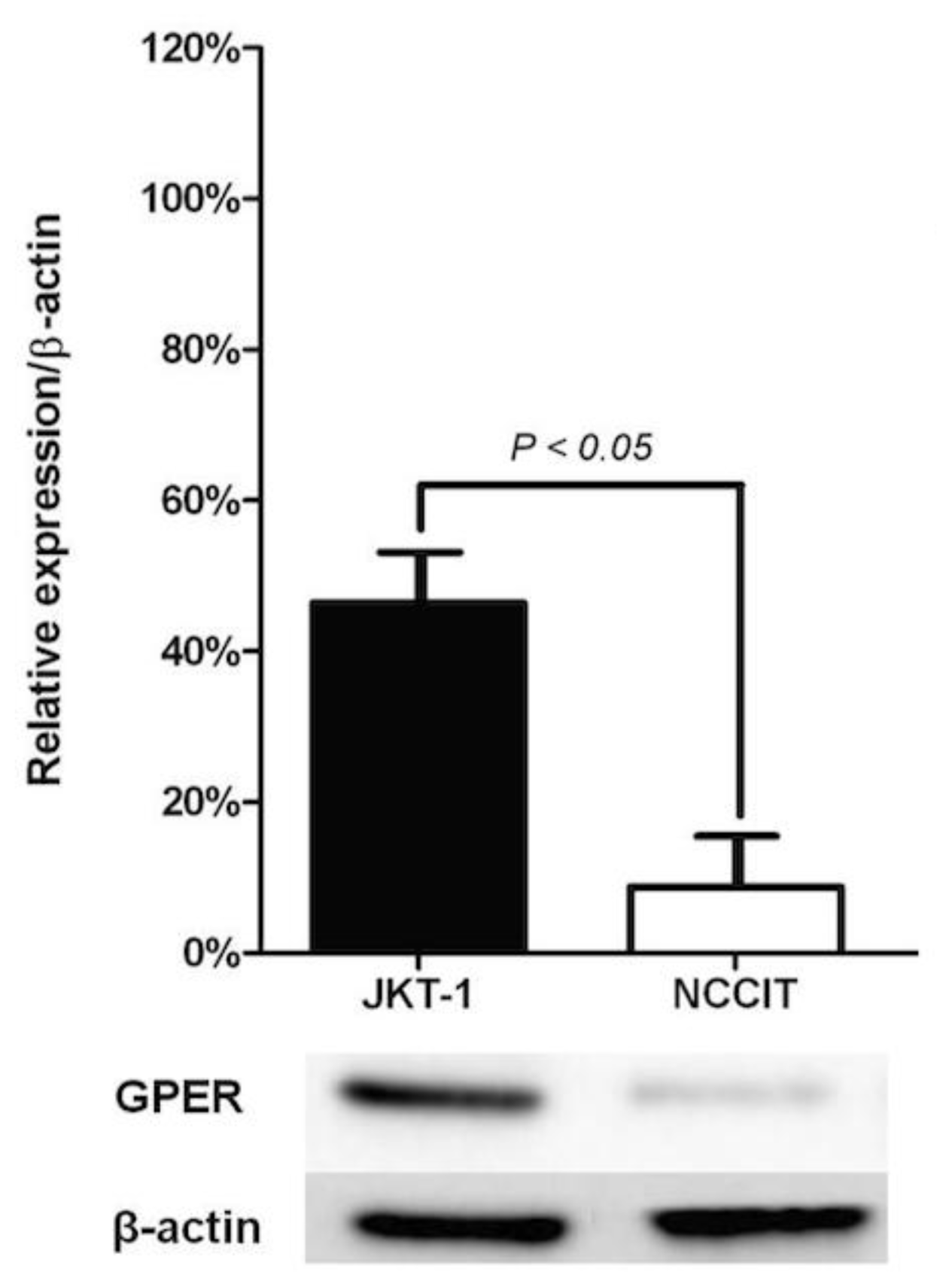
2.3.1. Relative Expression of GPER in Distinct TGCT
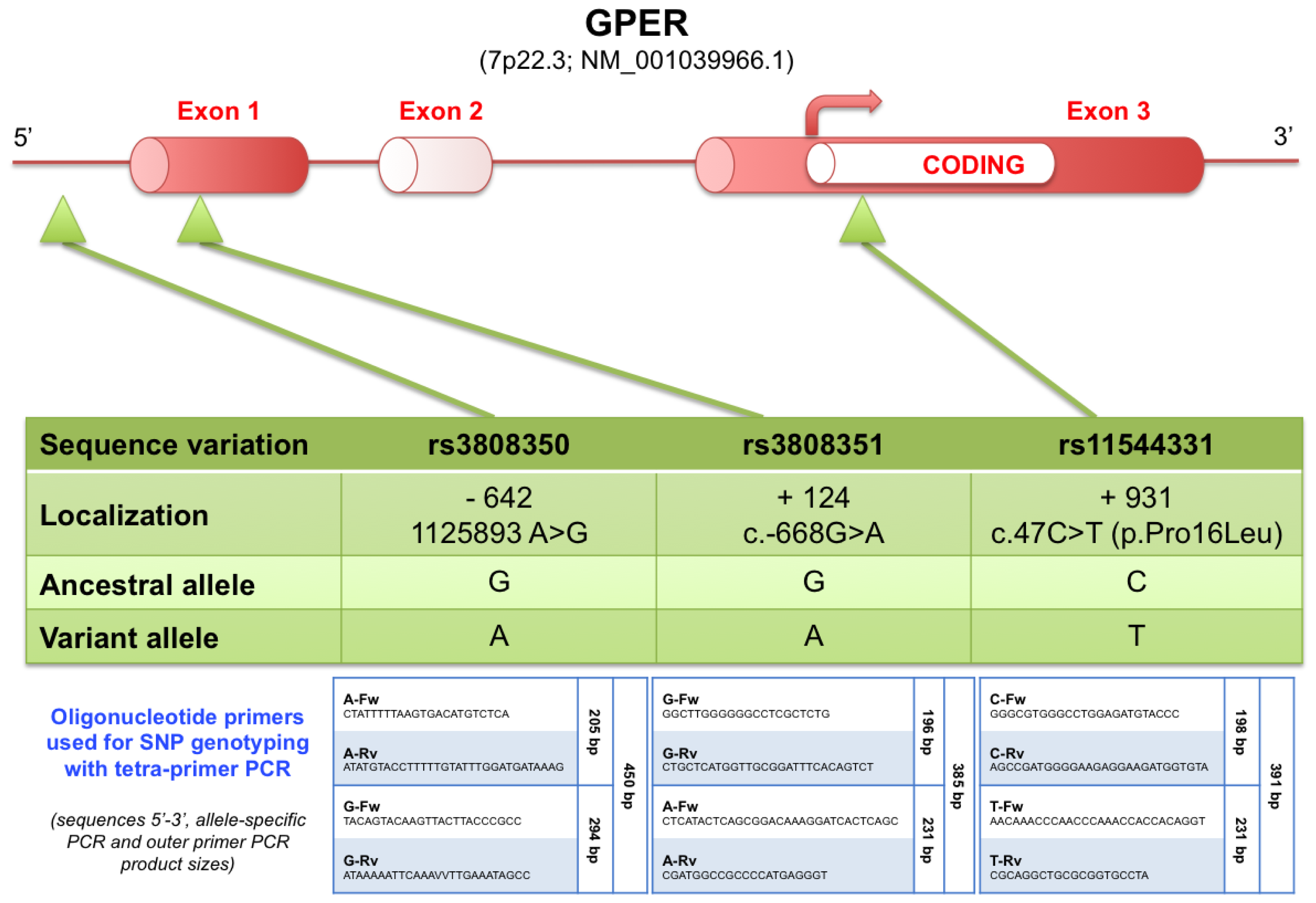
2.3.2. Mechanisms Leading to Overexpression of GPER in Human Seminomas
3. Experimental Section
3.1. Cell Culture
3.2. Subcellular Fractionation
3.3. Cell Proliferation Assay
3.4. Protein Purification and Western Blotting of Cells and Tumor Samples
3.5. SNP Analysis of Tumor Samples
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Richiardi, L.; Ekbom, A.; Pukkala, E.; Cuninkova, M.; Moller, H. Trends in testicular cancer incidence and mortality in 22 European countries: Continuing increases in incidence and declines in mortality. Int. J. Cancer 2006, 118, 3099–3111. [Google Scholar]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Jorgensen, N.; Carlsen, E.; Petersen, P.M.; Giwercman, A.; Andersen, A.G.; Jensen, T.K.; Andersson, A.M.; Muller, J. Germ cell cancer and disorders of spermatogenesis: An environmental connection? APMIS 1998, 106, 3–11, discussion 12. [Google Scholar]
- Hardell, L.; van Bavel, B.; Lindstrom, G.; Carlberg, M.; Dreifaldt, A.C.; Wijkstrom, H.; Starkhammar, H.; Eriksson, M.; Hallquist, A.; Kolmert, T. Increased concentrations of polychlorinated biphenyls, hexachlorobenzene, and chlordanes in mothers of men with testicular cancer. Environ. Health Perspect 2003, 111, 930–934. [Google Scholar]
- Rajpert-De Meyts, E. Developmental model for the pathogenesis of testicular carcinoma in situ: Genetic and environmental aspects. Hum. Reprod. Update 2006, 12, 303–323. [Google Scholar]
- Jones, M.E.; Simpson, E.R. Oestrogens in male reproduction. Bailliere’s Best Pract. Res. Clin. Endocrinol. Metab 2000, 14, 505–516. [Google Scholar]
- Carreau, S.; Bourguiba, S.; Lambard, S.; Galeraud-Denis, I.; Genissel, C.; Levallet, J. Reproductive system: Aromatase and estrogens. Mol. Cell. Endocrinol 2002, 193, 137–143. [Google Scholar]
