Oximes: Inhibitors of Human Recombinant Acetylcholinesterase. A Structure-Activity Relationship (SAR) Study



Abstract
:1. Introduction
2. Results and Discussion
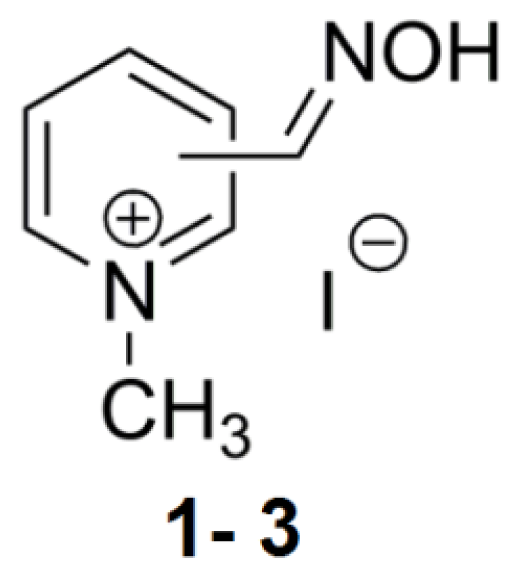
2.1. Mono-Pyridinium Reactivators with Different Oxime Group Positions
2.2. Bis-Pyridinium Reactivators with Various Connecting Chain Lengths
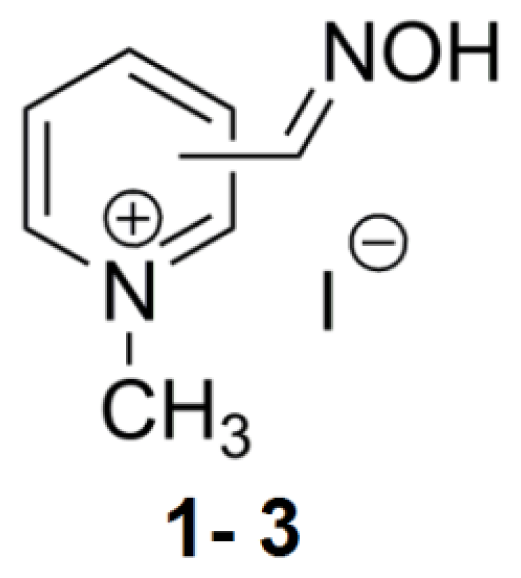
2.3. Bis-Pyridinium Reactivators with 3 or 4 Carbon Connecting Linker and Various Positions of the Oxime Group on the Pyridinium Ring
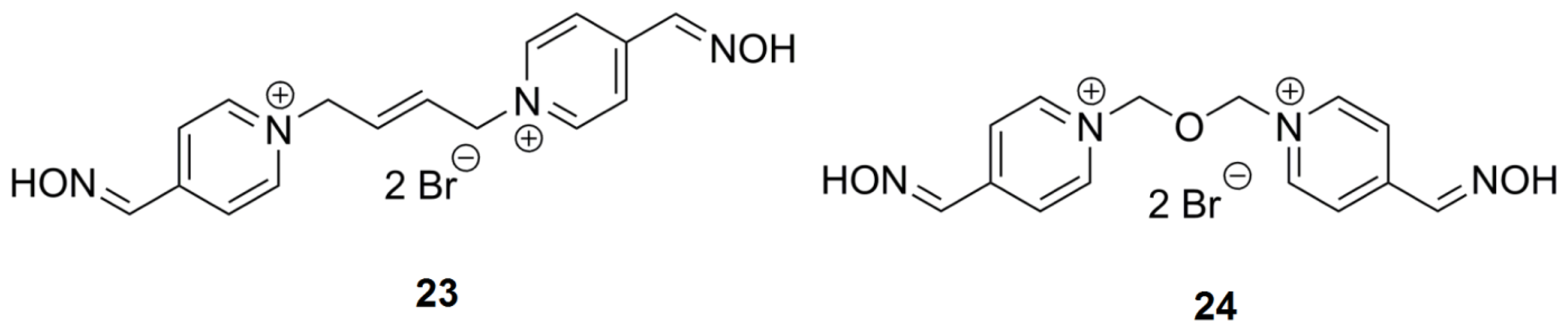
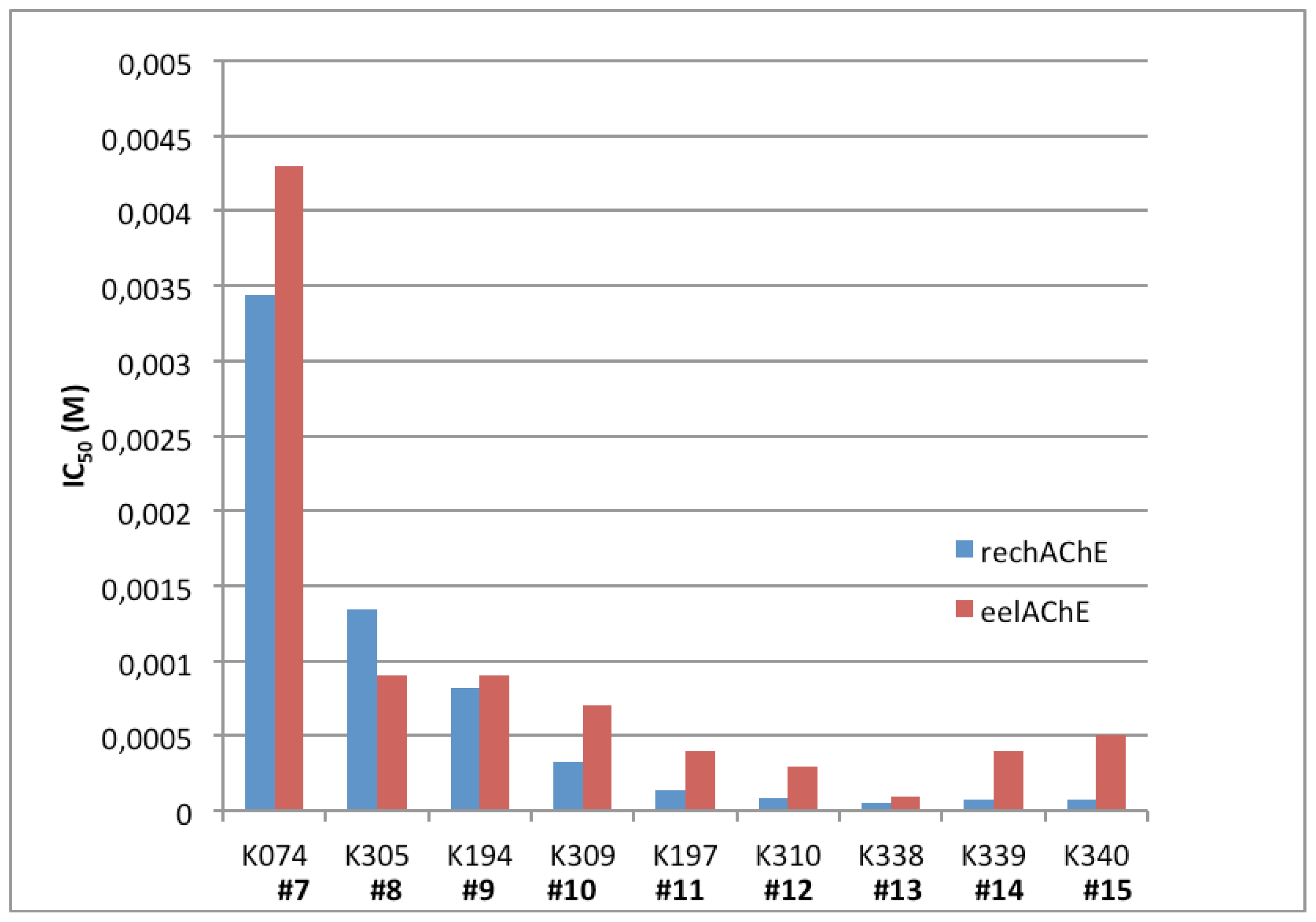
2.4. Bis-Pyridinium AChE Reactivators—Substitutions and Double Bond in Linker
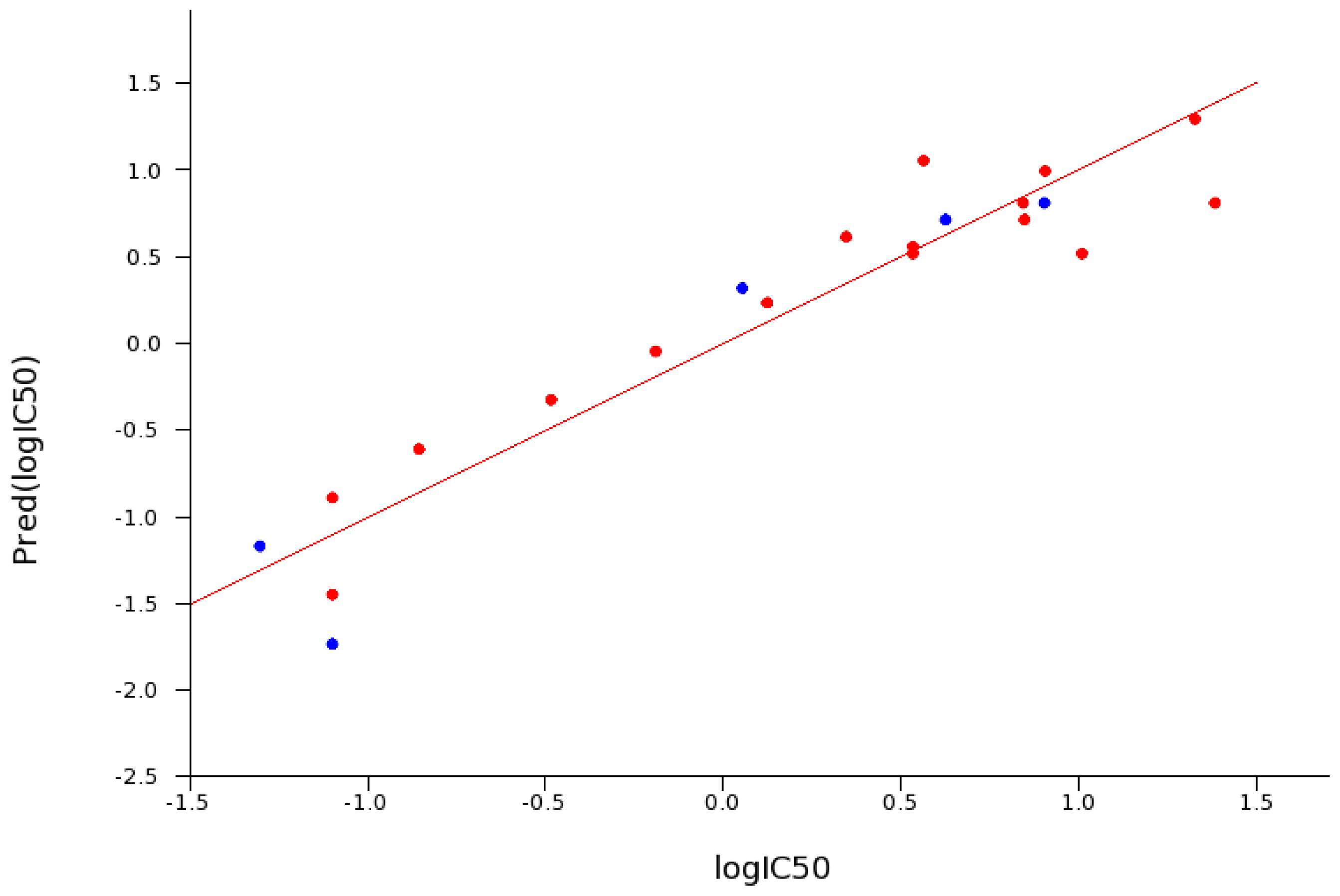
2.5. SAR for Reversible Inhibition of hrAChE
2.6. Reversible Inhibition of EeAChE versus hrAChE
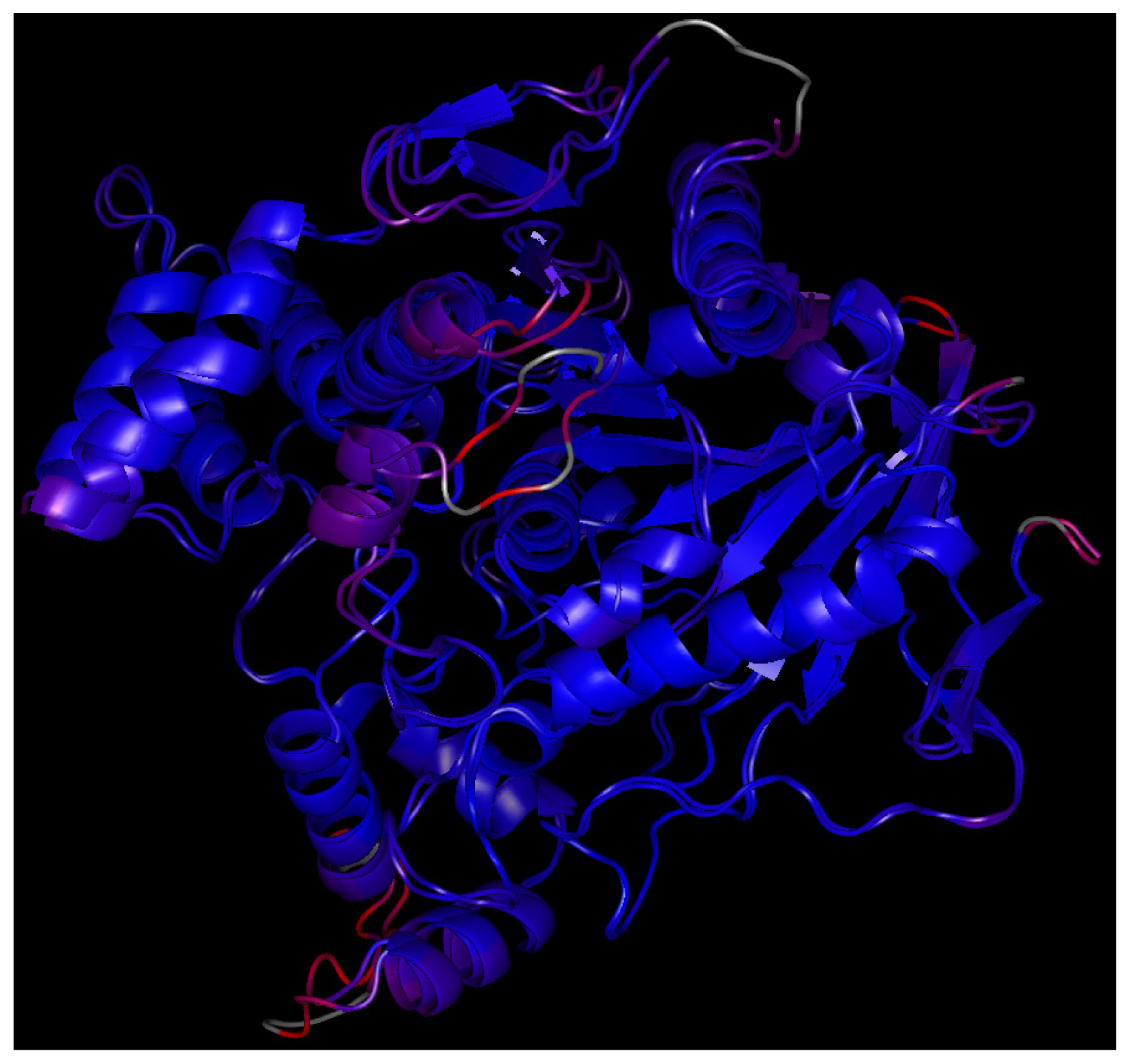
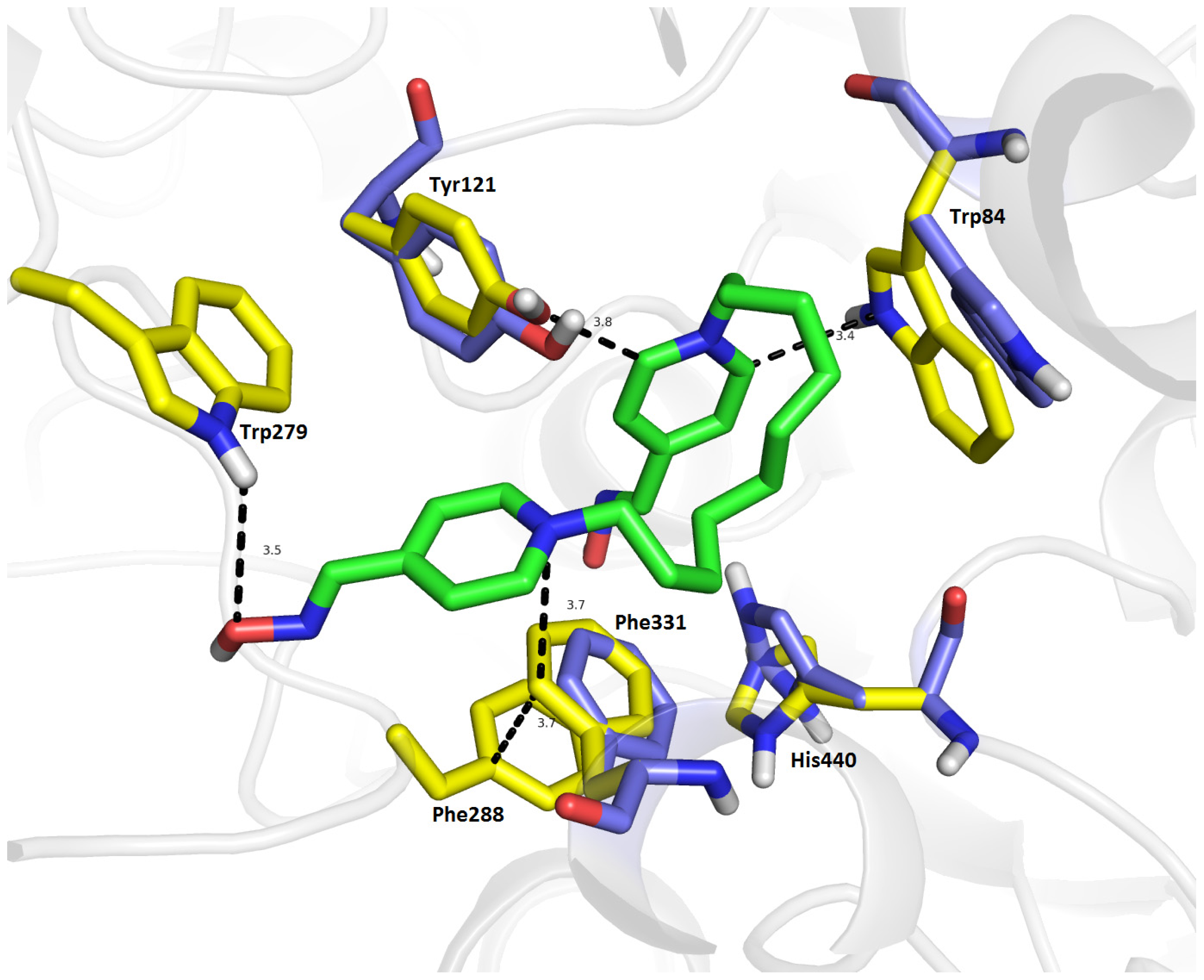
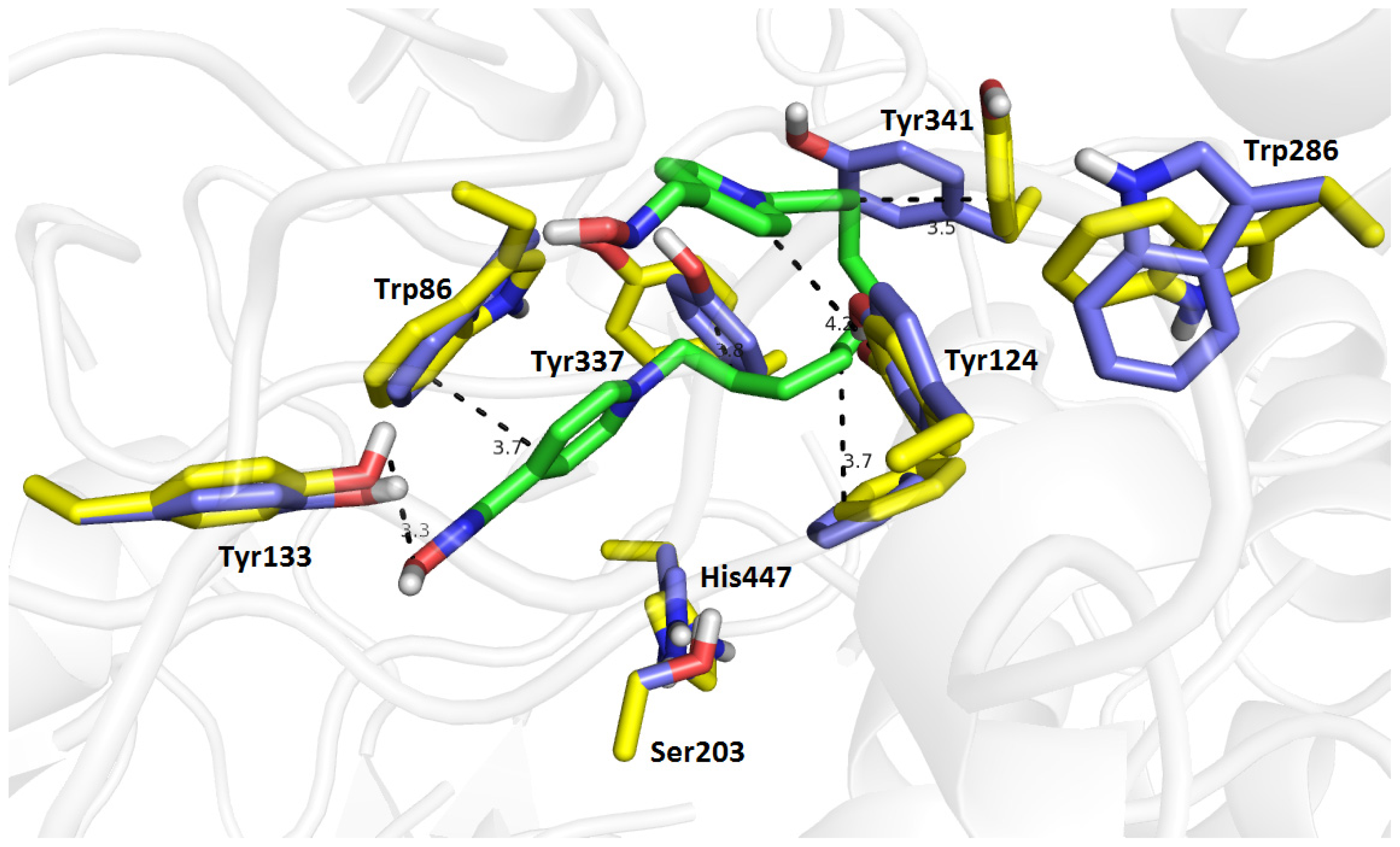
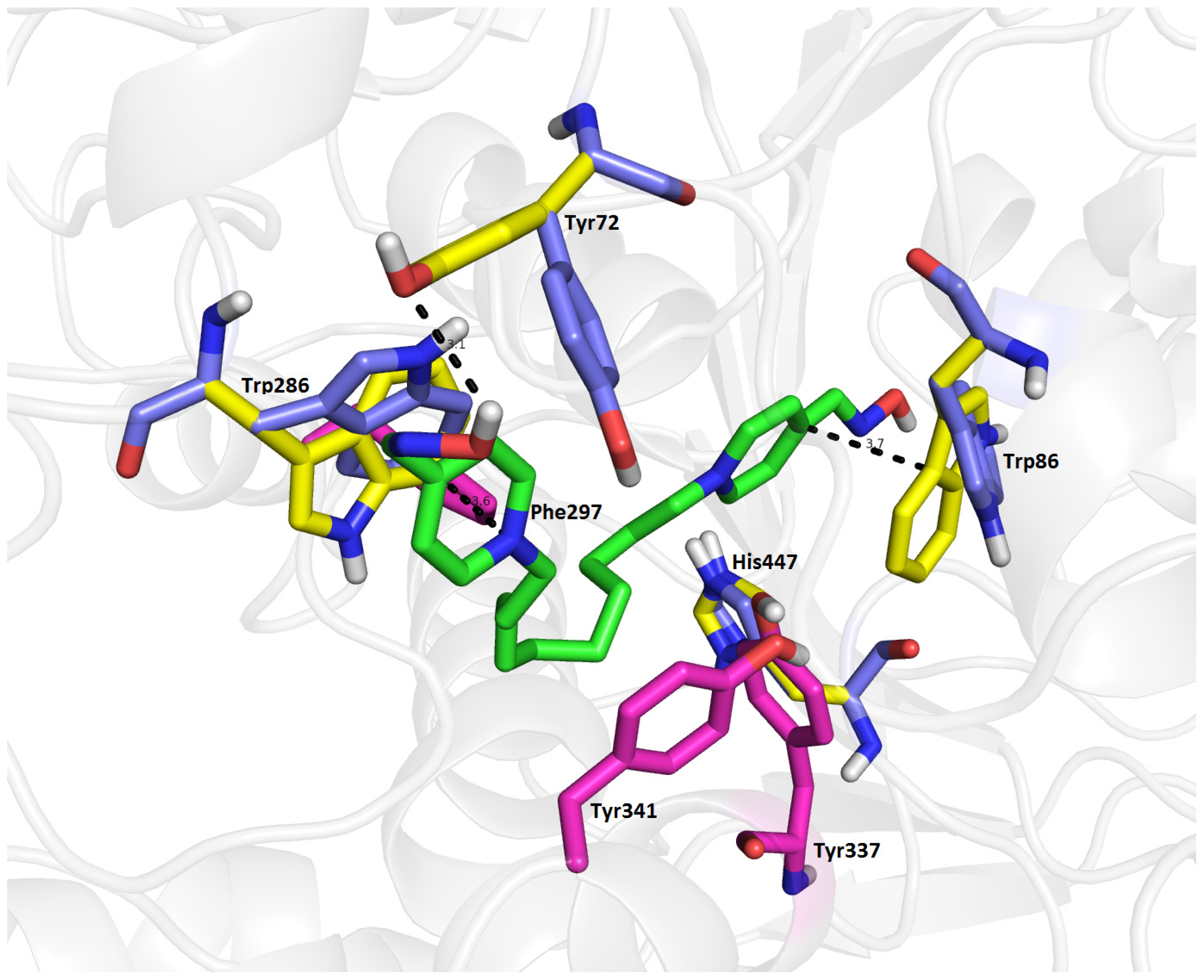
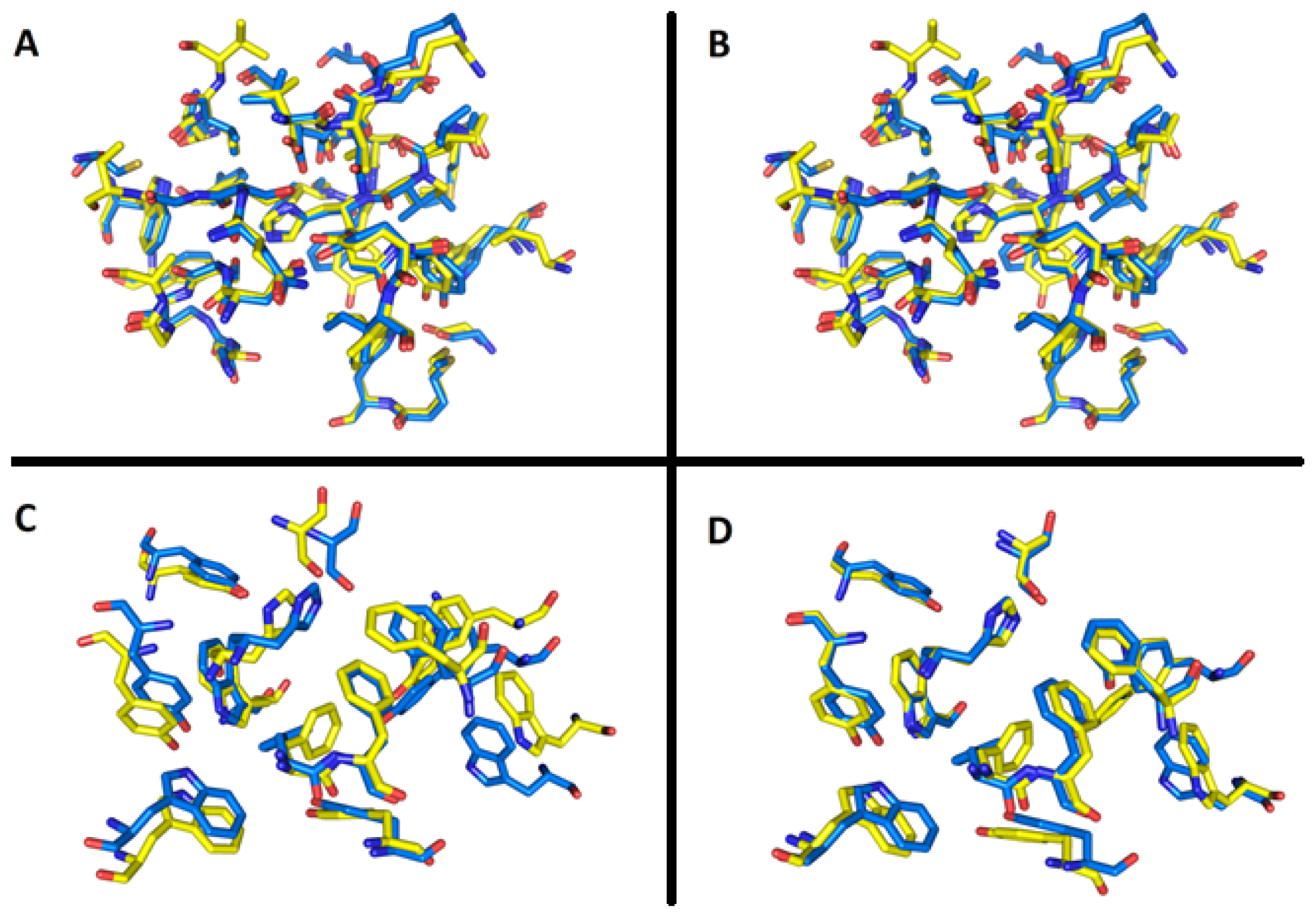
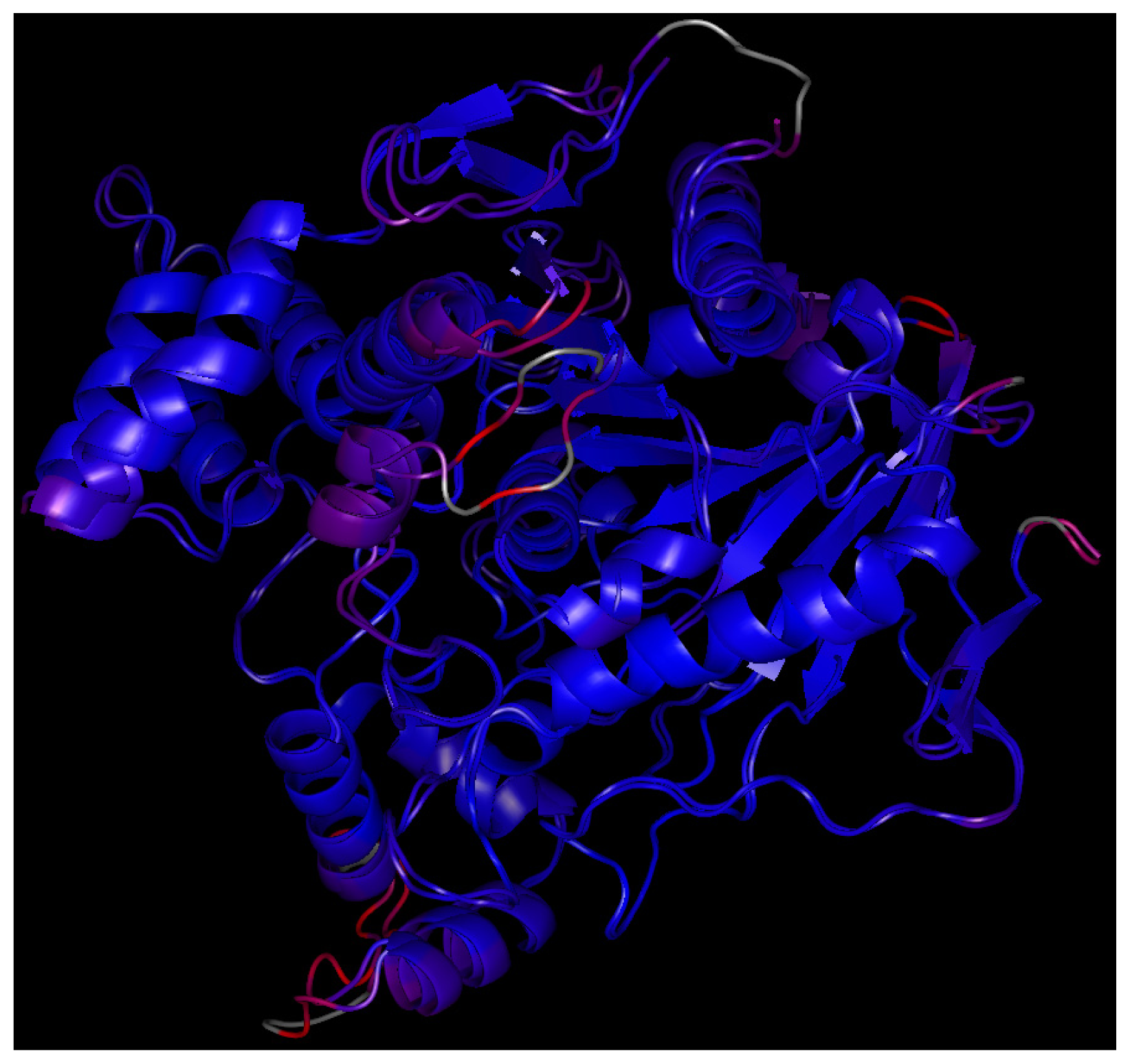
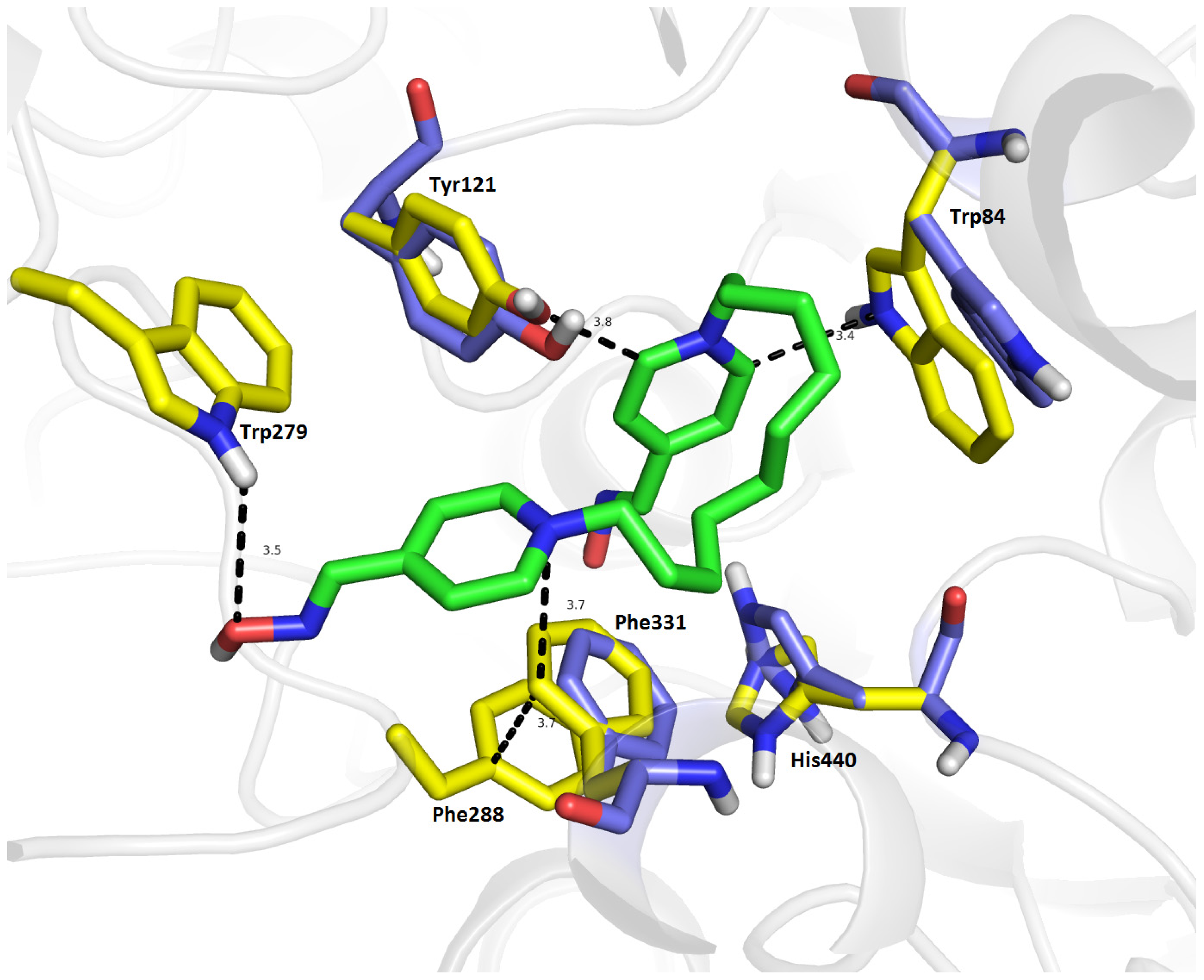
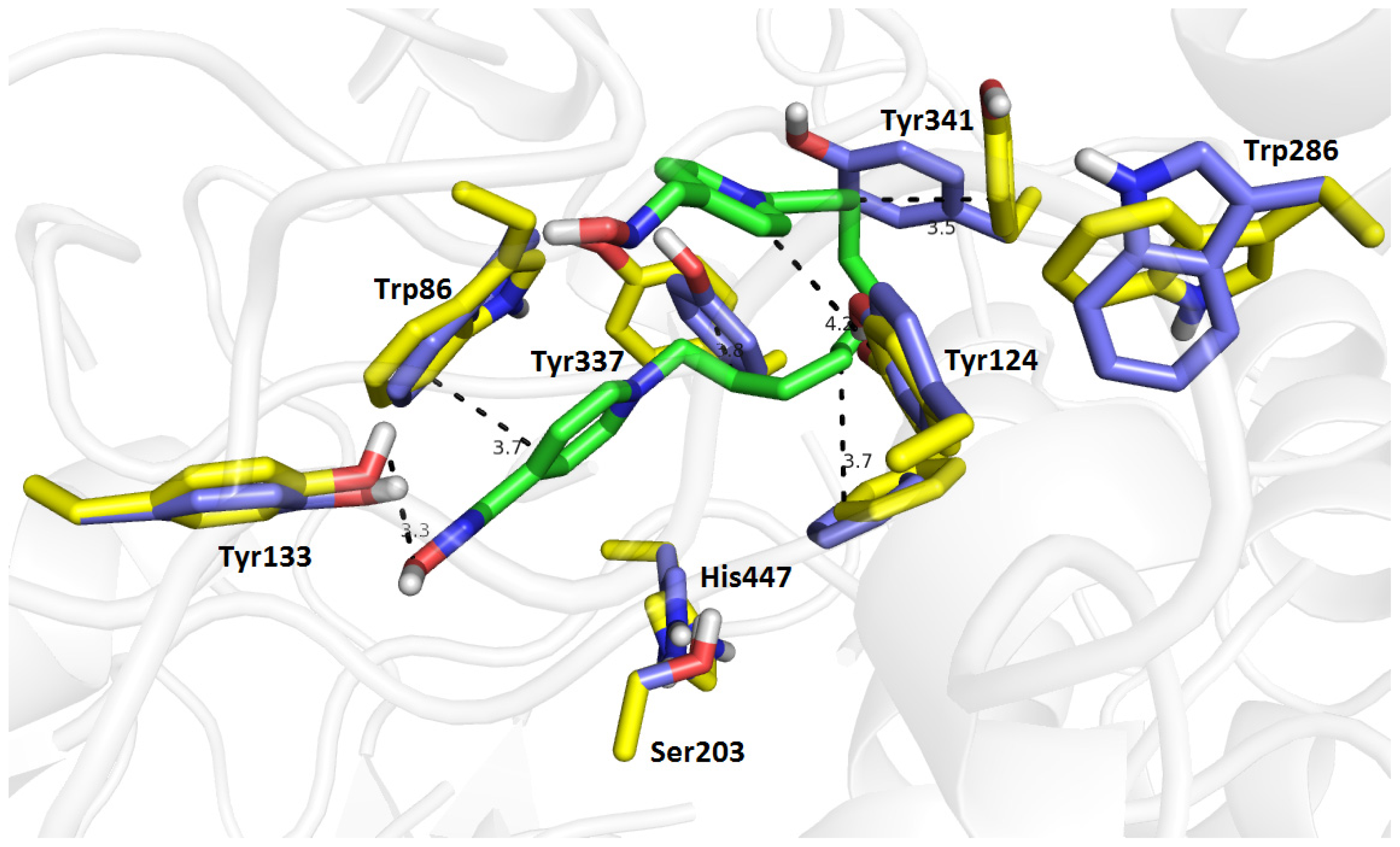
2.7. Molecular Docking
3. Experimental Section
3.1. Chemicals
3.2. The Measurement of IC50 of hrAChE
3.3. Docking Study
