The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
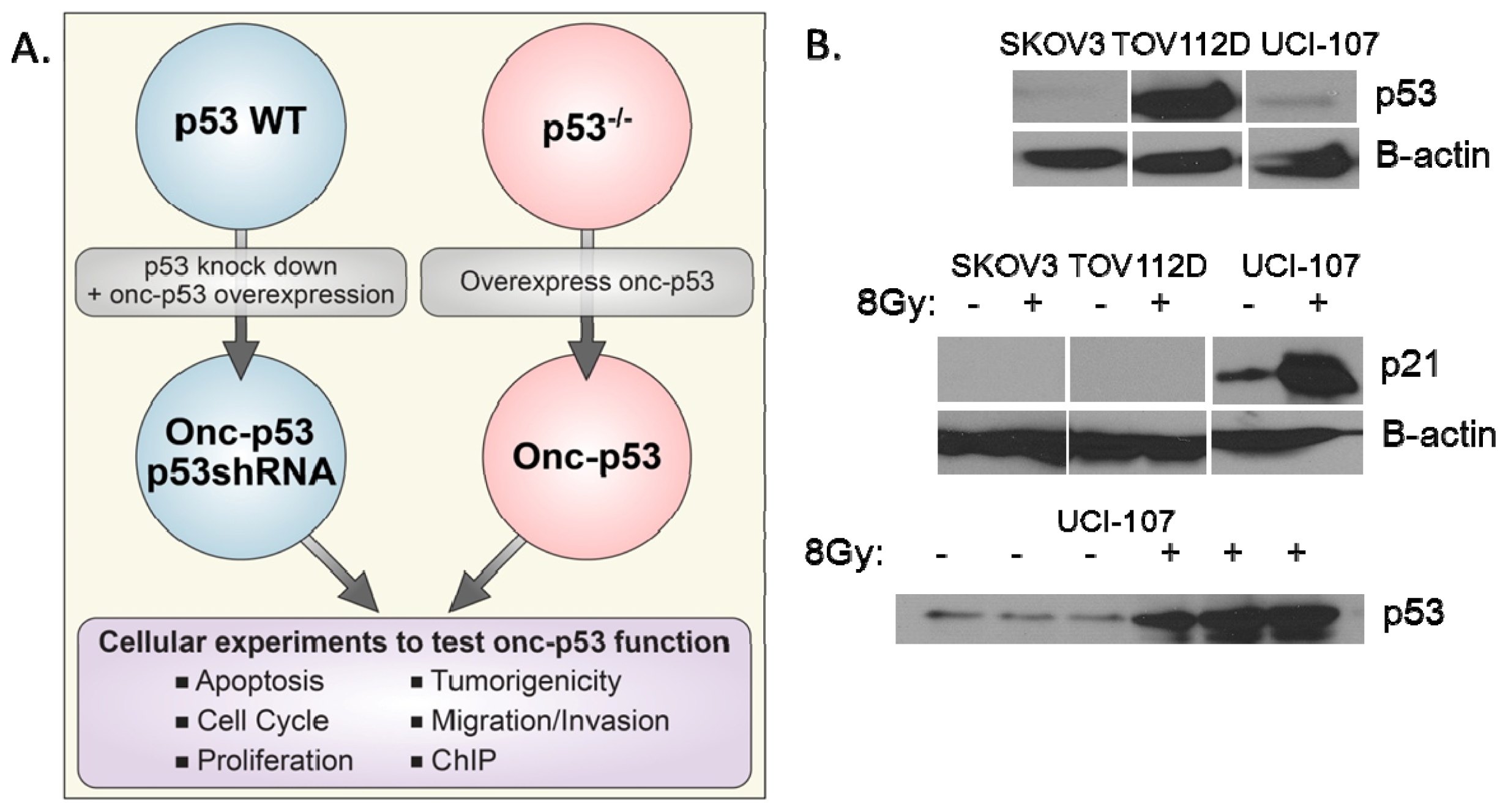
:1. Introduction
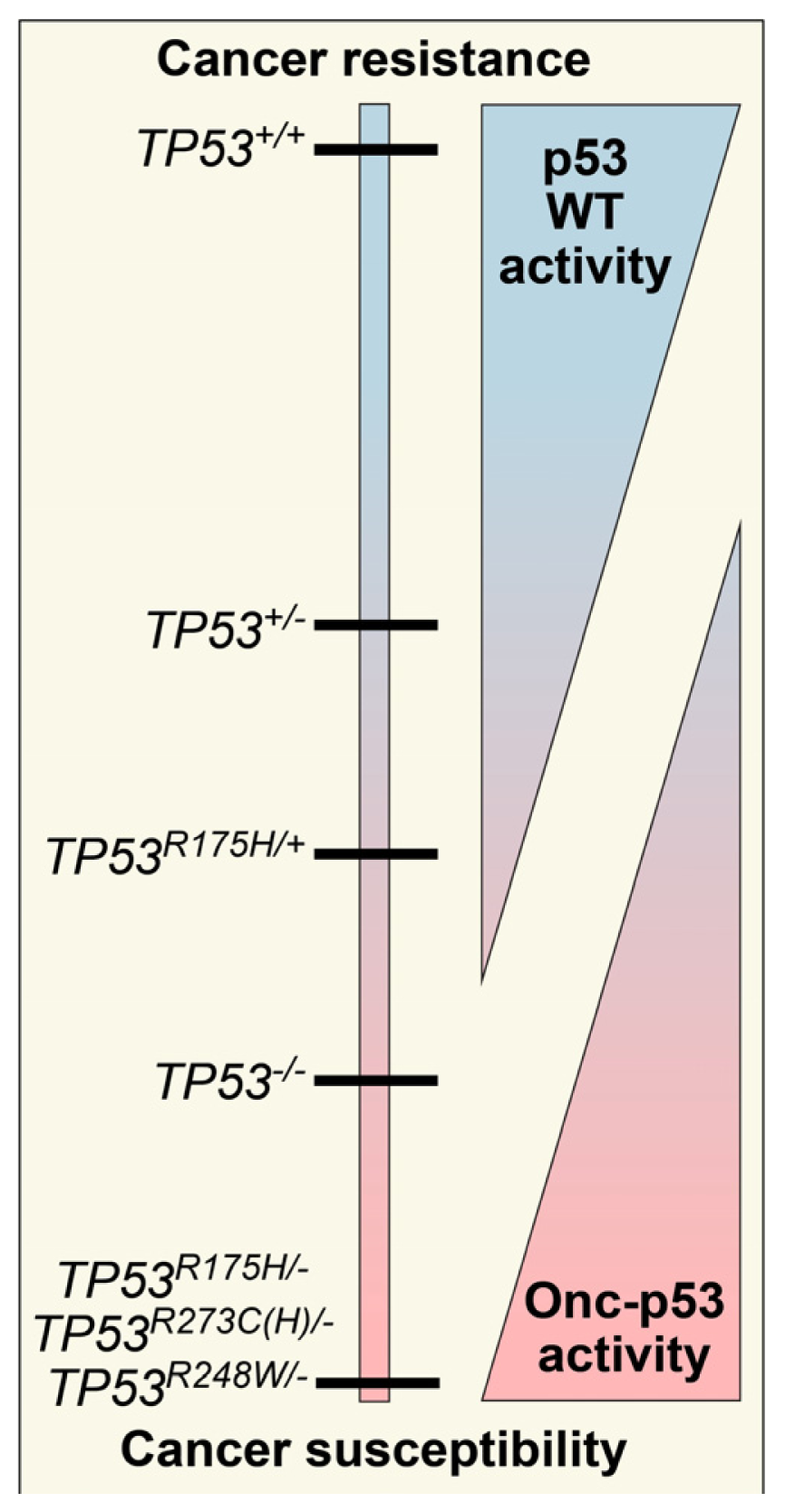
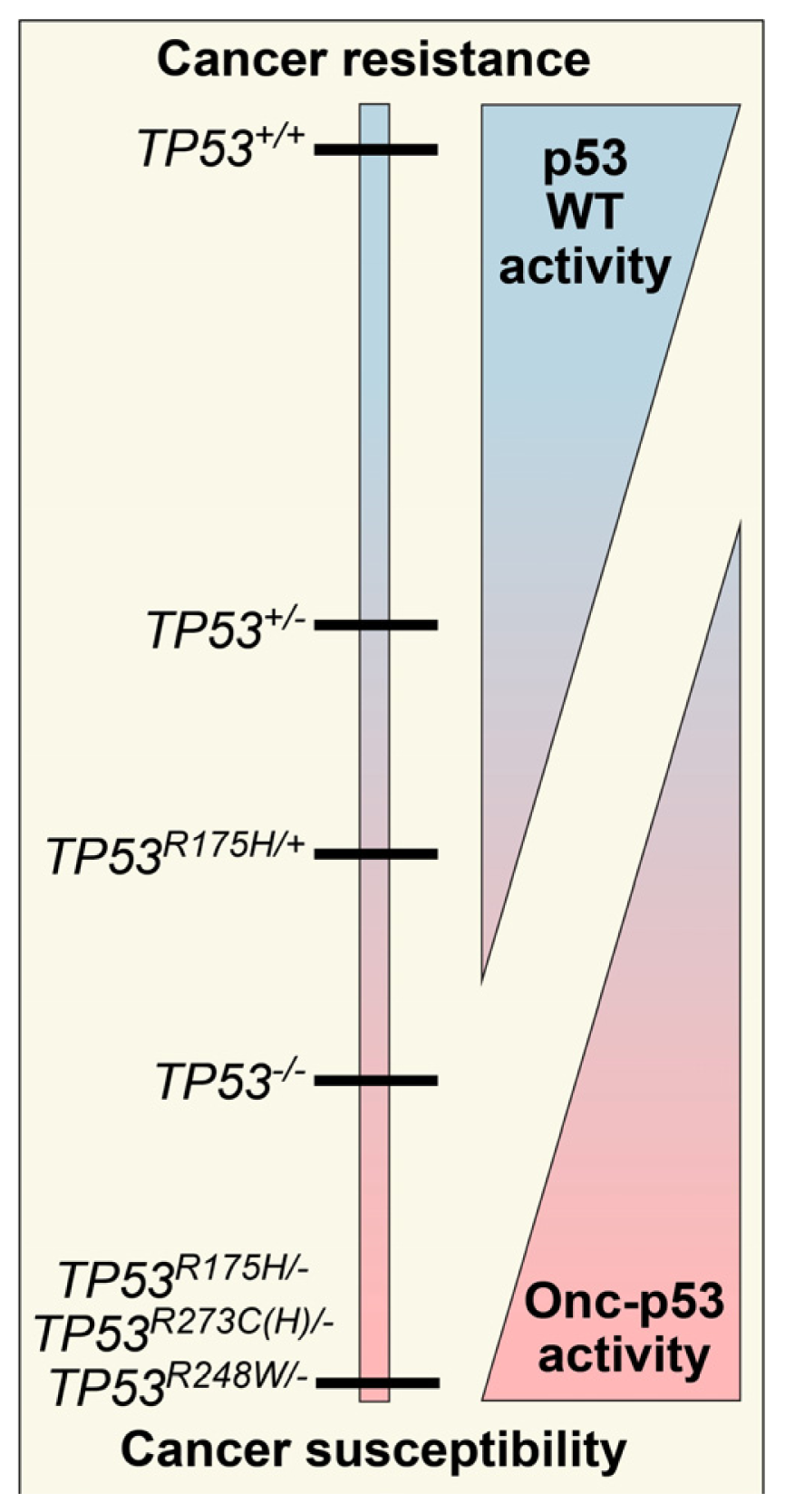
2. Defining TP53 Mutations
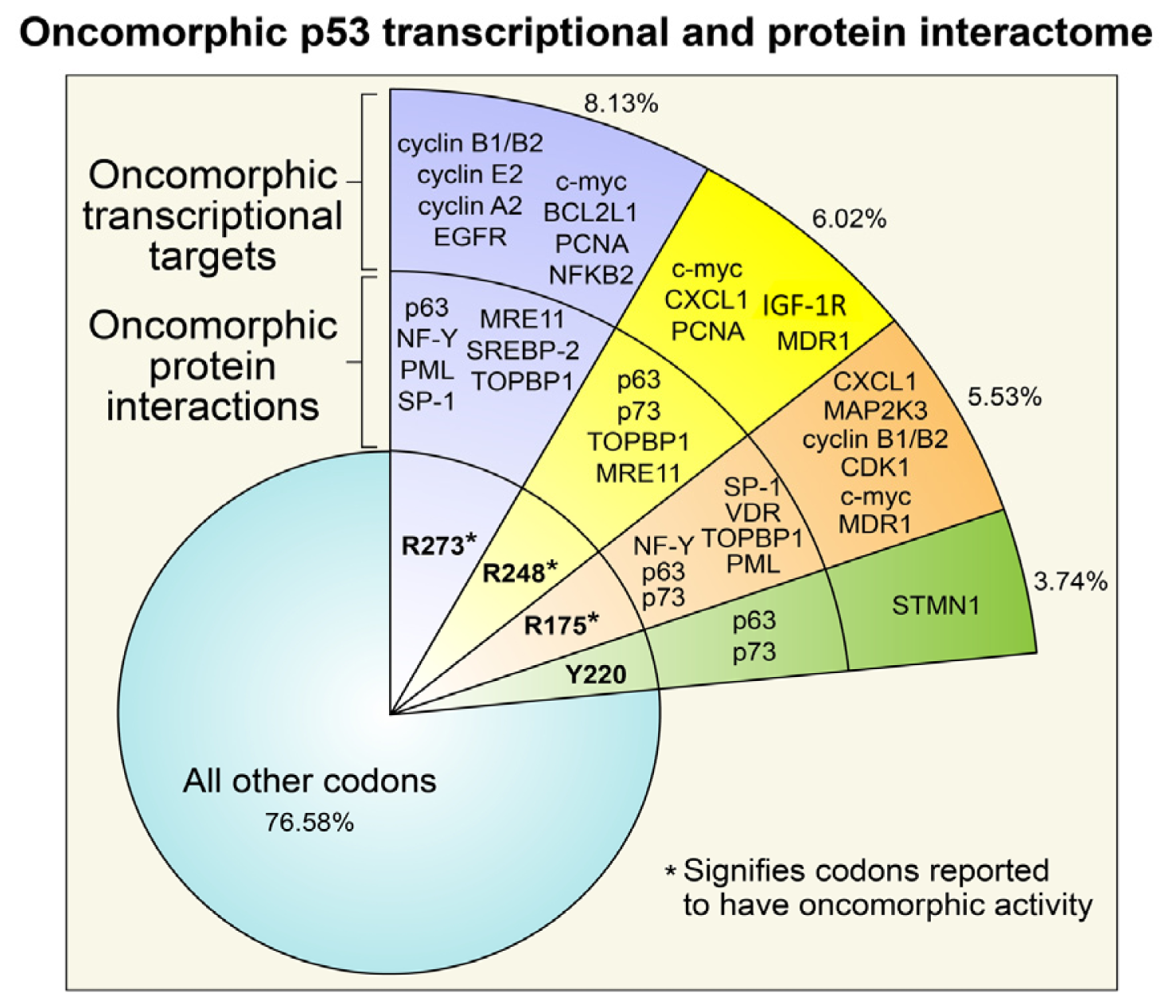
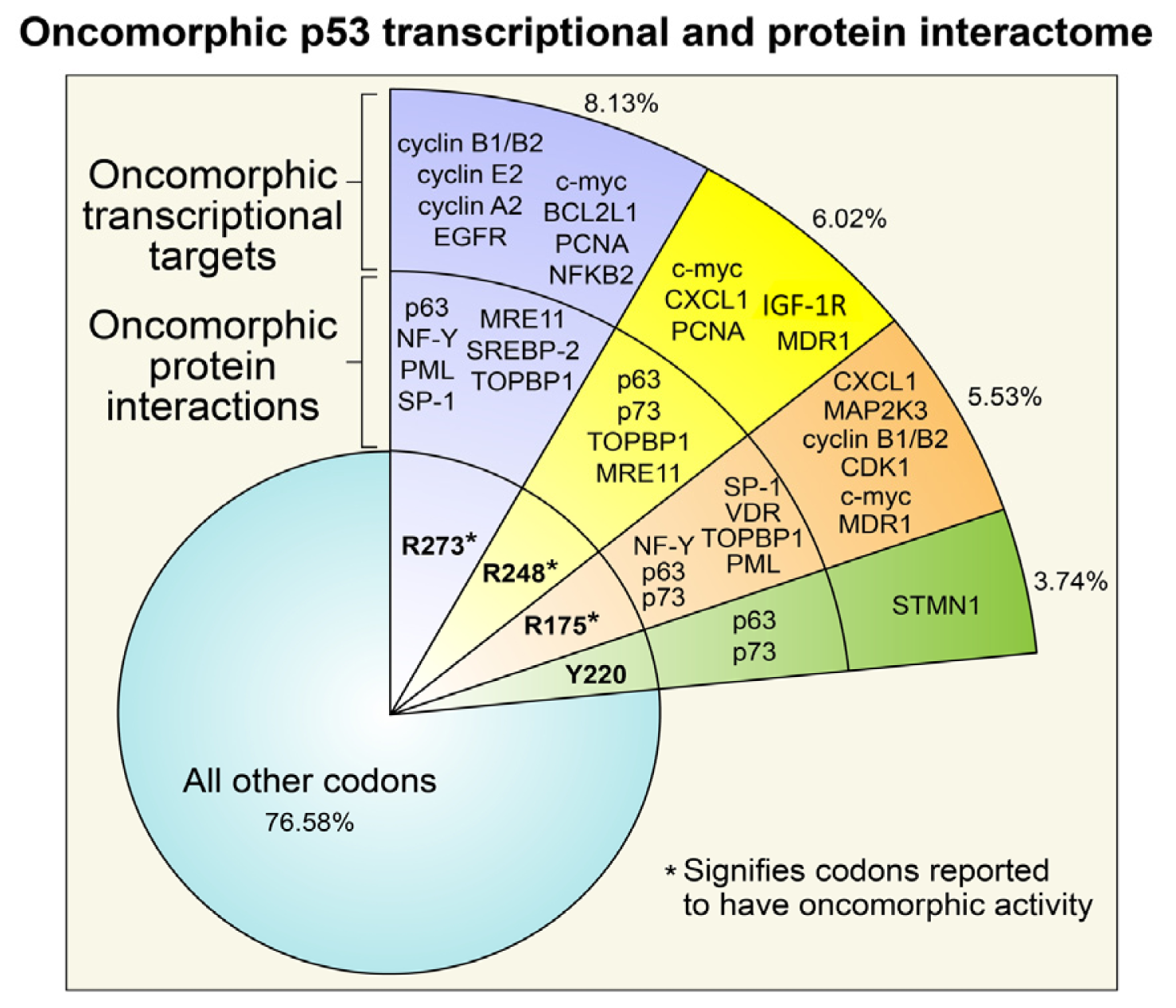
3. Oncomorphic p53 Mutations
3.1. R273 p53
3.2. R248 p53
3.3. R175 p53
3.4. Y220C p53
4. Methods of Identifying and Understanding Oncomorphisms
5. Conclusions and Consequences for the Future
Acknowledgments
Conflicts of Interest
References
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med 1996, 334, 1–6. [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.[Green Version]
- Weinberg, R.A. Tumor suppressor genes. Science 1991, 254, 1138–1146. [Google Scholar]
- Gadducci, A.; di Cristofano, C.; Zavaglia, M.; Giusti, L.; Menicagli, M.; Cosio, S.; Naccarato, A.G.; Genazzani, A.R.; Bevilacqua, G.; Cavazzana, A.O. p53 gene status in patients with advanced serous epithelial ovarian cancer in relation to response to paclitaxel- plus platinum-based chemotherapy and long-term clinical outcome. Anticancer Res 2006, 26, 687–693. [Google Scholar]
- Smith-Sorensen, B.; Kaern, J.; Holm, R.; Dorum, A.; Trope, C.; Borresen-Dale, A.L. Therapy effect of either paclitaxel or cyclophosphamide combination treatment in patients with epithelial ovarian cancer and relation to TP53 gene status. Br. J. Cancer 1998, 78, 375–381. [Google Scholar]
- Bosari, S.; Viale, G.; Radaelli, U.; Bossi, P.; Bonoldi, E.; Coggi, G. p53 accumulation in ovarian carcinomas and its prognostic implications. Hum. Pathol 1993, 24, 1175–1179. [Google Scholar]
- Berker, B.; Dunder, I.; Ensari, A.; Cengiz, S.D.; Simsek, E. Prognostic significance of apoptotic index and bcl-2 and p53 expression in epithelial ovarian carcinoma. Eur. J. Gynaecol. Oncol 2002, 23, 505–510. [Google Scholar]
- Berker, B.; Dunder, I.; Ensari, A.; Cengiz, S.D. Prognostic value of p53 accumulation in epithelial ovarian carcinomas. Arch. Gynecol. Obstet 2002, 266, 205–209. [Google Scholar]
- Hawes, D.; Liu, P.Y.; Muggia, F.M.; Wilczynski, S.; Cote, R.; Felix, J.; Terada, K.; Belt, R.J.; Alberts, D.S. Correlation of p53 immunostaining in primary and residual ovarian cancer at the time of positive second-look laparotomy and its prognostic role: A Southwest Oncology Group ancillary study. Gynecol. Oncol 2002, 87, 17–23. [Google Scholar]