- Lambard, S.; Galeraud-Denis, I.; Saunders, P.T.; Carreau, S. Human immature germ cells and ejaculated spermatozoa contain aromatase and oestrogen receptors. J. Mol. Endocrinol 2004, 32, 279–289. [Google Scholar]
- Li, H.; Papadopoulos, V.; Vidic, B.; Dym, M.; Culty, M. Regulation of rat testis gonocyte proliferation by platelet-derived growth factor and estradiol: Identification of signaling mechanisms involved. Endocrinology 1997, 138, 1289–1298. [Google Scholar]
- Pentikainen, V.; Erkkila, K.; Suomalainen, L.; Parvinen, M.; Dunkel, L. Estradiol acts as a germ cell survival factor in the human testis in vitro. J. Clin. Endocrinol. Metab. 2000, 85, 2057–2067. [Google Scholar]
- Pais, V.; Leav, I.; Lau, K.M.; Jiang, Z.; Ho, S.M. Estrogen receptor-beta expression in human testicular germ cell tumors. Clin. Cancer Res 2003, 9, 4475–4482. [Google Scholar]
- Roger, C.; Lambard, S.; Bouskine, A.; Mograbi, B.; Chevallier, D.; Nebout, M.; Pointis, G.; Carreau, S.; Fenichel, P. Estrogen-induced growth inhibition of human seminoma cells expressing estrogen receptor beta and aromatase. J. Mol. Endocrinol 2005, 35, 191–199. [Google Scholar]
- Guido, C.; Panza, S.; Santoro, M.; Avena, P.; Panno, M.L.; Perrotta, I.; Giordano, F.; Casaburi, I.; Catalano, S.; de Amicis, F.; et al. Estrogen receptor beta (ERbeta) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle 2012, 11, 2911–2921. [Google Scholar]
- Kinugawa, K.; Hyodo, F.; Matsuki, T.; Jo, Y.; Furukawa, Y.; Ueki, A.; Tanaka, H. Establishment and characterization of a new human testicular seminoma cell line, JKT-1. Int. J. Urol 1998, 5, 282–287. [Google Scholar]
- Delbes, G.; Levacher, C.; Pairault, C.; Racine, C.; Duquenne, C.; Krust, A.; Habert, R. Estrogen receptor beta-mediated inhibition of male germ cell line development in mice by endogenous estrogens during perinatal life. Endocrinology 2004, 145, 3395–3403. [Google Scholar]