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wilson, I.B.; Ginsburg, S. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim. Biophys. Acta 1955, 18, 168–170. [Google Scholar]
- Wilson, I.B.; Ginsburg, S. Reactivation of acetylcholinesterase inhibited by alkylphosphates. Arch. Biochem. Biophys 1955, 54, 569–571. [Google Scholar]
- Childs, A.F.; Davies, D.R.; Green, A.L.; Rutland, J.P. The reactivation by oximes and hydroxamic acids of cholinesterase inhibited by organo-phosphorus compounds. Br. J. Pharmacol. Chemother 1955, 10, 462–465. [Google Scholar]
- Bajgar, J. Organophosphates/nerve agent poisoning: Mechanism of action, diagnosis, prophylaxis, and treatment. Adv. Clin. Chem 2004, 38, 151–216. [Google Scholar]
- Macllwain, C. Study proves Iraq used nerve gas. Nature 1993, 363, 3. [Google Scholar]
- Delfino, R.T.; Ribeiro, T.S.; Figueroa-Villar, J.D. Organophosphorus compounds as chemical warfare agents: A review. J. Braz. Chem. Soc 2009, 20, 407–428. [Google Scholar]
- Nagao, M.; Takatori, T.; Matsuda, Y.; Nakajima, M.; Iwase, H.; Iwadate, K. Definitive evidence for the acute sarin poisoning diagnosis in the Tokyo subway. Toxicol. Appl. Pharmacol 1997, 144, 198–203. [Google Scholar]
- Jokanovic, M.; Stojiljkovic, M.P. Current understanding of the application of pyridinium oximes as cholinesterase reactivators in treatment of organophosphate poisoning. Eur. J. Pharmacol 2006, 553, 10–17. [Google Scholar]