- Niwa, K.; Itoh, M.; Murase, T.; Morishita, S.; Itoh, N.; Mori, H.; Tamaya, T. Alteration of p53 gene in ovarian carcinoma: Clinicopathological correlation and prognostic significance. Br. J. Cancer 1994, 70, 1191–1197. [Google Scholar]
- Herod, J.J.; Eliopoulos, A.G.; Warwick, J.; Niedobitek, G.; Young, L.S.; Kerr, D.J. The prognostic significance of Bcl-2 and p53 expression in ovarian carcinoma. Cancer Res 1996, 56, 2178–2184. [Google Scholar]
- Marx, D.; Meden, H.; Ziemek, T.; Lenthe, T.; Kuhn, W.; Schauer, A. Expression of the p53 tumour suppressor gene as a prognostic marker in platinum-treated patients with ovarian cancer. Eur. J. Cancer 1998, 34, 845–850. [Google Scholar]
- Levesque, M.A.; Katsaros, D.; Massobrio, M.; Genta, F.; Yu, H.; Richiardi, G.; Fracchioli, S.; Durando, A.; Arisio, R.; Diamandis, E.P. Evidence for a dose-response effect between p53 (but not p21WAF1/Cip1) protein concentrations, survival, and responsiveness in patients with epithelial ovarian cancer treated with platinum-based chemotherapy. Clin. Cancer Res 2000, 6, 3260–3270. [Google Scholar]
- Shahin, M.S.; Hughes, J.H.; Sood, A.K.; Buller, R.E. The prognostic significance of p53 tumor suppressor gene alterations in ovarian carcinoma. Cancer 2000, 89, 2006–2017. [Google Scholar]
- Rohlke, P.; Milde-Langosch, K.; Weyland, C.; Pichlmeier, U.; Jonat, W.; Loning, T. p53 is a persistent and predictive marker in advanced ovarian carcinomas: Multivariate analysis including comparison with Ki67 immunoreactivity. J. Cancer Res. Clin. Oncol 1997, 123, 496–501. [Google Scholar]
- Van der Zee, A.G.; Hollema, H.; Suurmeijer, A.J.; Krans, M.; Sluiter, W.J.; Willemse, P.H.; Aalders, J.G.; de Vries, E.G. Value of P-glycoprotein, glutathione S-transferase pi, c-erbB-2, and as prognostic factors in ovarian carcinomas. J. Clin. Oncol 1995, 13, 70–78. [Google Scholar]
- Petty, R.; Evans, A.; Duncan, I.; Kurbacher, C.; Cree, I. Drug resistance in ovarian cancer—The role of p53. Pathol. Oncol. Res 1998, 4, 97–102. [Google Scholar]
- Laframboise, S.; Chapman, W.; McLaughlin, J.; Andrulis, I.L. p53 mutations in epithelial ovarian cancers: Possible role in predicting chemoresistance. Cancer J 2000, 6, 302–308. [Google Scholar]
- Baekelandt, M.; Kristensen, G.B.; Nesland, J.M.; Trope, C.G.; Holm, R. Clinical significance of apoptosis-related factors p53, Mdm2, and Bcl-2 in advanced ovarian cancer. J. Clin. Oncol 1999, 17, 2061. [Google Scholar]