- Bouskine, A.; Nebout, M.; Mograbi, B.; Brucker-Davis, F.; Roger, C.; Fenichel, P. Estrogens promote human testicular germ cell cancer through a membrane-mediated activation of extracellular regulated kinase and protein kinase A. Endocrinology 2008, 149, 565–573. [Google Scholar]
- Chevalier, N.; Vega, A.; Bouskine, A.; Siddeek, B.; Michiels, J.F.; Chevallier, D.; Fenichel, P. GPR30, the non-classical membrane G protein related estrogen receptor, is overexpressed in human seminoma and promotes seminoma cell proliferation. PLoS One 2012, 7, e34672. [Google Scholar]
- HUGO Gene Nomenclature Committee Home page. Available online: http://www.genenames.org (accessed on 24 February 2012).
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar]
- Huang, G.S.; Gunter, M.J.; Arend, R.C.; Li, M.; Arias-Pulido, H.; Prossnitz, E.R.; Goldberg, G.L.; Smith, H.O. Co-expression of GPR30 and ERbeta and their association with disease progression in uterine carcinosarcoma. Am. J. Obstet. Gynecol 2010, 203. [Google Scholar]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol 2009, 114, 465–471. [Google Scholar]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res 2006, 12, 6359–6366. [Google Scholar]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Ando, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res 2007, 67, 1859–1866. [Google Scholar]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: Its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; de Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect 2012, 120, 1177–1182. [Google Scholar]
- Albanito, L.; Lappano, R.; Madeo, A.; Chimento, A.; Prossnitz, E.R.; Cappello, A.R.; Dolce, V.; Abonante, S.; Pezzi, V.; Maggiolini, M. G-protein-coupled receptor 30 and estrogen receptor-alpha are involved in the proliferative effects induced by atrazine in ovarian cancer cells. Environ. Health Perspect 2008, 116, 1648–1655. [Google Scholar]