- Lorke, D.E.; Kalasz, H.; Petroianu, G.A.; Tekes, K. Entry of oximes into the brain: A review. Curr. Med. Chem 2008, 15, 743–753. [Google Scholar]
- Karasova, J.Z.; Pohanka, M.; Musilek, K.; Zemek, F.; Kuca, K. Passive diffusion of acetylcholinesterase oxime reactivators through the blood-brain barrier: Influence of molecular structure. Toxicol. in Vitro 2010, 24, 1838–1844. [Google Scholar]
- Palin, R.; Clark, J.K.; Cowley, P.; Muir, A.W.; Pow, E.; Prosser, A.B.; Taylor, R.; Zhang, M.Q. Novel piperidinium and pyridinium agents as water-soluble acetylcholinesterase inhibitors for the reversal of neuromuscular blockade. Bioorg. Med. Chem. Lett 2002, 12, 2569–2572. [Google Scholar]
- Petrova, I.; Bielavsky, J. An overview of the syntheses of cholinesterase reactivators from 1980 to 1992. Mil. Med. Sci. Lett 2001, 70, 63–73. [Google Scholar]
- Sepsova, V.; Karasova, J.; Zemek, F.; Bennion, B.J.; Kuca, K. Oximes as inhibitors of acetylcholinesterase—A structure-activity relationship (SAR) study. Mil. Med. Sci. Lett 2011, 80, 178–186. [Google Scholar]
- Oh, K.A.; Park, N.J.; Park, N.S.; Kuca, K.; Jun, D.; Jung, Y.S. Reactivation of DFP- and paraoxon-inhibited acetylcholinesterases by pyridinium oximes. Chem. Biol. Interact 2008, 175, 365–367. [Google Scholar]