- Righetti, S.C.; Della Torre, G.; Pilotti, S.; Menard, S.; Ottone, F.; Colnaghi, M.I.; Pierotti, M.A.; Lavarino, C.; Cornarotti, M.; Oriana, S.; et al. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res 1996, 56, 689–693. [Google Scholar]
- Wen, W.H.; Reles, A.; Runnebaum, I.B.; Sullivan-Halley, J.; Bernstein, L.; Jones, L.A.; Felix, J.C.; Kreienberg, R.; el-Naggar, A.; Press, M.F. p53 mutations and expression in ovarian cancers: Correlation with overall survival. Int. J. Gynecol. Pathol 1999, 18, 29–41. [Google Scholar]
- Sagarra, R.A.; Andrade, L.A.; Martinez, E.Z.; Pinto, G.A.; Syrjanen, K.J.; Derchain, S.F. p53 and Bcl-2 as prognostic predictors in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2002, 12, 720–727. [Google Scholar]
- Lane, D.P. On the expression of the p53 protein in human cancer. Mol. Biol. Rep 1994, 19, 23–29. [Google Scholar]
- Hall, J.; Paul, J.; Brown, R. Critical evaluation of p53 as a prognostic marker in ovarian cancer. Expert. Rev. Mol. Med 2004, 6, 1–20. [Google Scholar]
- Hall, P.A.; Lane, D.P. p53 in tumour pathology: Can we trust immunohistochemistry?—Revisited! J. Pathol 1994, 172, 1–4. [Google Scholar]
- Kern, A.; Taubert, H.; Scheele, J.; Rudroff, C.; Mothes, H.; Kappler, M.; Bartel, F.; Richter, K.K. Association of p53 mutations, microvessel density and neoangiogenesis in pairs of colorectal cancers and corresponding liver metastases. Int. J. Oncol 2002, 21, 243–249. [Google Scholar]
- Wadayama, B.; Toguchida, J.; Yamaguchi, T.; Sasaki, M.S.; Yamamuro, T. p53 expression and its relationship to DNA alterations in bone and soft tissue sarcomas. Br. J. Cancer 1993, 68, 1134–1139. [Google Scholar]
- Jolly, K.W.; Malkin, D.; Douglass, E.C.; Brown, T.F.; Sinclair, A.E.; Look, A.T. Splice-site mutation of the p53 gene in a family with hereditary breast-ovarian cancer. Oncogene 1994, 9, 97–102. [Google Scholar]
- Magnusson, K.P.; Sandstrom, M.; Stahlberg, M.; Larsson, M.; Flygare, J.; Hellgren, D.; Wiman, K.G.; Ljungquist, S. p53 splice acceptor site mutation and increased HsRAD51 protein expression in Bloom’s syndrome GM1492 fibroblasts. Gene 2000, 246, 247–254. [Google Scholar]
- Holmila, R.; Fouquet, C.; Cadranel, J.; Zalcman, G.; Soussi, T. Splice mutations in the p53 gene: Case report and review of the literature. Hum. Mutat 2003, 21, 101–102. [Google Scholar]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zoric, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative splicing of p53 and p73: The novel p53 splice variant p53delta is an independent prognostic marker in ovarian cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar]
