Inhibition of Stromal PlGF Suppresses the Growth of Prostate Cancer Xenografts

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Results
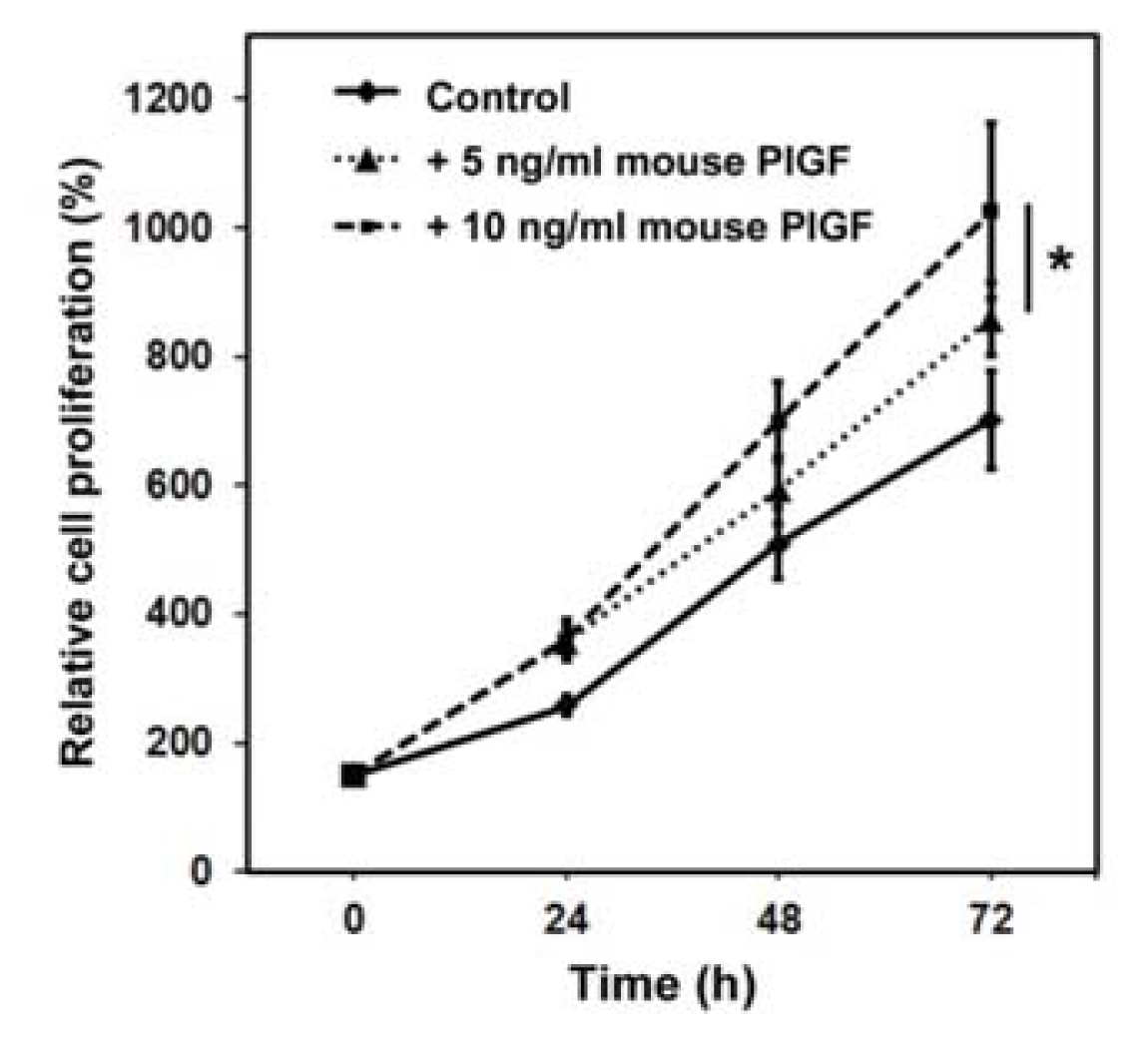
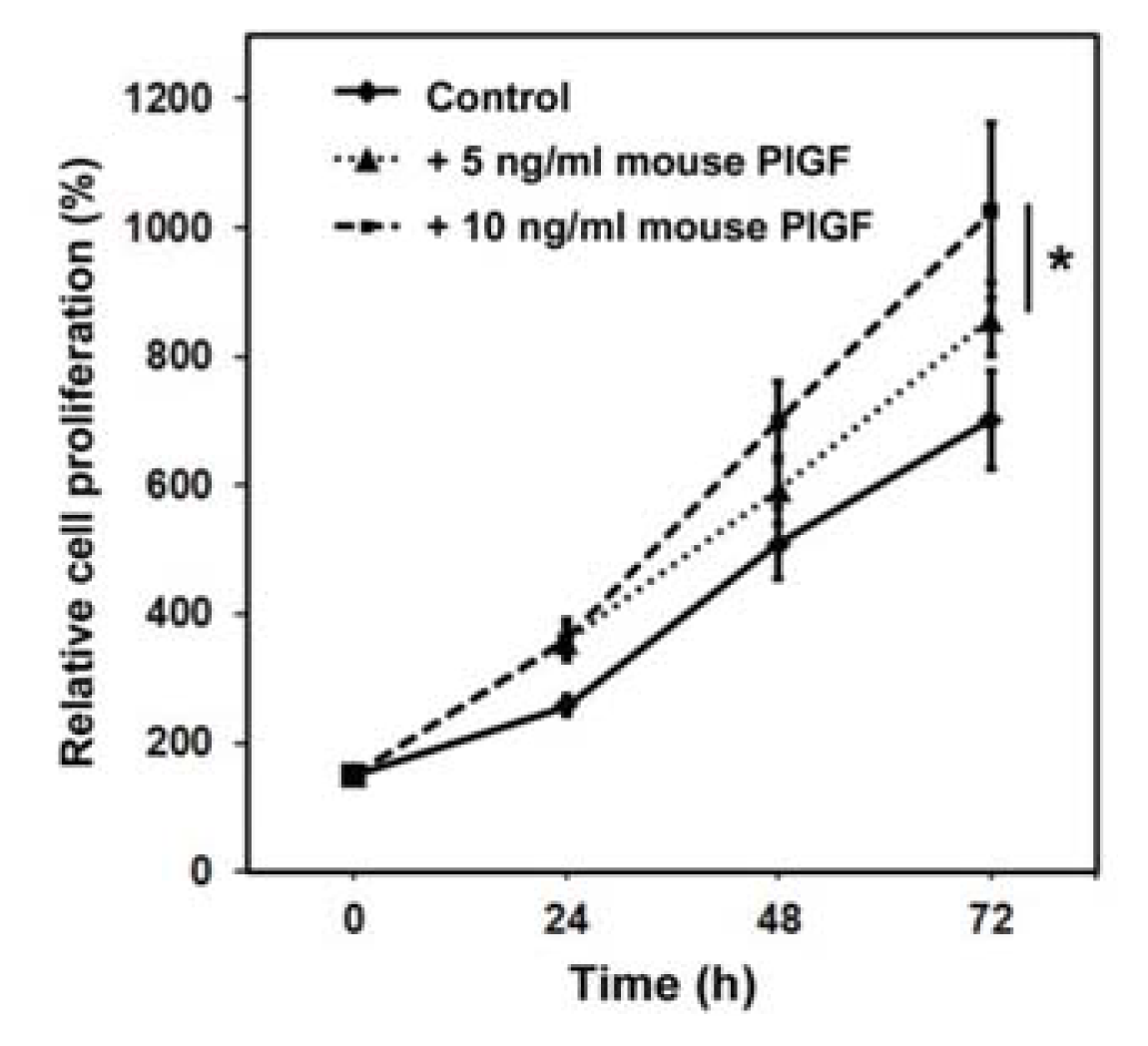
2.1.1. Murine PlGF Stimulates the Proliferation of PC-3 Cells in Vitro
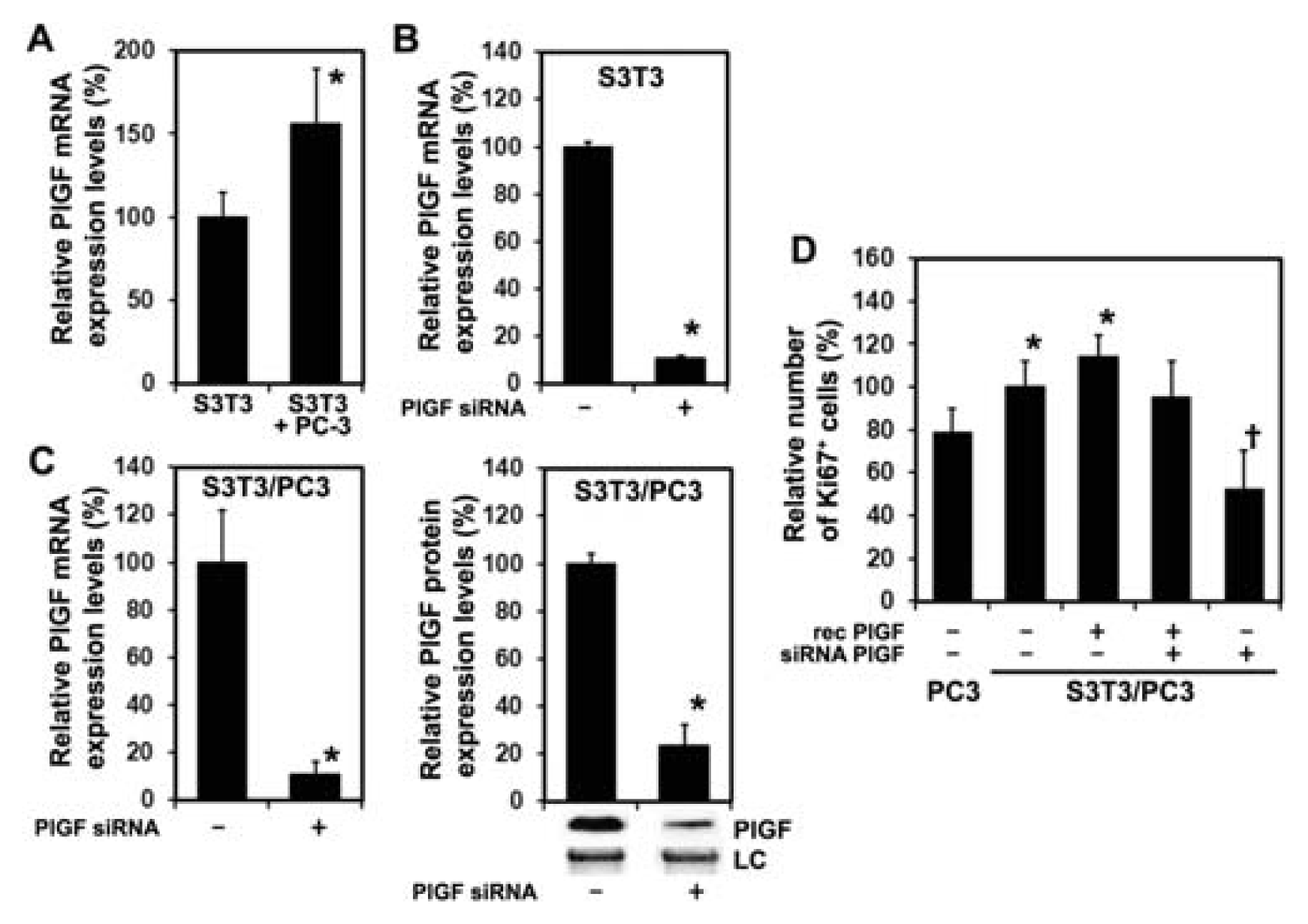
2.1.2. Inhibition of PlGF Expression in Co-Cultured PC-3 and S3T3 Fibroblasts
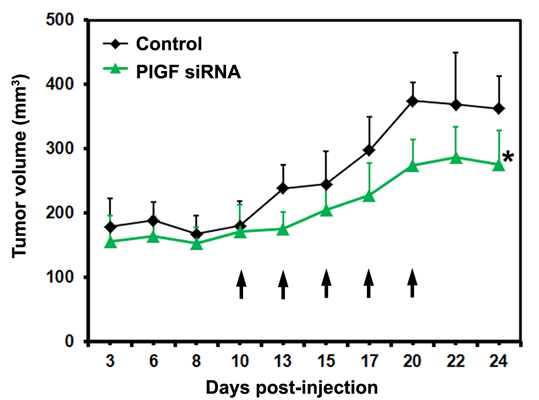
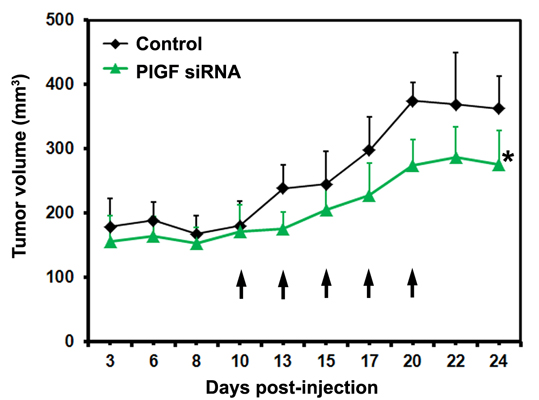
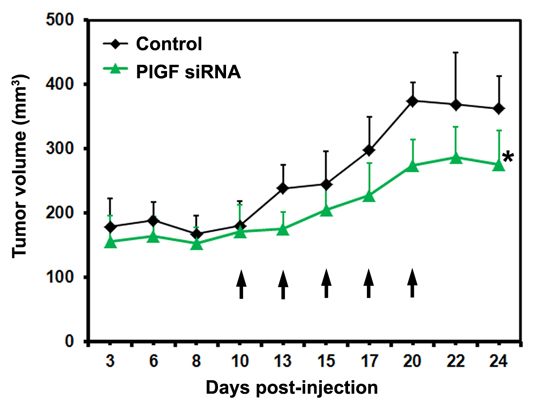
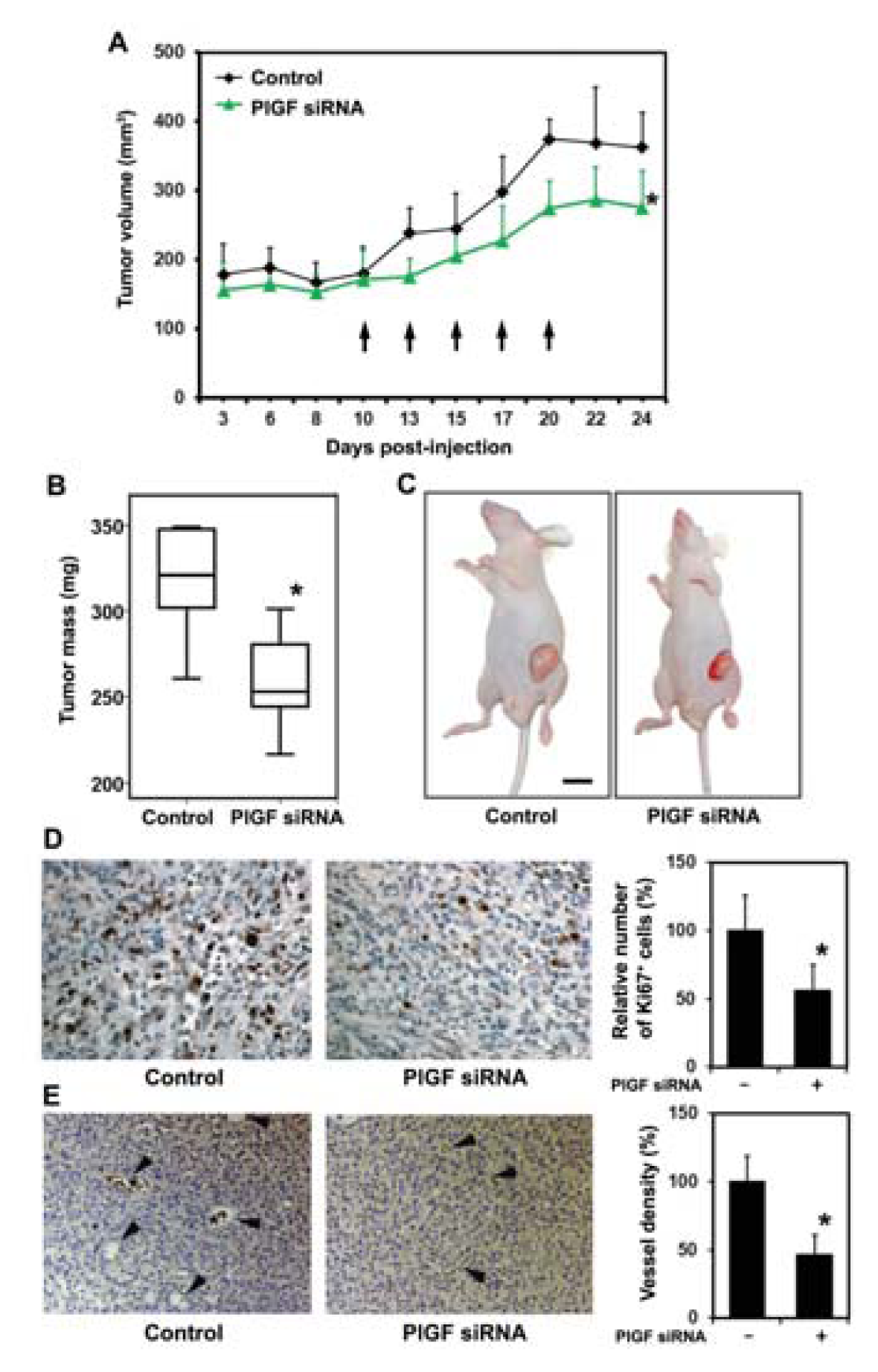
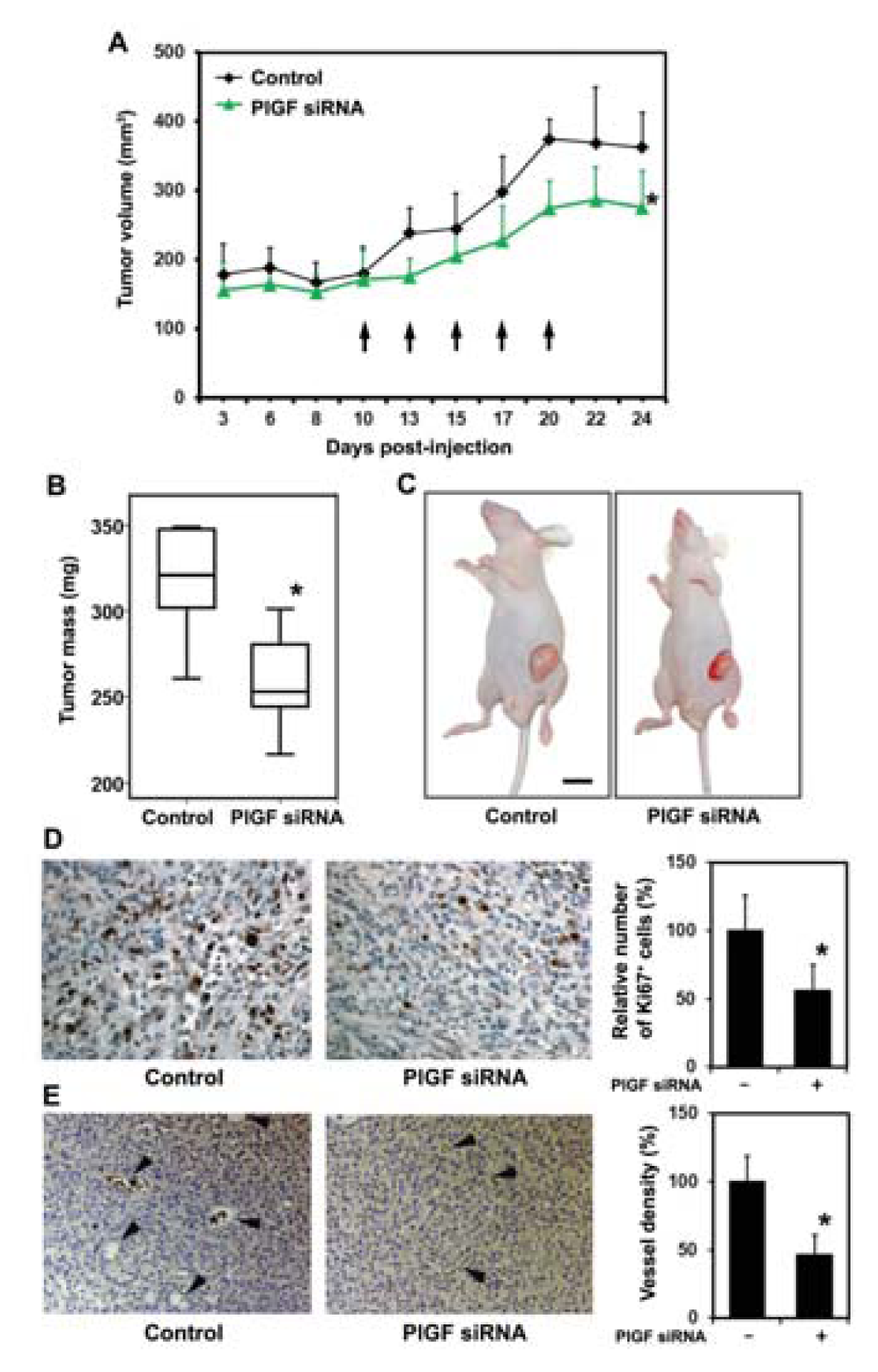
2.1.3. siRNA Mediated Blockade of Host PlGF Attenuates PC-3 Prostate Cancer Growth in Vivo
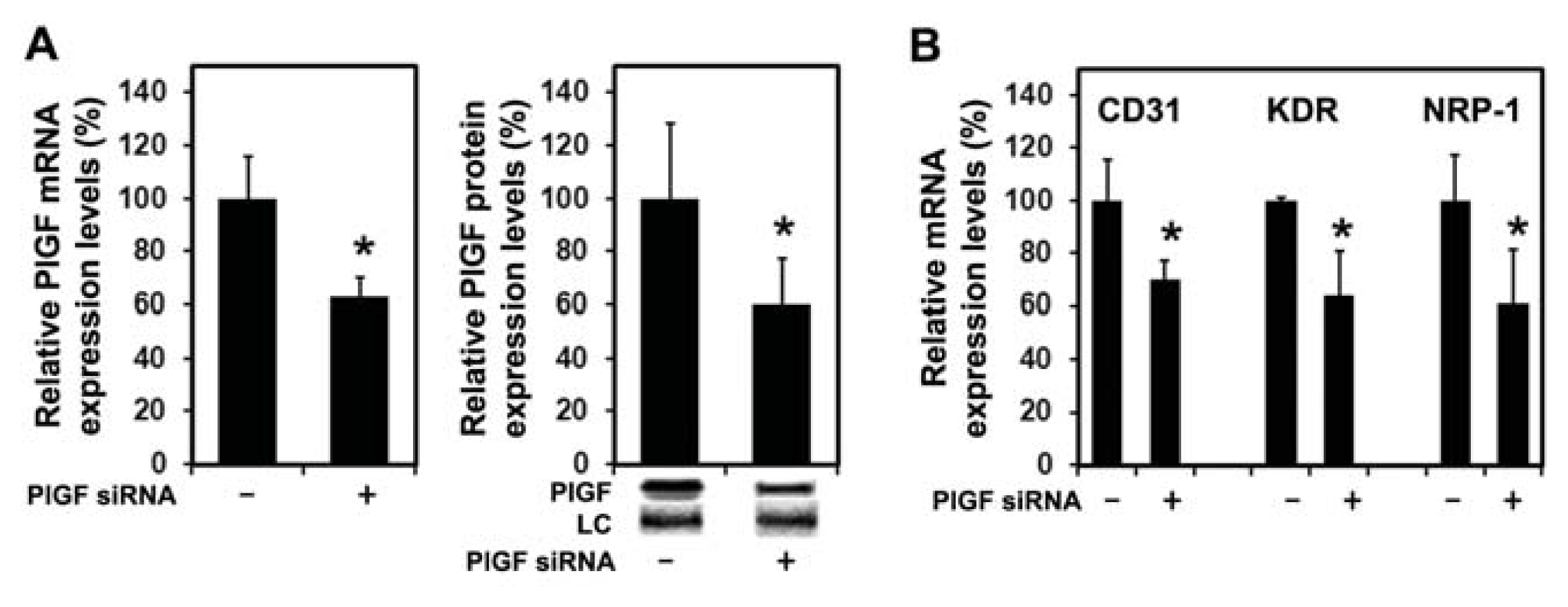
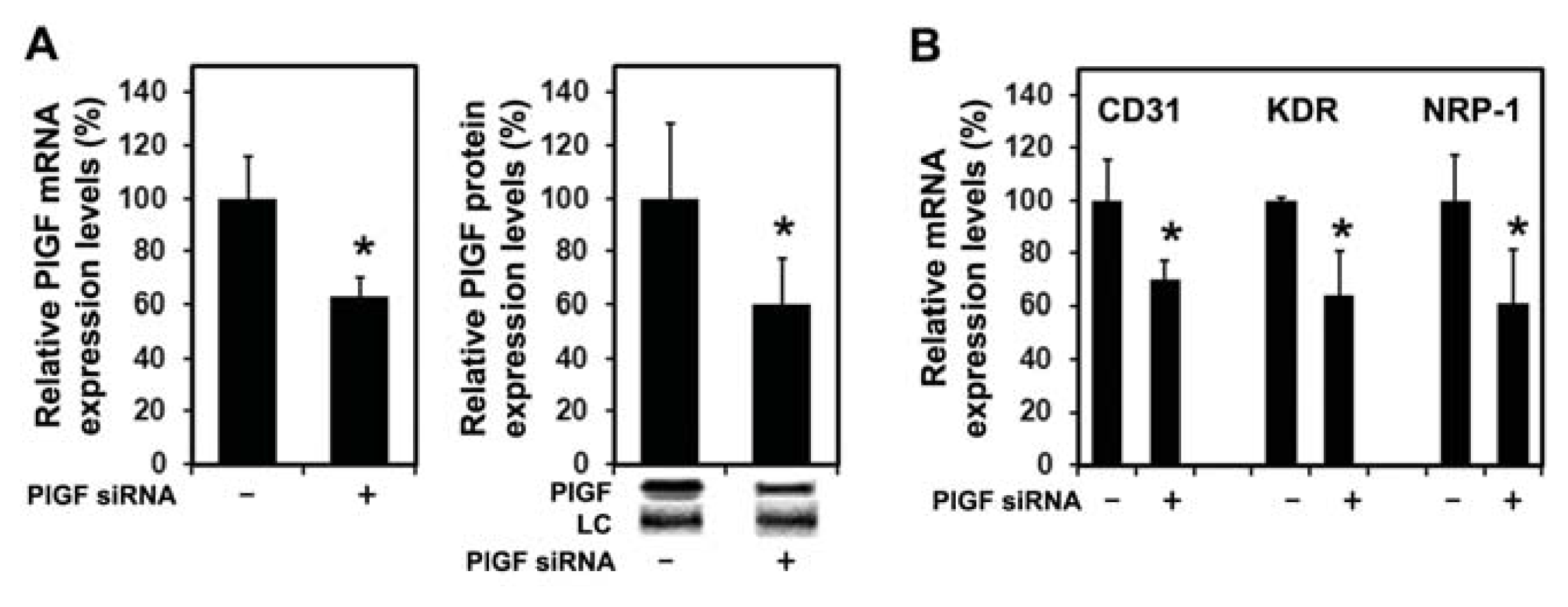
2.1.4. PlGF siRNA Treated Tumors Reduce Expression of Angiogenesis-Related Factors in Host Tissue
2.2. Discussion
3. Experimental Section
3.1. Cell Culture and Transfection
3.2. Cell Proliferation Assays
3.3. Real Time RT-PCR
3.4. Western Blotting
3.5. Histology and Immunohistochemistry, Immunofluorescence
3.6. Xenograft Model
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA: A Cancer J. Clin 2013, 63, 11–30. [Google Scholar]