- Kuca, K.; Kassa, J. A comparison of the ability of a new bispyridinium oxime-1-(4- hydroxyiminomethylpyridinium)-4-(4-carbamoylpyridinium)butane dibromide and currently used oximes to reactivate nerve agent-inhibited rat brain acetylcholinesterase by in vitro methods. J. Enzyme Inhib. Med. Chem 2003, 18, 529–535. [Google Scholar]
- Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Design, evaluation and structure-activity relationship studies of the AChE reactivators against organophosphorus pesticides. Med. Res. Rev 2011, 31, 548–575. [Google Scholar]
- Kassa, J.; Kuca, K.; Bartosova, L.; Kunesova, G. The development of new structural analogues of oximes for the antidotal treatment of poisoning by nerve agents and the comparison of their reactivating and therapeutic efficacy with currently available oximes. Curr. Org. Chem 2007, 11, 267–283. [Google Scholar]
- Jin, G.Y.; He, X.C.; Zhang, H.Y.; Bai, D.L. Synthesis of alkylene-linked dieters of (−)-huperzine A. Chin. Chem. Lett 2002, 13, 23–26. [Google Scholar]
- Kassa, J.; Jun, D.; Kuca, K.; Bajgar, J. Comparison of reactivating and therapeutic efficacy of two salts of the oxime HI-6 against tabun, soman and cyclosarin in rats. Basic Clin. Pharmacol. Toxicol. 2007, 101, 328–332. [Google Scholar]
