Dopamine D4 Receptor Counteracts Morphine-Induced Changes in µ Opioid Receptor Signaling in the Striosomes of the Rat Caudate Putamen




Abstract
:1. Introduction
2. Results
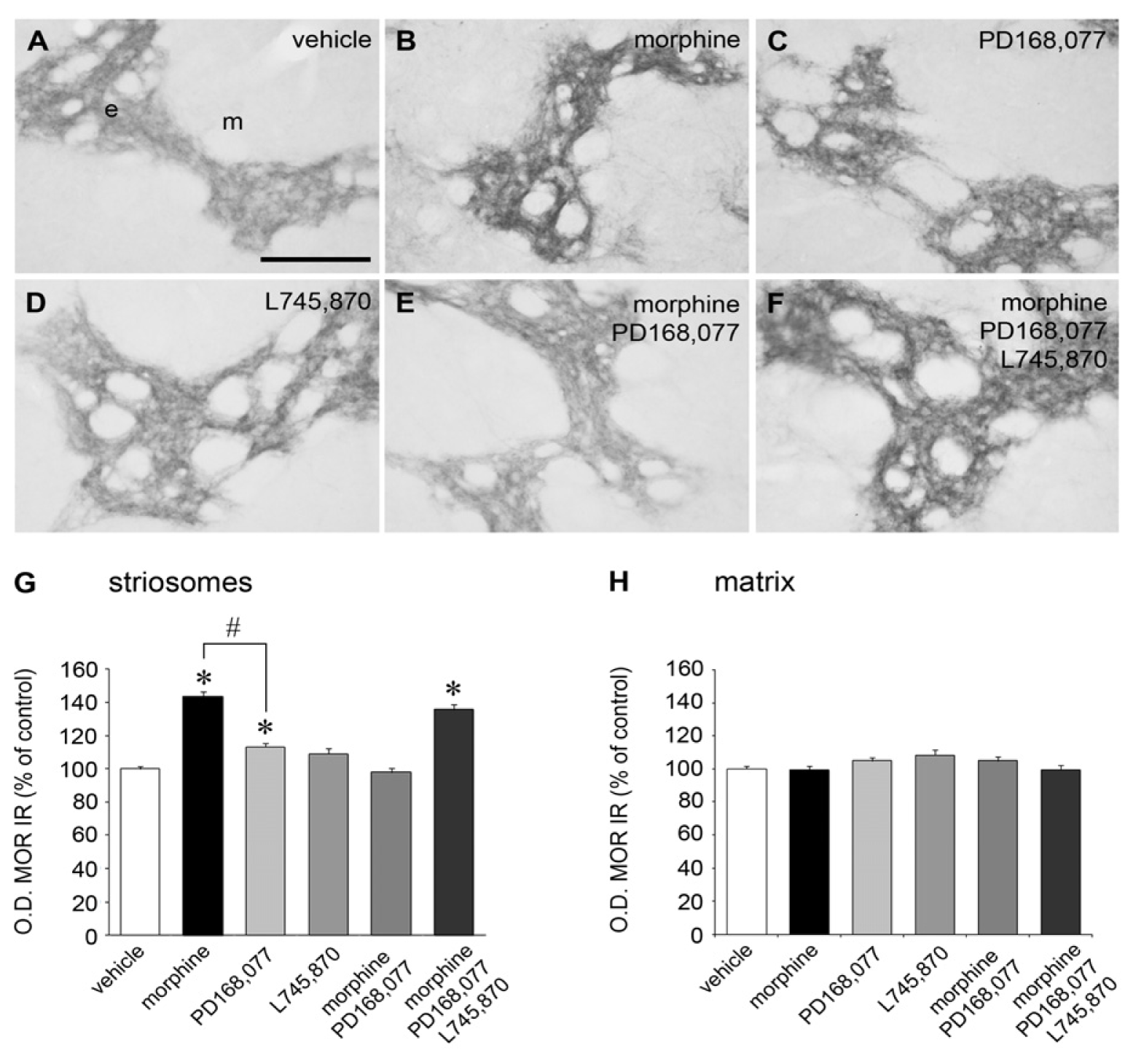
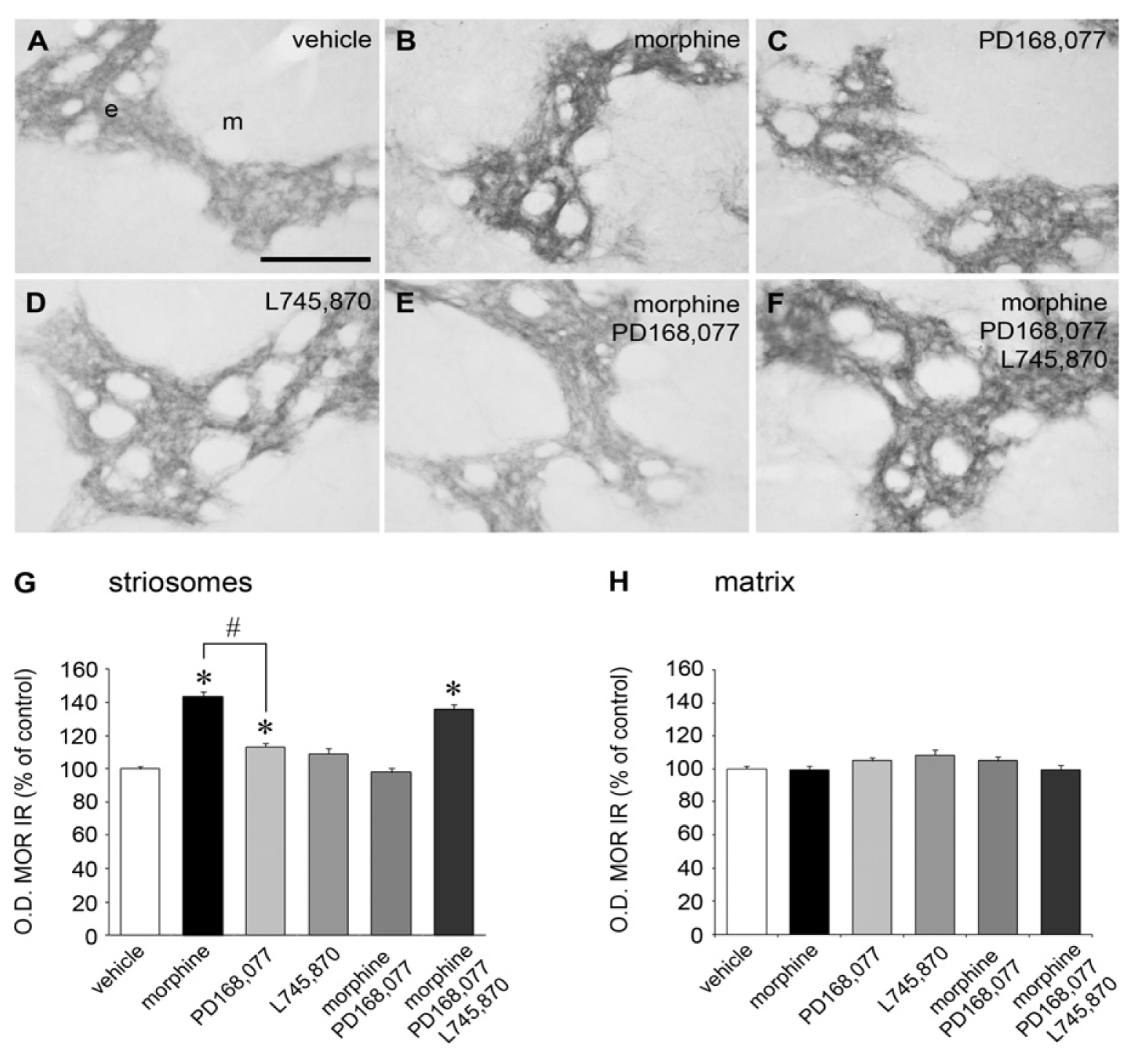
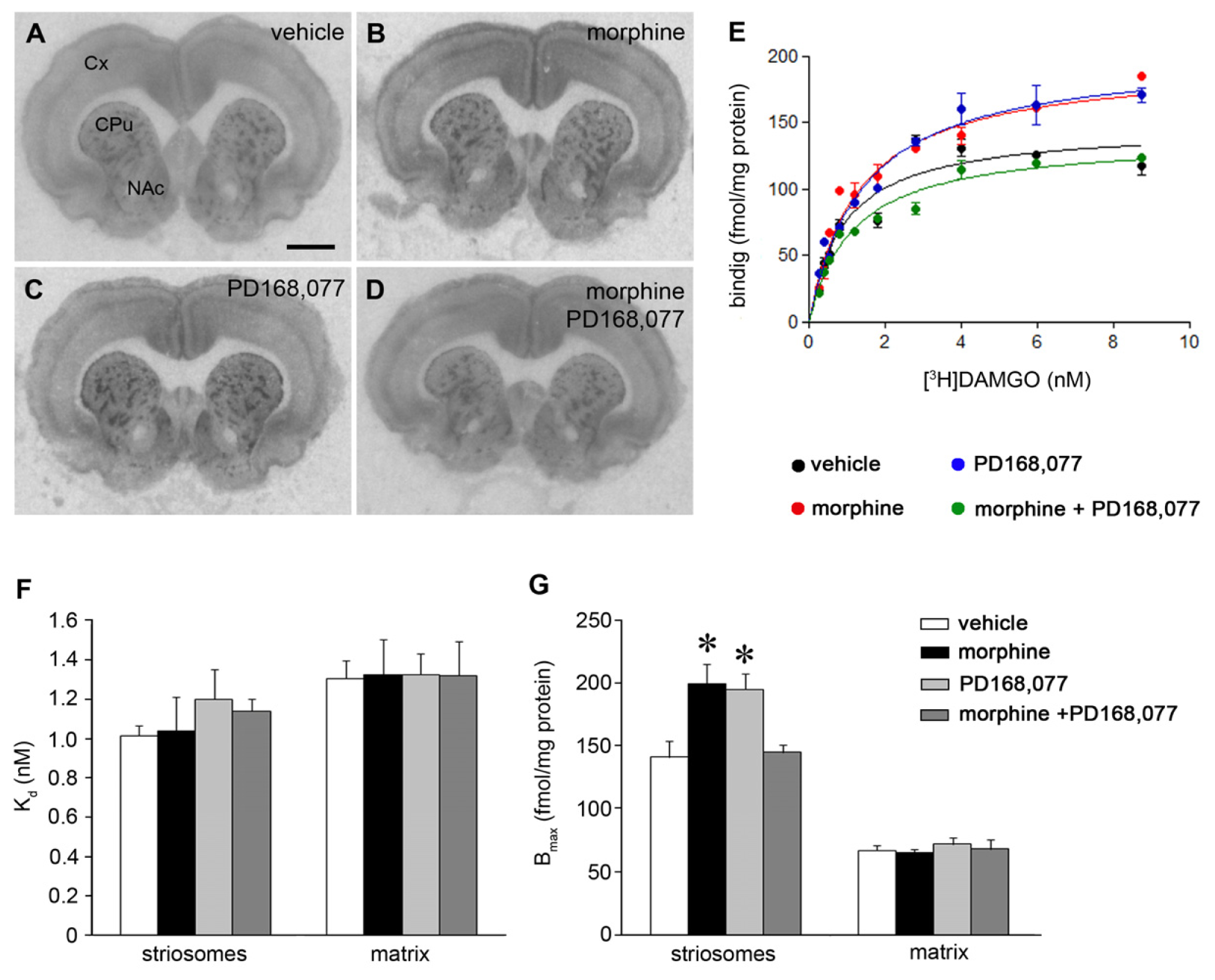
2.1. Cross-Inhibition of MOR IR Expression after Continuous Co-Administration of Morphine and the D4R Agonist PD168,077
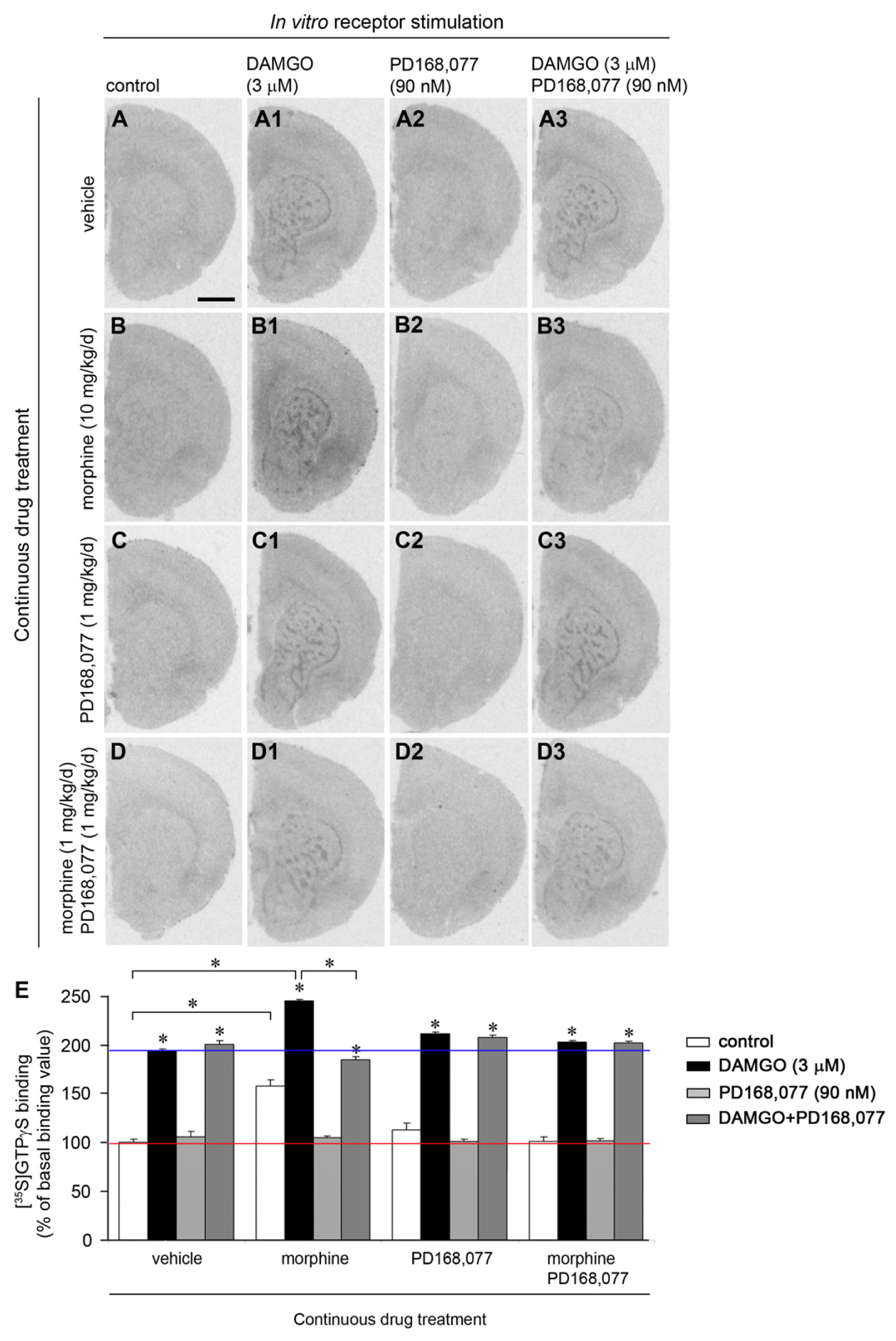
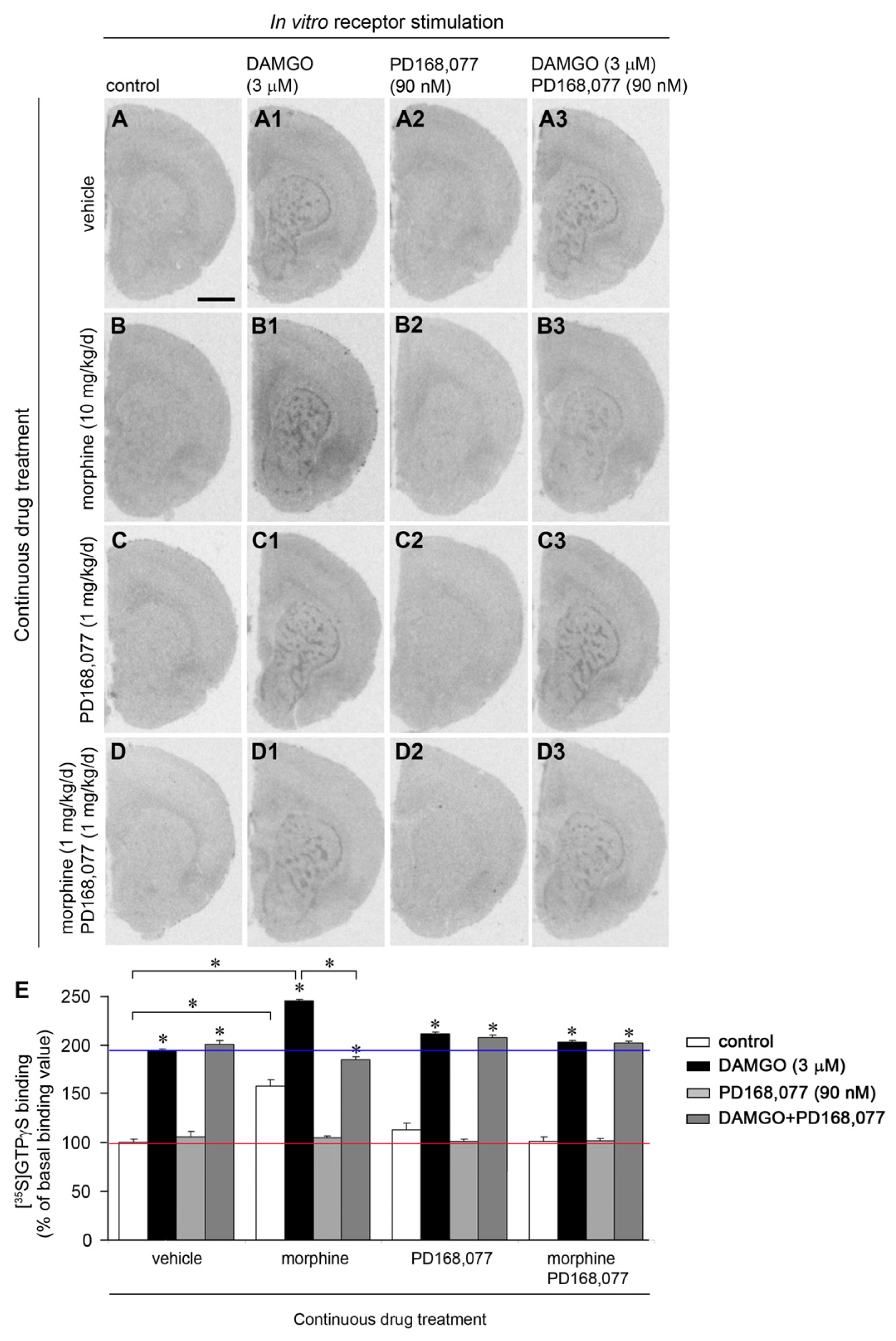
2.2. D4R Activation Counteracts the Increase of MOR Recognition and Signaling Induced by the Continuous Treatment with Morphine
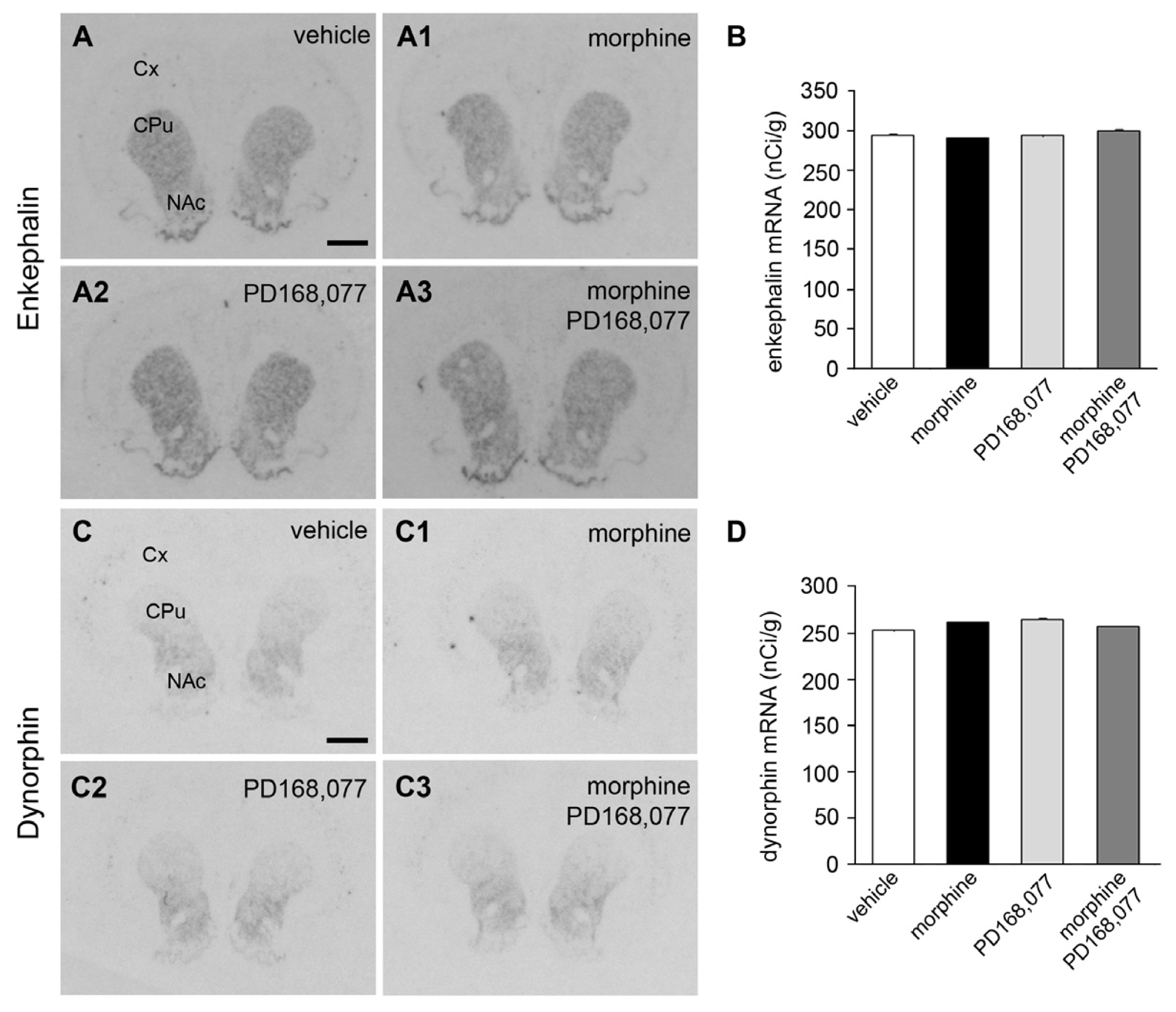
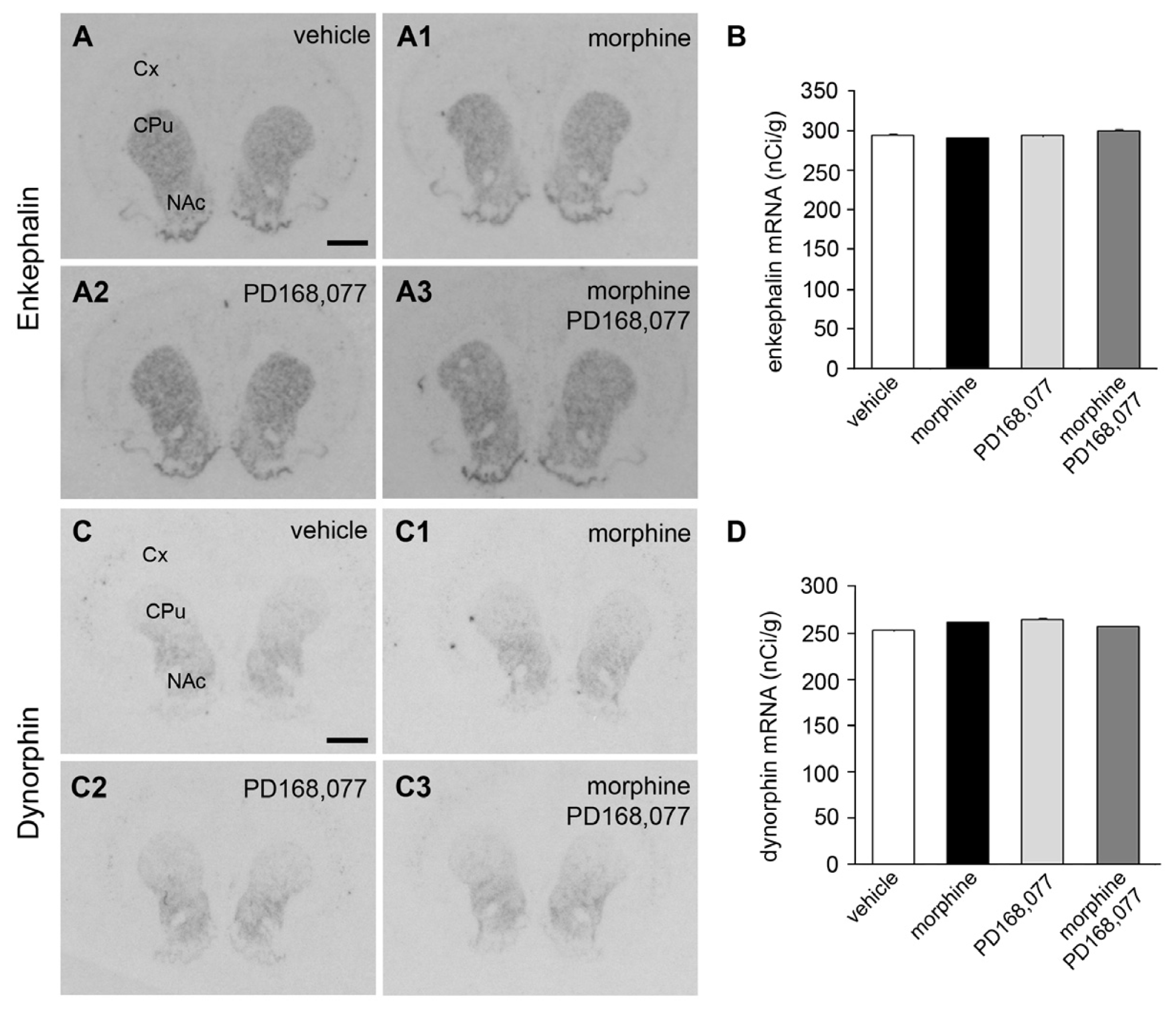
2.3. Absence of Changes in Enk and Dyn mRNA Levels in the CPu after the Continuous Administration of Morphine and/or PD168,077
3. Discussion
4. Experimental Section
4.1. Animals
4.2. Drugs
4.3. Drug Administration
4.4. Immunohistochemistry
4.5. μ Opioid Receptor Autoradiography
4.6. Agonist-Stimulated [35S]GTPγS Binding in Autoradiography
4.7. In Situ Hybridization
4.8. Statistical Analysis
5. Conclusions
Supplementary Information
1. Methods
1.1. Cell Culture and Transfection
1.2. ERK1/2 Phosphorylation Assay on Western Blot
1.3. In-Cell Western ERK1/2 Phosphorylation Assay
2. Results
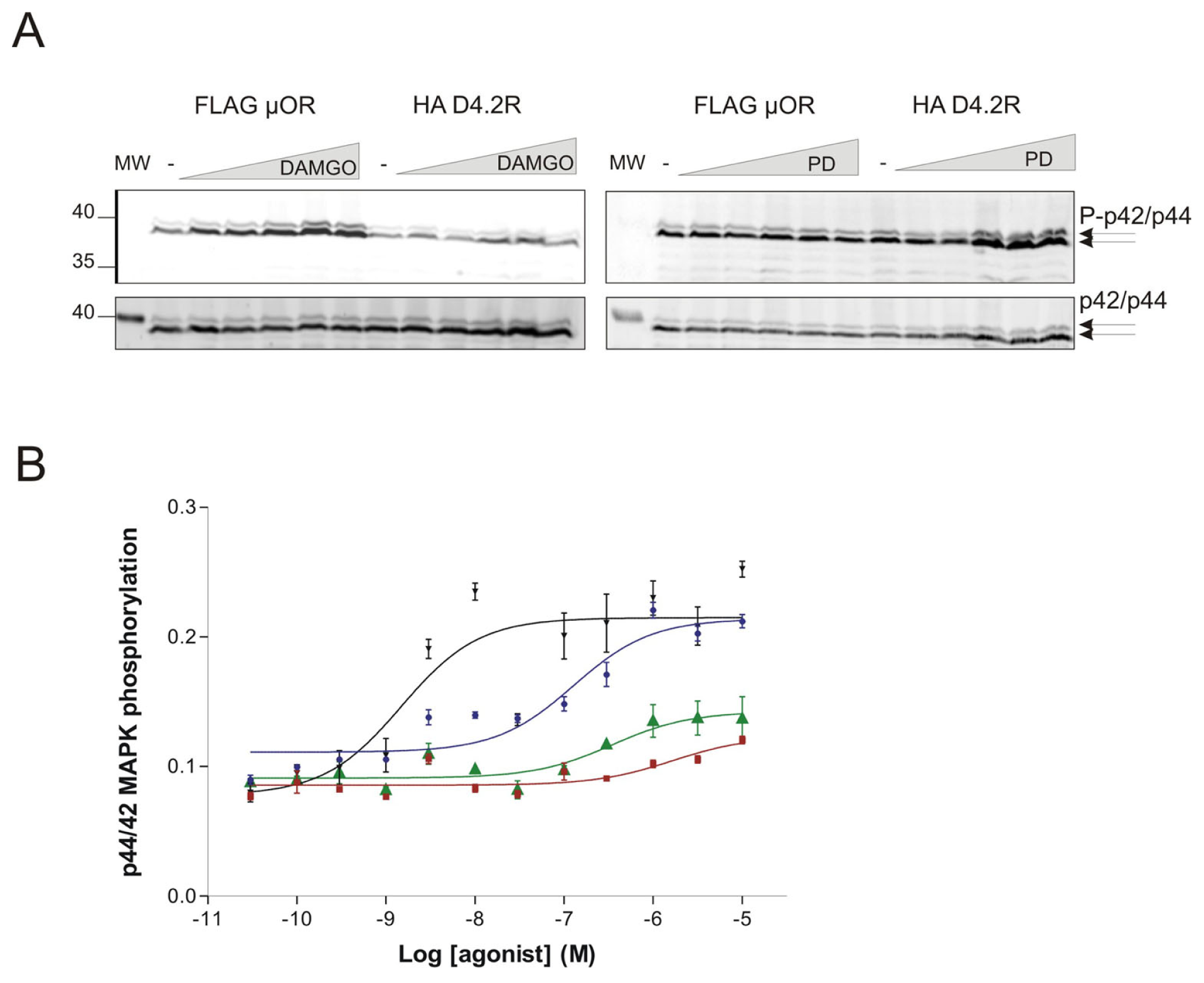
2.1. Specificity of the Drug PD168.077 for the Dopamine D4 Receptor
Acknowledgments
Conflicts of Interest
References
- Bloodworth, D. Issues in opioid management. Am. J. Phys. Med. Rehabil 2005, 84, S42–S55. [Google Scholar]
- Hyman, S.E.; Malenka, R.C.; Nestler, E.J. Neural mechanisms of addiction: The role of reward-related learning and memory. Annu. Rev. Neurosci 2006, 29, 565–598. [Google Scholar]