- Beltran, H.; Beer, T.M.; Carducci, M.A.; de Bono, J.; Gleave, M.; Hussain, M.; Kelly, W.K.; Saad, F.; Sternberg, C.; Tagawa, S.T.; et al. New therapies for castration-resistant prostate cancer: Efficacy and safety. Eur. Urol 2011, 60, 279–290. [Google Scholar]
- Pagliarulo, V.; Bracarda, S.; Eisenberger, M.A.; Mottet, N.; Schroder, F.H.; Sternberg, C.N.; Studer, U.E. Contemporary role of androgen deprivation therapy for prostate cancer. Eur. Urol 2012, 61, 11–25. [Google Scholar]
- Carles, J.; Castellano, D.; Climent, M.A.; Maroto, P.; Medina, R.; Alcaraz, A. Castration-resistant metastatic prostate cancer: Current status and treatment possibilities. Clin. Transl. Oncol 2012, 14, 169–176. [Google Scholar]
- Weidner, N.; Carroll, P.R.; Flax, J.; Blumenfeld, W.; Folkman, J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am. J. Pathol 1993, 143, 401–409. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Cunha, G.R.; Hayward, S.W.; Wang, Y.Z. Role of stroma in carcinogenesis of the prostate. Differentiation 2002, 70, 473–485. [Google Scholar]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res 2011, 1, 482–497. [Google Scholar]
- Franco, O.E.; Hayward, S.W. Targeting the tumor stroma as a novel therapeutic approach for prostate cancer. Adv. Pharmacol 2012, 65, 267–313. [Google Scholar]