- Rowan, S.; Ludwig, R.L.; Haupt, Y.; Bates, S.; Lu, X.; Oren, M.; Vousden, K.H. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J 1996, 15, 827–838. [Google Scholar]
- IARC TP53 Database R16 Version. Available online: http://p53.iarc.fr/ (accessed on 3 January 2013).
- Petitjean, A.M.E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat 2007, 28, 622–629. [Google Scholar]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar]
- Yoshikawa, K.; Hamada, J.; Tada, M.; Kameyama, T.; Nakagawa, K.; Suzuki, Y.; Ikawa, M.; Hassan, N.M.; Kitagawa, Y.; Moriuchi, T. Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomed. Res 2010, 31, 401–411. [Google Scholar]
- Chan, K.T.; Lung, M.L. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother. Pharmacol 2004, 53, 519–526. [Google Scholar]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res 2000, 60, 6788–6793. [Google Scholar]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet 1993, 4, 42–46. [Google Scholar]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar]
- Chang, F.L.; Lai, M.D. Various forms of mutant p53 confer sensitivity to cisplatin and doxorubicin in bladder cancer cells. J. Urol 2001, 166, 304–310. [Google Scholar]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar]
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar]
- Donehower, L.A. Does p53 affect organismal aging? J. Cell. Physiol 2002, 192, 23–33. [Google Scholar]
- Liu, G.; McDonnell, T.J.; Montes de Oca Luna, R.; Kapoor, M.; Mims, B.; El-Naggar, A.K.; Lozano, G. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4174–4179. [Google Scholar]
- Lee, M.K.; Sabapathy, K. The R246S hot-spot p53 mutant exerts dominant-negative effects in embryonic stem cells in vitro and in vivo. J. Cell. Sci 2008, 121, 1899–1906. [Google Scholar]
- Lozano, G. Mouse models of p53 functions. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Donehower, L.A.; Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 2009, 9, 831–841. [Google Scholar]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol 2013, 15, 2–8. [Google Scholar]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev 2012, 26, 1268–1286. [Google Scholar]
- Frazier, M.W.; He, X.; Wang, J.; Gu, Z.; Cleveland, J.L.; Zambetti, G.P. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol 1998, 18, 3735–3743. [Google Scholar]