- Stewart, J.; Badiani, A. Tolerance and sensitization to the behavioral effects of drugs. Behav. Pharmacol 1993, 4, 289–312. [Google Scholar]
- Bodnar, R.J.; Klein, G.E. Endogenous opiates and behaviour: 2005. Peptides 2006, 27, 3391–3478. [Google Scholar]
- Gavériaux-Ruff, C.; Kieffer, B.L. Opioid receptor genes inactivated in mice: The highlights. Neuropeptides 2002, 36, 62–71. [Google Scholar]
- Lutz, P.E.; Kieffer, B.L. The multiple facets of opioid receptor function: Implications for addiction. Curr. Opin. Neurobiol 2013, 23, 473–479. [Google Scholar]
- Matthes, H.W.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature 1996, 383, 819–823. [Google Scholar]
- Margolis, E.B.; Hjelmstad, G.O.; Bonci, A.; Fields, H.L. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J. Neurosci 2003, 23, 9981–9986. [Google Scholar]
- Wee, S.; Koob, G.F. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology 2010, 210, 121–135. [Google Scholar]
- Le Merrer, J.; Plaza-Zabala, A.; del Boca, C.; Matifas, A.; Maldonado, R.; Kieffer, B.L. Deletion of the δ opioid receptor gene impairs place conditioning but preserves morphine reinforcement. Biol. Psychiatry 2011, 69, 700–703. [Google Scholar]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Rivera, A.; van Craenenbroeck, K.; Tarakanov, A.O.; Agnati, L.F.; Fuxe, K. On the G-protein-coupled receptor heteromers and their allosteric receptor-receptor interactions in the central nervous system: Focus on their role in pain modulation. Evid. Based Complement. Alternat. Med 2013, 2013, 563716. [Google Scholar]