- Chevalier, N.; Bouskine, A.; Fenichel, P. Bisphenol A promotes testicular seminoma cell proliferation through GPER/GPR30. Int. J. Cancer 2012, 130, 241–242. [Google Scholar]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.I.; Sklar, L.A.; Hathaway, H.J. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol 2008, 70, 165–190. [Google Scholar]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar]
- Maggiolini, M.; Picard, D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol 2010, 204, 105–114. [Google Scholar]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol 2002, 16, 70–84. [Google Scholar]
- Cheng, S.B.; Graeber, C.T.; Quinn, J.A.; Filardo, E.J. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids 2011, 76, 892–896. [Google Scholar]
- Franco, R.; Boscia, F.; Gigantino, V.; Marra, L.; Esposito, F.; Ferrara, D.; Pariante, P.; Botti, G.; Caraglia, M.; Minucci, S.; et al. GPR30 is overexpressed in post-puberal testicular germ cell tumors. Cancer Biol. Ther 2011, 11, 609–613. [Google Scholar]
- Rago, V.; Romeo, F.; Giordano, F.; Maggiolini, M.; Carpino, A. Identification of the estrogen receptor GPER in neoplastic and non-neoplastic human testes. Reprod. Biol. Endocrinol 2011, 9, 135. [Google Scholar]
- Bouskine, A.; Nebout, M.; Brucker-Davis, F.; Benahmed, M.; Fenichel, P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ. Health Perspect 2009, 117, 1053–1058. [Google Scholar]
- Ye, S.; Dhillon, S.; Ke, X.; Collins, A.R.; Day, I.N. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res 2001, 29, e88. [Google Scholar]
- Ferlin, A.; Ganz, F.; Pengo, M.; Selice, R.; Frigo, A.C.; Foresta, C. Association of testicular germ cell tumor with polymorphisms in estrogen receptor and steroid metabolism genes. Endocr.-Relat. Cancer 2010, 17, 17–25. [Google Scholar]
- dbSNp Home page. Available online: http://www.ncbi.nlm.nih.gov/SNP (accessed on 24 February 2012).