- Tammela, T.; Enholm, B.; Alitalo, K.; Paavonen, K. The biology of vascular endothelial growth factors. Cardiovasc Res 2005, 65, 550–563. [Google Scholar]
- Tjwa, M.; Luttun, A.; Autiero, M.; Carmeliet, P. VEGF and PlGF: Two pleiotropic growth factors with distinct roles in development and homeostasis. Cell Tissue Res 2003, 314, 5–14. [Google Scholar]
- Luttun, A.; Autiero, M.; Tjwa, M.; Carmeliet, P. Genetic dissection of tumor angiogenesis: Are PlGF and VEGFR-1 novel anti-cancer targets? Biochim Biophys. Acta 2004, 1654, 79–94. [Google Scholar]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med 2012, 44, 1–9. [Google Scholar]
- Neufeld, G.; Kessler, O.; Herzog, Y. The interaction of Neuropilin-1 and Neuropilin-2 with tyrosine-kinase receptors for VEGF. Adv. Exp. Med. Biol 2002, 515, 81–90. [Google Scholar]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; de Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med 2003, 9, 936–943. [Google Scholar]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; de Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med 2001, 7, 575–583. [Google Scholar]
- Dewerchin, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect. Med. 2012. [Google Scholar] [CrossRef]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; de Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar]
- Xu, L.; Jain, R.K. Down-regulation of placenta growth factor by promoter hypermethylation in human lung and colon carcinoma. Mol. Cancer Res 2007, 5, 873–880. [Google Scholar]
- Snuderl, M.; Batista, A.; Kirkpatrick, N.D.; Ruiz de Almodovar, C.; Riedemann, L.; Walsh, E.C.; Anolik, R.; Huang, Y.; Martin, J.D.; Kamoun, W.; et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 2013, 152, 1065–1076. [Google Scholar]
- Schmidt, T.; Kharabi Masouleh, B.; Loges, S.; Cauwenberghs, S.; Fraisl, P.; Maes, C.; Jonckx, B.; de Keersmaecker, K.; Kleppe, M.; Tjwa, M.; et al. Loss or inhibition of stromal-derived PlGF prolongs survival of mice with imatinib-resistant Bcr-Abl1(+) leukemia. Cancer Cell 2011, 19, 740–753. [Google Scholar]
- Xu, L.; Cochran, D.M.; Tong, R.T.; Winkler, F.; Kashiwagi, S.; Jain, R.K.; Fukumura, D. Placenta growth factor overexpression inhibits tumor growth, angiogenesis, and metastasis by depleting vascular endothelial growth factor homodimers in orthotopic mouse models. Cancer Res 2006, 66, 3971–3977. [Google Scholar]