- Costantino, C.M.; Gomes, I.; Stockton, S.D.; Lim, M.P.; Devi, L.A. Opioid receptor heteromers in analgesia. Expert. Rev. Mol. Med 2012, 14, e9. [Google Scholar]
- Kabli, N.; Martin, N.; Fan, T.; Nguyen, T.; Hasbi, A.; Balboni, G.; O’Dowd, B.F.; George, S.R. Agonists at the δ-opioid receptor modify the binding of μ-receptor agonists to the μ-δ receptor hetero-oligomer. Br. J. Pharmacol 2010, 161, 1122–1136. [Google Scholar]
- Gerrits, M.A.; Lesscher, H.B.; van Ree, J.M. Drug dependence and the endogenous opioid system. Eur. Neuropsychopharmacol 2003, 13, 424–434. [Google Scholar]
- Badiani, A.; Belin, D.; Epstein, D.; Calu, D.; Shaham, Y. Opiate versus psychostimulant addiction: The differences do matter. Nat. Rev. Neurosci 2011, 12, 685–700. [Google Scholar]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar]
- Johnson, S.W.; North, R.A. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci 1992, 12, 483–488. [Google Scholar]
- Margolis, E.B.; Toy, B.; Himmels, P.; Morales, M.; Fields, H.L. Identification of rat ventral tegmental area GABAergic neurons. PLoS One 2012, 7, e42365. [Google Scholar]
- Arvidsson, U.; Riedl, M.; Chakrabarti, S.; Lee, J.H.; Nakano, A.H.; Dado, R.J.; Loh, H.H.; Law, P.Y.; Wessendorf, M.W.; Elde, R. Distribution and targeting of a μ-opioid receptor (MOR1) in brain and spinal cord. J. Neurosci 1995, 15, 3328–3341. [Google Scholar]
- Fujiyama, F.; Sohn, J.; Nakano, T.; Furuta, T.; Nakamura, K.C.; Matsuda, W.; Kaneko, T. Exclusive and common targets of neostriatofugal projections of rat striosome neurons: A single neuron-tracing study using a viral vector. Eur. J. Neurosci 2011, 33, 668–677. [Google Scholar]