- Kakinuma, N.; Sato, M.; Yamada, T.; Kohu, K.; Nakajima, M.; Akiyama, T.; Ohwada, S.; Shibanaka, Y. Cloning of novel LERGU mRNAs in GPR30 3′ untranslated region and detection of 2 bp-deletion polymorphism in gastric cancer. Cancer Sci 2005, 96, 191–196. [Google Scholar]
- Giess, M.; Lattrich, C.; Springwald, A.; Goerse, R.; Ortmann, O.; Treeck, O. GPR30 gene polymorphisms are associated with progesterone receptor status and histopathological characteristics of breast cancer patients. J. Steroid Biochem. Mol. Biol 2010, 118, 7–12. [Google Scholar]
- Chevalier, N.; Barlier, A.; Roche, C.; Mograbi, B.; Camparo, P.; Devouassoux-Shisheboran, M.; Michiels, J.F.; Nebout, M.; Chevallier, D.; Benahmed, M.; et al. RET gene mutations are not involved in the origin of human testicular seminoma. Int. J. Androl 2010, 33, 848–852. [Google Scholar]
- Bouskine, A.; Vega, A.; Nebout, M.; Benahmed, M.; Fenichel, P. Expression of embryonic stem cell markers in cultured JKT-1, a cell line derived from a human seminoma. Int. J. Androl 2010, 33, 54–63. [Google Scholar]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol 2009, 5, 421–427. [Google Scholar]
- Roger, C.; Mograbi, B.; Chevallier, D.; Michiels, J.F.; Tanaka, H.; Segretain, D.; Pointis, G.; Fenichel, P. Disrupted traffic of connexin 43 in human testicular seminoma cells: Overexpression of Cx43 induces membrane location and cell proliferation decrease. J. Pathol 2004, 202, 241–246. [Google Scholar]
- Filardo, E.J. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: A novel signaling pathway with potential significance for breast cancer. J. Steroid Biochem. Mol. Biol 2002, 80, 231–238. [Google Scholar]
- Vivacqua, A.; Bonofiglio, D.; Recchia, A.G.; Musti, A.M.; Picard, D.; Ando, S.; Maggiolini, M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol. Endocrinol 2006, 20, 631–646. [Google Scholar]