- Matsumoto, K.; Suzuki, K.; Koike, H.; Hasumi, M.; Matsui, H.; Okugi, H.; Shibata, Y.; Ito, K.; Yamanaka, H. Placental growth factor gene expression in human prostate cancer and benign prostate hyperplasia. Anticancer Res 2003, 23, 3767–3773. [Google Scholar]
- Marrony, S.; Bassilana, F.; Seuwen, K.; Keller, H. Bone morphogenetic protein 2 induces placental growth factor in mesenchymal stem cells. Bone 2003, 33, 426–433. [Google Scholar]
- Tu, H.J.; Lin, T.H.; Chiu, Y.C.; Tang, C.H.; Yang, R.S.; Fu, W.M. Enhancement of placenta growth factor expression by oncostatin M in human rheumatoid arthritis synovial fibroblasts. J. Cell Physiol 2013, 228, 983–990. [Google Scholar]
- Fujii, T.; Yonemitsu, Y.; Onimaru, M.; Inoue, M.; Hasegawa, M.; Kuwano, H.; Sueishi, K. VEGF function for upregulation of endogenous PlGF expression during FGF-2-mediated therapeutic angiogenesis. Atherosclerosis 2008, 200, 51–57. [Google Scholar]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar]
- Shangguan, L.; Ti, X.; Krause, U.; Hai, B.; Zhao, Y.; Yang, Z.; Liu, F. Inhibition of TGF-beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 2012, 30, 2810–2819. [Google Scholar]
