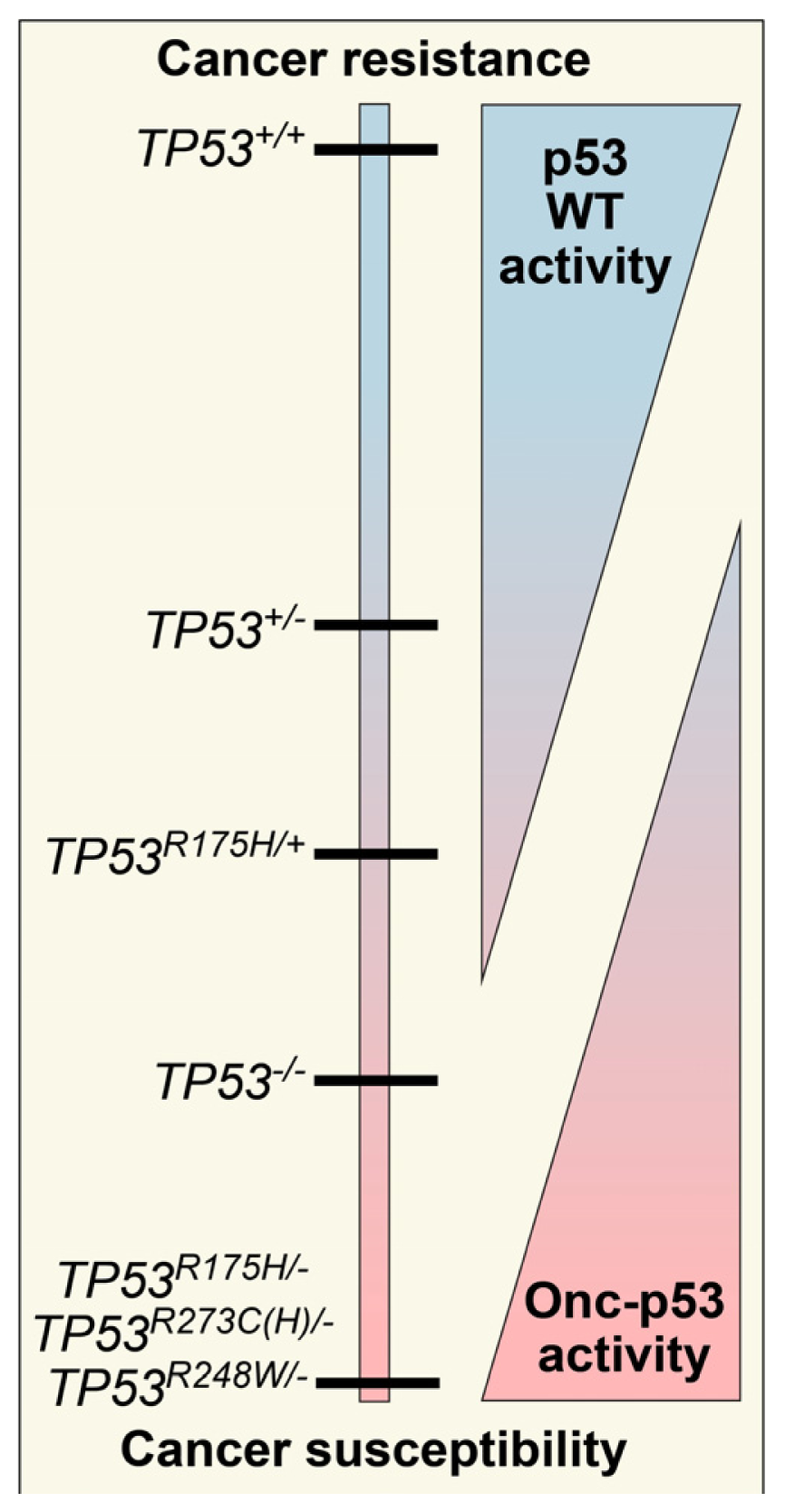
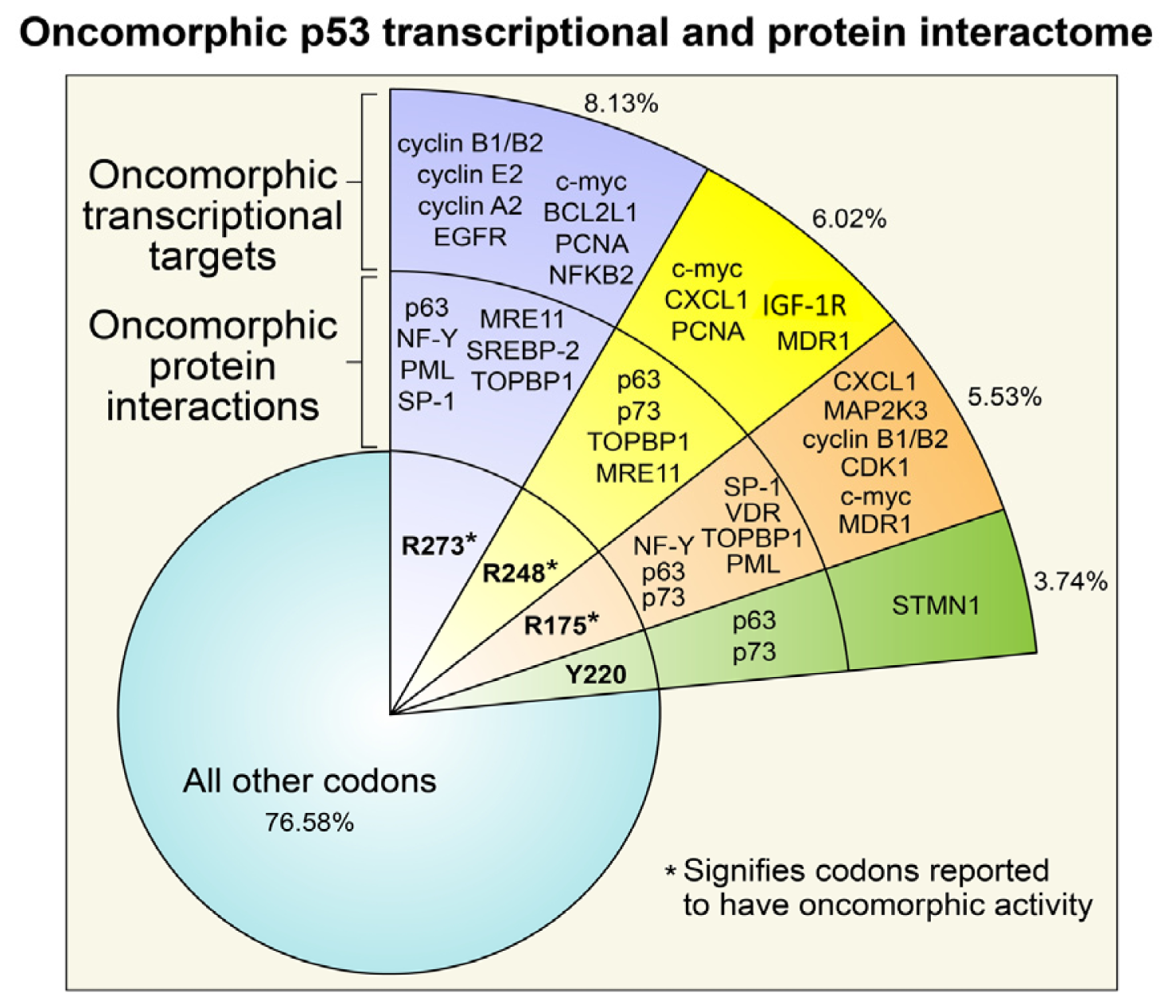
The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Brachova, P.; Thiel, K.W.; Leslie, K.K. The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 19257-19275. https://doi.org/10.3390/ijms140919257
Brachova P, Thiel KW, Leslie KK. The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. International Journal of Molecular Sciences. 2013; 14(9):19257-19275. https://doi.org/10.3390/ijms140919257
Chicago/Turabian StyleBrachova, Pavla, Kristina W. Thiel, and Kimberly K. Leslie. 2013. "The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer" International Journal of Molecular Sciences 14, no. 9: 19257-19275. https://doi.org/10.3390/ijms140919257
APA StyleBrachova, P., Thiel, K. W., & Leslie, K. K. (2013). The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. International Journal of Molecular Sciences, 14(9), 19257-19275. https://doi.org/10.3390/ijms140919257