- Musilek, K.; Holas, O.; Hambalek, J.; Kuca, K.; Jun, D.; Dohnal, V.; Dolezal, M. Synthesis of bispyridinium compounds bearing propane linker and evaluation of their reactivation activity against tabun- and paraoxon-inhibited acetylcholinesterase. Lett. Org. Chem 2006, 3, 831–835. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.3; Schrödinger, LLC, 2010. [Google Scholar]
- Bolea, I.; Juarez-Jimenez, J.; de Los Rios, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem 2011, 54, 8251–8270. [Google Scholar]
- Musilek, K.; Komloova, M.; Holas, O.; Hrabinova, M.; Pohanka, M.; Dohnal, V.; Nachon, F.; Dolezal, M.; Kuca, K. Preparation and in vitro screening of symmetrical bis-isoquinolinium cholinesterase inhibitors bearing various connecting linkage—Implications for early Myasthenia gravis treatment. Eur. J. Med. Chem 2011, 46, 811–818. [Google Scholar]
- Komloova, M.; Musilek, K.; Horova, A.; Holas, O.; Dohnal, V.; Gunn-Moore, F.; Kuca, K. Preparation, in vitro screening and molecular modelling of symmetrical bis-quinolinium cholinesterase inhibitors-implications for early Myasthenia gravis treatment. Bioorg. Med. Chem. Lett 2011, 21, 2505–2509. [Google Scholar]