- Ziche, M.; Maglione, D.; Ribatti, D.; Morbidelli, L.; Lago, C.T.; Battisti, M.; Paoletti, I.; Barra, A.; Tucci, M.; Parise, G.; et al. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab. Invest 1997, 76, 517–531. [Google Scholar]
- Bais, C.; Wu, X.; Yao, J.; Yang, S.; Crawford, Y.; McCutcheon, K.; Tan, C.; Kolumam, G.; Vernes, J.M.; Eastham-Anderson, J.; et al. PlGF blockade does not inhibit angiogenesis during primary tumor growth. Cell 2010, 141, 166–177. [Google Scholar]
- Yao, J.; Wu, X.; Zhuang, G.; Kasman, I.M.; Vogt, T.; Phan, V.; Shibuya, M.; Ferrara, N.; Bais, C. Expression of a functional VEGFR-1 in tumor cells is a major determinant of anti-PlGF antibodies efficacy. Proc. Natl. Acad. Sci. USA 2011, 108, 11590–11595. [Google Scholar]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar]
- Dawson, M.R.; Duda, D.G.; Fukumura, D.; Jain, R.K. VEGFR1-activity-independent metastasis formation. Nature 2009, 461, E4. [Google Scholar]
- Jackson, D.E. The unfolding tale of PECAM-1. FEBS Lett 2003, 540, 7–14. [Google Scholar]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell. Res 2006, 312, 549–560. [Google Scholar]
- Migdal, M.; Huppertz, B.; Tessler, S.; Comforti, A.; Shibuya, M.; Reich, R.; Baumann, H.; Neufeld, G. Neuropilin-1 is a placenta growth factor-2 receptor. J. Biol. Chem 1998, 273, 22272–22278. [Google Scholar]