- Watabe-Uchida, M.; Zhu, L.; Ogawa, S.K.; Vamanrao, A.; Uchida, N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 2012, 74, 858–873. [Google Scholar]
- Kieffer, B.L.; Evans, C.J. Opioid tolerance in search of the Holy Grail. Cell 2002, 108, 587–590. [Google Scholar]
- Brady, L.S.; Herkenham, M.; Long, J.B.; Rothman, R.B. Chronic morphine increases μ-opiate receptor binding in rat brain: A quantitative autoradiographic study. Brain Res 1989, 477, 382–386. [Google Scholar]
- Goodman, C.B.; Emilien, B.; Becketts, K.; Cadet, J.L.; Rothman, R.B. Downregulation of mu-opioid binding sites following chronic administration of neuropeptide FF (NPFF) and morphine. Peptides 1996, 17, 389–397. [Google Scholar]
- Viganò, D.; Rubino, T.; di Chiara, G.; Ascari, I.; Massi, P.; Parolaro, D. Mu opioid receptor signaling in morphine sensitization. Neuroscience 2003, 117, 921–929. [Google Scholar]
- Gago, B.; Fuxe, K.; Agnati, L.; Penafiel, A.; de la Calle, A.; Rivera, A. Dopamine D(4) receptor activation decreases the expression of μ-opioid receptors in the rat striatum. J. Comp. Neurol 2007, 502, 358–366. [Google Scholar]
- Rivera, A.; Cuellar, B.; Giron, F.J.; Grandy, D.K.; de la Calle, A.; Moratalla, R. Dopamine D4 receptors are heterogeneously distributed in the striosomes/matrix compartments of the striatum. J. Neurochem 2002, 80, 219–229. [Google Scholar]
- Gago, B.; Suárez-Boomgaard, D.; Fuxe, K.; Brené, S.; Reina-Sánchez, M.D.; Rodríguez-Pérez, L.M.; Agnati, L.F.; de la Calle, A.; Rivera, A. Effect of acute and continuous morphine treatment on transcription factor expression in subregions of the rat caudate putamen. Marked modulation by D4 receptor activation. Brain Res 2011, 1407, 47–61. [Google Scholar]