- Musilek, K.; Holas, O.; Kuca, K.; Jun, D.; Dohnal, V.; Opletalova, V.; Dolezal, M. Novel series of bispyridinium compounds bearing a (Z)-but-2-ene linker—Synthesis and evaluation of their reactivation activity against tabun and paraoxon-inhibited acetylcholinesterase. Bioorg. Med. Chem. Lett 2007, 17, 3172–3176. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol 1961, 7, 88–90. [Google Scholar]
- Pohanka, M.; Hrabinova, M.; Kuca, K. Diagnosis of intoxication by the organophosphate VX: Comparison between an electrochemical sensor and Ellman’s photometric method. Sensors 2008, 8, 5229–5237. [Google Scholar]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem 2007, 370, 223–227. [Google Scholar]
- Duan, J.X.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model 2010, 29, 157–170. [Google Scholar]
- Trott, O.; Olson, A.J. Software news and update autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar]
- Kryger, G.; Harel, M.; Giles, K.; Toker, L.; Velan, B.; Lazar, A.; Kronman, C.; Barak, D.; Ariel, N.; Shafferman, A.; et al. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr. D 2000, 56, 1385–1394. [Google Scholar]
- Harel, M.; Schalk, I.; Ehretsabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand-binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar]
- Bourne, Y.; Grassi, J.; Bougis, P.E.; Marchot, P. Conformational flexibility of the acetylcholinesterase tetramer suggested by X-ray crystallography. J. Biol. Chem 1999, 274, 30370–30376. [Google Scholar]