- Yan, W.; Chen, X. Identification of GRO1 as a critical determinant for mutant p53 gain of function. J. Biol. Chem 2009, 284, 12178–12187. [Google Scholar]
- Bossi, G.; Marampon, F.; Maor-Aloni, R.; Zani, B.; Rotter, V.; Oren, M.; Strano, S.; Blandino, G.; Sacchi, A. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle 2008, 7, 1870–1879. [Google Scholar]
- Gurtner, A.; Starace, G.; Norelli, G.; Piaggio, G.; Sacchi, A.; Bossi, G. Mutant p53-induced up-regulation of mitogen-activated protein kinase kinase 3 contributes to gain of function. J. Biol. Chem 2010, 285, 14160–14169. [Google Scholar]
- Preuss, U.; Kreutzfeld, R.; Scheidtmann, K.H. Tumor-derived p53 mutant C174Y is a gain-of-function mutant which activates the fos promoter and enhances colony formation. Int. J. Cancer 2000, 88, 162–171. [Google Scholar]
- Deb, S.; Jackson, C.T.; Subler, M.A.; Martin, D.W. Modulation of cellular and viral promoters by mutant human p53 proteins found in tumor cells. J. Virol 1992, 66, 6164–6170. [Google Scholar]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar]
- Sankala, H.; Vaughan, C.; Wang, J.; Deb, S.; Graves, P.R. Upregulation of the mitochondrial transport protein, Tim50, by mutant p53 contributes to cell growth and chemoresistance. Arch. Biochem. Biophys 2011, 512, 52–60. [Google Scholar]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol 2009, 16, 1086–1093. [Google Scholar]
- Scian, M.J.; Stagliano, K.E.; Deb, D.; Ellis, M.A.; Carchman, E.H.; Das, A.; Valerie, K.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce oncogenesis by transactivating growth-promoting genes. Oncogene 2004, 23, 4430–4443. [Google Scholar]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol 2011, 31, 4464–4481. [Google Scholar]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol 2001, 21, 1874–1887. [Google Scholar]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem 2002, 277, 18817–18826. [Google Scholar]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol 1999, 19, 1438–1449. [Google Scholar]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet 2000, 25, 47–54. [Google Scholar]
- Bargonetti, J.; Chicas, A.; White, D.; Prives, C. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell. Mol. Biol 1997, 43, 935–949. [Google Scholar]
- Chicas, A.; Molina, P.; Bargonetti, J. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem. Biophys. Res. Commun 2000, 279, 383–390. [Google Scholar]