- Gago, B.; Fuxe, K.; Brené, S.; Díaz-Cabiale, Z.; Reina-Sánchez, M.D.; Suárez-Boomgaard, D.; Roales-Buján, R.; Valderrama-Carvajal, A.; de la Calle, A.; Rivera, A. Early modulation by the dopamine D4 receptor of morphine-induced changes in the opioid peptide systems in the rat caudate putamen. J. Neurosci. Res 2013, 91, 1533–1540. [Google Scholar]
- Fuxe, K.; Marcellino, D.; Rivera, A.; Diaz-Cabiale, Z.; Filip, M.; Gago, B.; Roberts, D.C.; Langel, U.; Genedani, S.; Ferraro, L.; de la Calle, A.; et al. Receptor-receptor interactions within receptor mosaics. Impact on neuropsychopharmacology. Brain Res. Rev 2008, 58, 415–452. [Google Scholar]
- Winzer-Serhan, U.H.; Chen, Y.; Leslie, F.M. Expression of opioid peptides and receptors in striatum and substantia nigra during rat brain development. J. Chem. Neuroanat 2003, 26, 17–36. [Google Scholar]
- Tajima, K.; Fukuda, T. Region-specific diversity of striosomes in the mouse striatum revealed by the differential immunoreactivities for mu-opioid receptor, substance P, and enkephalin. Neuroscience 2013, 241, 215–228. [Google Scholar]
- Miura, M.; Saino-Saito, S.; Masuda, M.; Kobayashi, K.; Aosaki, T. Compartment-specific modulation of GABAergic synaptic transmission by mu-opioid receptor in the mouse striatum with green fluorescent protein-expressing dopamine islands. J. Neurosci 2007, 27, 9721–9728. [Google Scholar]
- De Vries, T.J.; Tjon, Tien; Ril, G.H.; van der Laan, J.W.; Mulder, A.H.; Schoffelmeer, A.N. Chronic exposure to morphine and naltrexone induces changes in catecholaminergic neurotransmission in rat brain without altering μ-opioid receptor sensitivity. Life Sci 1993, 52, 1685–1693. [Google Scholar]