{kind=link}
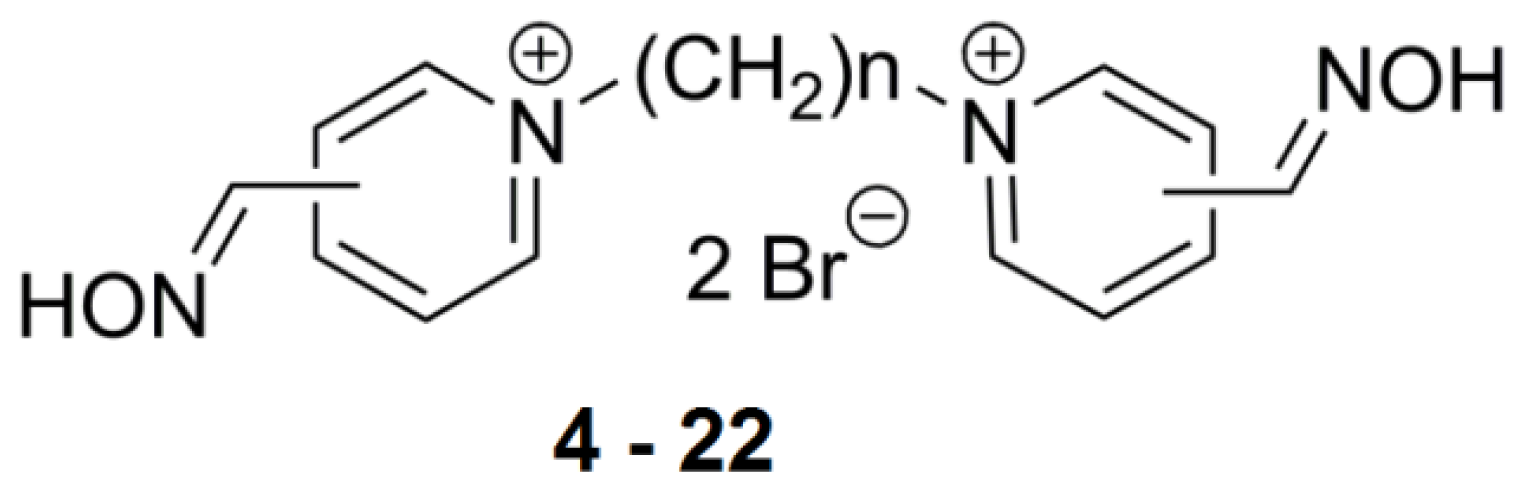{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name of oxime | Oxime position | IC50 values EeAChE (mM) | IC50 values hrAChE (mM) | 95% confidence intervals (mM) |
|---|---|---|---|---|---|
| 1 | 2PAM | 2 | 54.9 | 45.1 | 26.5–160.6 |
| 2 | 3PAM | 3 | 27.1 | 41.6 | 8.96–192.8 |
| 3 | 4PAM | 4 | 64.3 | - | - |
| Compound | Name of oxime | n | Oxime position | IC50 values EeAChE (mM) | IC50 values rec-hrAChE (mM) | 95% confidence intervals (mM) |
|---|---|---|---|---|---|---|
| 4 | K154 | 1 | 4,4′ | 227.4 | 21.38 | - |
| 5 | K191 | 2 | 4,4′ | 124.6 | 3.69 | 0.94–10.75 |
| 6 | K018 | 3 | 4,4′ | 51.9 | 24.37 | 13.84–42.92 |
| 7 | K074 | 4 | 4,4′ | 4.3 | 3.44 | 1.92–6.15 |
| 8 | K305 | 5 | 4,4′ | 0.9 | 1.34 | 0.66–2.72 |
| 9 | K194 | 6 | 4,4′ | 0.9 | 0.65 | 0.64–1.07 |
| 10 | K309 | 7 | 4,4′ | 0.7 | 0.33 | 0.27–0.42 |
| 11 | K197 | 8 | 4,4′ | 0.4 | 0.14 | 0.09–0.21 |
| 12 | K310 | 9 | 4,4′ | 0.3 | 0.08 | 0.04–0.19 |
| 13 | K338 | 10 | 4,4′ | 0.1 | 0.05 | 0.03–0.08 |
| 14 | K339 | 11 | 4,4′ | 0.4 | 0.08 | 0.04–0.13 |
| 15 | K340 | 12 | 4,4′ | 0.5 | 0.08 | 0.06–0.11 |
| 16 | K005 | 3 | 2,2′ | 0.4 | 2.23 | 1.76–2.83 |
| 17 | K099 | 3 | 3,3′ | 19.5 | 7.01 | 3.63–13.51 |