- Torgeman, A.; Mor-Vaknin, N.; Zelin, E.; Ben-Aroya, Z.; Lochelt, M.; Flugel, R.M.; Aboud, M. Sp1-p53 heterocomplex mediates activation of HTLV-I long terminal repeat by 12-O-tetradecanoylphorbol-13-acetate that is antagonized by protein kinase C. Virology 2001, 281, 10–20. [Google Scholar]
- Hwang, C.I.; Choi, J.; Zhou, Z.; Flesken-Nikitin, A.; Tarakhovsky, A.; Nikitin, A.Y. MET-dependent cancer invasion may be preprogrammed by early alterations of p53-regulated feedforward loop and triggered by stromal cell-derived HGF. Cell Cycle 2011, 10, 3834–3840. [Google Scholar]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell. Biol 2007, 9, 573–580. [Google Scholar]
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res 2009, 69, 4818–4826. [Google Scholar]
- Wong, A.S.; Maines-Bandiera, S.L.; Rosen, B.; Wheelock, M.J.; Johnson, K.R.; Leung, P.C.; Roskelley, C.D.; Auersperg, N. Constitutive and conditional cadherin expression in cultured human ovarian surface epithelium: Influence of family history of ovarian cancer. Int. J. Cancer 1999, 81, 180–188. [Google Scholar]
- Joerger, A.C.; Rajagopalan, S.; Natan, E.; Veprintsev, D.B.; Robinson, C.V.; Fersht, A.R. Structural evolution of p53, p63, and p73: Implication for heterotetramer formation. Proc. Natl. Acad. Sci. USA 2009, 106, 17705–17710. [Google Scholar]
- Weisz, L.; Zalcenstein, A.; Stambolsky, P.; Cohen, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res 2004, 64, 8318–8327. [Google Scholar]
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol. Cell. Biol 2005, 25, 10097–10110. [Google Scholar]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar]
- Gualberto, A.; Aldape, K.; Kozakiewicz, K.; Tlsty, T.D. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 5166–5171. [Google Scholar]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar]
- Imbriano, C.; Gurtner, A.; Cocchiarella, F.; di Agostino, S.; Basile, V.; Gostissa, M.; Dobbelstein, M.; del Sal, G.; Piaggio, G.; Mantovani, R. Direct p53 transcriptional repression: In vivo analysis of CCAAT-containing G2/M promoters. Mol. Cell. Biol 2005, 25, 3737–3751. [Google Scholar]
- Wong, K.B.; deDecker, B.S.; Freund, S.M.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442. [Google Scholar]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ 2013, 20, 898–909. [Google Scholar]
- Werner, H.; Karnieli, E.; Rauscher, F.J.; LeRoith, D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8318–8323. [Google Scholar]