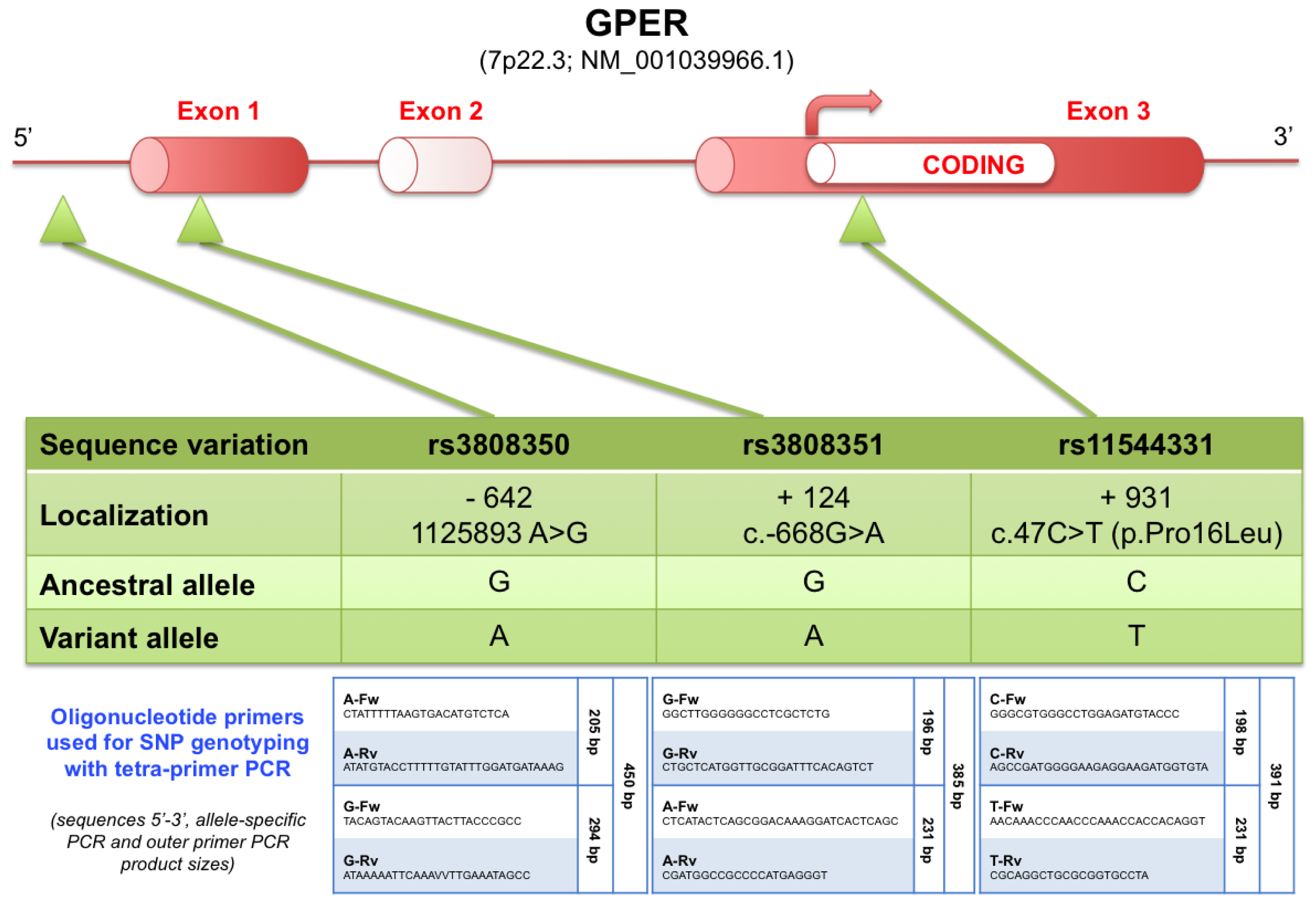

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype frequency | Allele frequency | |||||
|---|---|---|---|---|---|---|
| Sequence variation (localisation) | AA | AG | GG | A | G | |
| rs3808350 (1125893 A > G) | Reference population (n = 224) | 0.366 (n = 82) | 0.491 (n = 110) | 0.143 (n = 32) | 0.612 (n = 274) | 0.388 (n = 174) |
| Seminoma (somatic) (n = 81) | 0.531 (n = 43) | 0.444 (n = 36) | 0.025 (n = 2) | 0.753 (n = 122) | 0.247 (n = 40) | |
| p | 0.0122 | 0.5174 | 0.0032 | 0.0011 | ||
| Non seminoma (somatic) (n = 8) | 0.250 (n = 2) | 0.625 (n = 5) | 0.125 (n = 1) | 0.562 (n = 9) | 0.438 (n = 7) | |
| p | 0.7142 | 0.4973 | 1.0000 | 0.7956 | ||
| rs3808351 (c.-668 G > A) | Reference population (n = 222) | 0.054 (n = 12) | 0.514 (n = 114) | 0.432 (n = 96) | 0.311 (n = 138) | 0.689 (n = 306) |
| Seminoma (somatic) (n = 91) | 0.154 (n = 14) | 0.670 (n = 61) | 0.176 (n = 16) | 0.489 (n = 89) | 0.511 (n = 93) | |
| p | 0.0061 | 0.0123 | <0.0001 | <0.0001 | ||
| Non seminoma (somatic) (n = 9) | 0.111 (n = 1) | 0.889 (n = 8) | 0.000 (n = 0) | 0.555 (n = 10) | 0.445 (n = 8) | |
| p | 0.4119 | 0.0381 | 0.0114 | 0.0385 | ||
| Sequence variation (localisation) | CC | CT | TT | C | T | |
| rs11544331 (c.47 C > T; p.Pro16Leu) | Reference population (n = 4374) | 0.65 (n = 2843) | 0.302 (n = 1321) | 0.048 (n = 210) | 0.801 (n = 7007) | 0.199 (n = 1741) |
| Seminoma (somatic) (n = 115) | 0.617 (n = 71) | 0.365 (n = 42) | 0.018 (n = 2) | 0.800 (n = 184) | 0.200 (n = 46) | |
| p | 0.5328 | 0.1763 | 0.1918 | 0.9627 | ||
| Non seminoma (somatic) (n = 8) | 0.875 (n = 7) | 0.125 (n = 1) | 0.000 (n = 0) | 0.937 (n = 15) | 0.063 (n = 1) | |
| p | 0.3358 | 0.4812 | 0.8468 | 0.2921 | ||
| Age at diagnosis | Tumoral size | Tumoral Spread | |||||
|---|---|---|---|---|---|---|---|