- Bagley, R.G.; Ren, Y.; Weber, W.; Yao, M.; Kurtzberg, L.; Pinckney, J.; Bangari, D.; Nguyen, C.; Brondyk, W.; Kaplan, J.; et al. Placental growth factor upregulation is a host response to antiangiogenic therapy. Clin. Cancer Res 2011, 17, 976–988. [Google Scholar]
- Lassen, U.; Nielsen, D.L.; Sorensen, M.; Winstedt, L.; Niskanen, T.; Stenberg, Y.; Pakola, S.; Stassen, J.M.; Glazer, S. A phase I, dose-escalation study of TB-403, a monoclonal antibody directed against PlGF, in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 678–684. [Google Scholar]
- Martinsson-Niskanen, T.; Riisbro, R.; Larsson, L.; Winstedt, L.; Stenberg, Y.; Pakola, S.; Stassen, J.M.; Glazer, S. Monoclonal antibody TB-403: A first-in-human, Phase I, double-blind, dose escalation study directed against placental growth factor in healthy male subjects. Clin. Ther 2011, 33, 1142–1149. [Google Scholar]
- Singh, S. Nanomaterials as non-viral siRNA delivery agents for cancer therapy. Bioimpacts 2013, 3, 53–65. [Google Scholar]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol 2013, 31, 653–658. [Google Scholar]