- Stafford, K.; Gomes, A.B.; Shen, J.; Yoburn, B.C. Mu-opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol. Biochem. Behav. 2001, 69, 233–237. [Google Scholar]
- Danks, J.A.; Tortella, F.C.; Long, J.B.; Bykov, V.; Jacobson, A.E.; Rice, K.C.; Holaday, J.W.; Rothman, R.B. Chronic administration of morphine and naltrexone up-regulate [3H][d-Ala2, d-leu5]enkephalin binding sites by different mechanisms. Neuropharmacology 1988, 27, 965–974. [Google Scholar]
- Trujillo, K.A.; Kubota, K.S.; Warmoth, K.P. Continuous administration of opioids produces locomotor sensitization. Pharmacol. Biochem. Behav 2004, 79, 661–669. [Google Scholar]
- Narita, M.; Imai, S.; Nakamura, A.; Ozeki, A.; Asato, M.; Rahmadi, M.; Sudo, Y.; Hojo, M.; Uezono, Y.; Devi, L.A.; Kuzumaki, N.; Suzuki, T. Possible involvement of prolonging spinal μ-opioid receptor desensitization in the development of antihyperalgesic tolerance to μ-opioids under a neuropathic pain-like state. Addict. Biol 2013, 18, 614–622. [Google Scholar]
- Fábián, G.; Bozó, B.; Szikszay, M.; Horváth, G.; Coscia, C.J.; Szücs, M. Chronic morphine-induced changes in μ-opioid receptors and G proteins of different subcellular loci in rat brain. J. Pharmacol. Exp. Ther 2002, 302, 774–780. [Google Scholar]
- Juhasz, J.R.; Hasbi, A.; Rashid, A.J.; So, C.H.; George, S.R.; O’Dowd, B.F. Mu-opioid receptor heterooligomer formation with the dopamine D1 receptor as directly visualized in living cells. Eur. J. Pharmacol 2008, 581, 235–243. [Google Scholar]
- Léna, I.; Bradshaw, S.; Pintar, J.; Kitchen, I. Adaptive changes in the expression of central opioid receptors in mice lacking the dopamine D2 receptor gene. Neuroscience 2008, 153, 773–788. [Google Scholar]