| Sequence variation (localisation) | Age (years) | p | Size (mm) | p | ≥pT2 | p | |
| rs3808350 (1125893 A > G) | AA (n = 26) | 39.62 ± 9.26 | NS | 46.08 ± 20.20 | NS | 26.9% (7/26) | NS |
| AG (n = 19) | 39.68 ± 10.50 | NS | 41.00 ± 19.50 | NS | 31.6% (6/19) | NS | |
| GG (n = 1) | 38.00 ± 0.00 | NS | 60.00 ± 0.00 | NS | 100% (1/1) | NS | |
| rs3808351 (c.-668 G > A) | AA (n = 12) | 37.58 ± 7.87 | NS | 50.00 ± 21.10 | NS | 50.0% (6/12) | NS |
| AG (n = 32) | 39.16 ± 9.83 | NS | 43.56 ± 19.91 | NS | 28.1% (9/32) | NS | |
| GG (n = 8) | 40.13 ± 10.12 | NS | 43.75 ± 25.03 | NS | 25.0% (2/8) | NS | |
| rs11544331 (c.47 C > T; p.Pro16Leu) | CC (n = 36) | 37.31 ± 8.78 | NS | 49.83 ± 20.35 | NS | 36.1% (13/36) | NS |
| CT (n = 20) | 40.70 ± 11.01 | NS | 41.40 ± 19.35 | NS | 20.0% (4/20) | NS | |
| TT (n = 0) | - | - | - | - | - | - | |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chevalier, N.; Paul-Bellon, R.; Camparo, P.; Michiels, J.-F.; Chevallier, D.; Fénichel, P. Genetic Variants of GPER/GPR30, a Novel Estrogen-Related G Protein Receptor, Are Associated with Human Seminoma. Int. J. Mol. Sci. 2014, 15, 1574-1589. https://doi.org/10.3390/ijms15011574
Chevalier N, Paul-Bellon R, Camparo P, Michiels J-F, Chevallier D, Fénichel P. Genetic Variants of GPER/GPR30, a Novel Estrogen-Related G Protein Receptor, Are Associated with Human Seminoma. International Journal of Molecular Sciences. 2014; 15(1):1574-1589. https://doi.org/10.3390/ijms15011574
Chicago/Turabian StyleChevalier, Nicolas, Rachel Paul-Bellon, Philippe Camparo, Jean-François Michiels, Daniel Chevallier, and Patrick Fénichel. 2014. "Genetic Variants of GPER/GPR30, a Novel Estrogen-Related G Protein Receptor, Are Associated with Human Seminoma" International Journal of Molecular Sciences 15, no. 1: 1574-1589. https://doi.org/10.3390/ijms15011574