- Ludes-Meyers, J.H.; Subler, M.A.; Shivakumar, C.V.; Munoz, R.M.; Jiang, P.; Bigger, J.E.; Brown, D.R.; Deb, S.P.; Deb, S. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol 1996, 16, 6009–6019. [Google Scholar]
- Strano, S.; Munarriz, E.; Rossi, M.; Cristofanelli, B.; Shaul, Y.; Castagnoli, L.; Levine, A.J.; Sacchi, A.; Cesareni, G.; Oren, M.; et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J. Biol. Chem 2000, 275, 29503–29512. [Google Scholar]
- Li, B.; Murphy, K.L.; Laucirica, R.; Kittrell, F.; Medina, D.; Rosen, J.M. A transgenic mouse model for mammary carcinogenesis. Oncogene 1998, 16, 997–1007. [Google Scholar]
- Wang, X.J.; Greenhalgh, D.A.; Jiang, A.; He, D.; Zhong, L.; Brinkley, B.R.; Roop, D.R. Analysis of centrosome abnormalities and angiogenesis in epidermal-targeted p53172H mutant and p53-knockout mice after chemical carcinogenesis: Evidence for a gain of function. Mol. Carcinog 1998, 23, 185–192. [Google Scholar]
- Meng, X.; Laidler, L.L.; Kosmacek, E.A.; Yang, S.; Xiong, Z.; Zhu, D.; Wang, X.; Dai, D.; Zhang, Y.; Wang, X.; et al. Induction of mitotic cell death by overriding G2/M checkpoint in endometrial cancer cells with non-functional p53. Gynecol. Oncol 2012, 128, 461–469. [Google Scholar]
- Dong, P.; Xu, Z.; Jia, N.; Li, D.; Feng, Y. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol. Cancer 2009, 8, 103. [Google Scholar]
- Yan, W.; Chen, X. Characterization of functional domains necessary for mutant p53 gain of function. J. Biol. Chem 2010, 285, 14229–14238. [Google Scholar]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar]
- Lin, J.; Teresky, A.K.; Levine, A.J. Two critical hydrophobic amino acids in the N-terminal domain of the p53 protein are required for the gain of function phenotypes of human p53 mutants. Oncogene 1995, 10, 2387–2390. [Google Scholar]
- Singer, S.; Ehemann, V.; Brauckhoff, A.; Keith, M.; Vreden, S.; Schirmacher, P.; Breuhahn, K. Protumorigenic overexpression of stathmin/Op18 by gain-of-function mutation in p53 in human hepatocarcinogenesis. Hepatology 2007, 46, 759–768. [Google Scholar]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar]
- Suzuki, M.; Saito, S.; Saga, Y.; Ohwada, M.; Sato, I. Loss of heterozygosity on chromosome 6q27 and p53 mutations in epithelial ovarian cancer. Med. Oncol 1998, 15, 119–123. [Google Scholar]