- Canales, J.J. Stimulant-induced adaptations in neostriatal matrix and striosome systems: Transiting from instrumental responding to habitual behavior in drug addiction. Neurobiol. Learn. Mem 2005, 83, 93–103. [Google Scholar]
- Crittenden, J.R.; Graybiel, A.M. Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat 2011, 5, 59. [Google Scholar]
- White, N.M.; Hiroi, N. Preferential localization of self-stimulation sites in striosomes/patches in the rat striatum. Proc. Natl. Acad. Sci. USA 1998, 95, 6486–6491. [Google Scholar]
- Douglass, J.; McKinzie, A.A.; Pollock, K.M. Identification of multiple DNA elements regulating basal and protein kinase A-induced transcriptional expression of the rat prodynorphin gene. Mol. Endocrinol 1994, 8, 333–344. [Google Scholar]
- Zurawski, G.; Benedik, M.; Kamb, B.J.; Abrams, J.S.; Zurawski, S.M.; Lee, F.D. Activation of mouse T-helper cells induces abundant preproenkephalin mRNA synthesis. Science 1986, 232, 772–775. [Google Scholar]
- Skieterska, K.; Duchou, J.; Lintermans, B.; Van Craenenbroeck, K. Detection of G protein-coupled receptor (GPCR) dimerization by coimmunoprecipitation. Methods Cell Biol 2013, 117, 323–340. [Google Scholar]
- Van Craenenbroeck, K.; Clark, S.D.; Cox, M.J.; Oak, J.N.; Liu, F.; Van Tol, H.H. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J. Biol. Chem 2005, 280, 19350–19357. [Google Scholar]
- Pfeiffer, M.; Koch, T.; Schröder, H.; Laugsch, M.; Höllt, V.; Schulz, S. Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J. Biol. Chem 2002, 277, 19762–19772. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level of the CPu | Vehicle | Morphine | PD168,077 | L745,870 | Morphine + PD168,077 | Morphine + PD168,077 + L745,870 |