- Abraham, D.; Zins, K.; Sioud, M.; Lucas, T.; Aharinejad, S. Host CD147 blockade by small interfering RNAs suppresses growth of human colon cancer xenografts. Front. Biosci 2008, 13, 5571–5579. [Google Scholar]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res 2007, 67, 1038–1045. [Google Scholar]
- Romero-Calvo, I.; Ocon, B.; Martinez-Moya, P.; Suarez, M.D.; Zarzuelo, A.; Martinez-Augustin, O.; de Medina, F.S. Reversible ponceau staining as a loading control alternative to actin in Western blots. Anal. Biochem 2010, 401, 318–320. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zins, K.; Thomas, A.; Lucas, T.; Sioud, M.; Aharinejad, S.; Abraham, D. Inhibition of Stromal PlGF Suppresses the Growth of Prostate Cancer Xenografts. Int. J. Mol. Sci. 2013, 14, 17958-17971. https://doi.org/10.3390/ijms140917958
Zins K, Thomas A, Lucas T, Sioud M, Aharinejad S, Abraham D. Inhibition of Stromal PlGF Suppresses the Growth of Prostate Cancer Xenografts. International Journal of Molecular Sciences. 2013; 14(9):17958-17971. https://doi.org/10.3390/ijms140917958
Chicago/Turabian StyleZins, Karin, Anita Thomas, Trevor Lucas, Mouldy Sioud, Seyedhossein Aharinejad, and Dietmar Abraham. 2013. "Inhibition of Stromal PlGF Suppresses the Growth of Prostate Cancer Xenografts" International Journal of Molecular Sciences 14, no. 9: 17958-17971. https://doi.org/10.3390/ijms140917958