- Frank, T.S.; Bartos, R.E.; Haefner, H.K.; Roberts, J.A.; Wilson, M.D.; Hubbell, G.P. Loss of heterozygosity and overexpression of the p53 gene in ovarian carcinoma. Mod. Pathol 1994, 7, 3–8. [Google Scholar]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar]
- Buller, R.E.; Runnebaum, I.B.; Karlan, B.Y.; Horowitz, J.A.; Shahin, M.; Buekers, T.; Petrauskas, S.; Kreienberg, R.; Slamon, D.; Pegram, M. A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther 2002, 9, 553–566. [Google Scholar]
- Buller, R.E.; Shahin, M.S.; Horowitz, J.A.; Runnebaum, I.B.; Mahavni, V.; Petrauskas, S.; Kreienberg, R.; Karlan, B.; Slamon, D.; Pegram, M. Long term follow-up of patients with recurrent ovarian cancer after Ad p53 gene replacement with SCH 58500. Cancer Gene Ther 2002, 9, 567–572. [Google Scholar]
- Sepehrnia, B.; Paz, I.B.; Dasgupta, G.; Momand, J. Heat shock protein 84 forms a complex with mutant p53 protein predominantly within a cytoplasmic compartment of the cell. J. Biol. Chem 1996, 271, 15084–15090. [Google Scholar]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Agrawal, S.; Chen, J. Stabilization of the MDM2 oncoprotein by mutant p53. J. Biol. Chem 2001, 276, 6874–6878. [Google Scholar]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem 2001, 276, 40583–40590. [Google Scholar]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ 2011, 18, 1904–1913. [Google Scholar]
- Liu, H.; Xiao, F.; Serebriiskii, I.G.; O’Brien, S.W.; Maglaty, M.A.; Astsaturov, I.A.; Martin, L.; Litwin, S.; Proia, D.A.; Golemis, E.A.; et al. Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clin. Cancer Res. 2013, in press. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brachova, P.; Thiel, K.W.; Leslie, K.K. The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 19257-19275. https://doi.org/10.3390/ijms140919257
Brachova P, Thiel KW, Leslie KK. The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. International Journal of Molecular Sciences. 2013; 14(9):19257-19275. https://doi.org/10.3390/ijms140919257
Chicago/Turabian StyleBrachova, Pavla, Kristina W. Thiel, and Kimberly K. Leslie. 2013. "The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer" International Journal of Molecular Sciences 14, no. 9: 19257-19275. https://doi.org/10.3390/ijms140919257
APA StyleBrachova, P., Thiel, K. W., & Leslie, K. K. (2013). The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. International Journal of Molecular Sciences, 14(9), 19257-19275. https://doi.org/10.3390/ijms140919257