|---|---|---|---|---|---|---|
| Rostral | 100 ± 2.3 | 134.6 ± 3.2 | 109.6 ± 3.1 | 91.4 ± 3.4 | 99.4 ± 3.4 | 135.0 ± 4.1 |
| Middle | 100 ± 2.6 | 133.8 ± 3.6 | 104.2 ± 3.5 | 100.4 ± 4.0 | 98.0 ± 4.2 | 127.4 ± 5.0 |
| Caudal | 100 ± 2.9 | 169.4 ± 6.1 * | 117.6 ± 4.6 * | 111.5 ± 6.9 | 98.6 ± 5.7 | 149.0 ± 6.2* |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suárez-Boomgaard, D.; Gago, B.; Valderrama-Carvajal, A.; Roales-Buján, R.; Van Craenenbroeck, K.; Duchou, J.; Borroto-Escuela, D.O.; Medina-Luque, J.; De la Calle, A.; Fuxe, K.; et al. Dopamine D4 Receptor Counteracts Morphine-Induced Changes in µ Opioid Receptor Signaling in the Striosomes of the Rat Caudate Putamen. Int. J. Mol. Sci. 2014, 15, 1481-1498. https://doi.org/10.3390/ijms15011481
Suárez-Boomgaard D, Gago B, Valderrama-Carvajal A, Roales-Buján R, Van Craenenbroeck K, Duchou J, Borroto-Escuela DO, Medina-Luque J, De la Calle A, Fuxe K, et al. Dopamine D4 Receptor Counteracts Morphine-Induced Changes in µ Opioid Receptor Signaling in the Striosomes of the Rat Caudate Putamen. International Journal of Molecular Sciences. 2014; 15(1):1481-1498. https://doi.org/10.3390/ijms15011481
Chicago/Turabian StyleSuárez-Boomgaard, Diana, Belén Gago, Alejandra Valderrama-Carvajal, Ruth Roales-Buján, Kathleen Van Craenenbroeck, Jolien Duchou, Dasiel O. Borroto-Escuela, José Medina-Luque, Adelaida De la Calle, Kjell Fuxe, and et al. 2014. "Dopamine D4 Receptor Counteracts Morphine-Induced Changes in µ Opioid Receptor Signaling in the Striosomes of the Rat Caudate Putamen" International Journal of Molecular Sciences 15, no. 1: 1481-1498. https://doi.org/10.3390/ijms15011481