| 18 | K207 | 3 | 2,3′ | 17.7 | 7.09 | 4.08–12.36 |
| 19 | K208 | 3 | 2,4′ | 3.1 | 4.26 | 2.43–7.49 |
| 20 | K209 | 3 | 3,4′ | 13.8 | 8.06 | 6.36–10.22 |
| 21 | K033 | 4 | 2,2′ | 1.1 | 1.14 | 0.81–1.62 |
| 22 | K101 | 4 | 3,3′ | 4.8 | 10.29 | 7.19–14.73 |
| Compound | Name of oxime | n | Oxime position | IC50 values EeAChE (mM) | IC50 values hrAChE (mM) | 95% confidence intervals (mM) |
|---|---|---|---|---|---|---|
| 6 | K018 | 3 | 4,4′ | 51.9 | 24.37 | 13.84–42.92 |
| 23 | K318 | CC=CC | 4,4′ | 36.7 | 17.33 | 8.22–36.52 |
| 7 | K074 | 4 | 4,4′ | 4.3 | 3.44 | 1.92–6.15 |
| 24 | K075 | C-O-C | 4,4′ | 6.7 | 8.09 | 4.51–9.91 |
| Enzyme | Amino acid residues | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TcAChE | GLY 80 | MET 83 | TRP 84 | GLY 117 | GLY 118 | GLY 119 | GLU 199 | SER 200 | ALA 201 | GLN 225 | SER 226 | GLY 227 | CYS 231 | TRP 233 | PHE 288 |
| EeAChE | GLY 82 | MET 85 | TRP 86 | GLY 120 | GLY 121 | GLY 122 | GLU 202 | SER 203 | ALA 204 | GLN 228 | SER 229 | GLY 230 | THR 231 | TRP 236 | PHE 295 |
| TcAChE | PHE 290 | VAL 323 | ASN 324 | LYS 325 | ASP 326 | GLU 327 | GLY 328 | SER 329 | PHE 330 | PHE 331 | GLY 396 | ASN 399 | VAL 400 | TRP 432 | MET 436 |
| EeAChE | PHE 297 | VAL 330 | VAL 331 | LYS 332 | ASP 333 | GLU 334 | GLY 335 | SER 336 | TYR 337 | PHE 338 | GLY 403 | ASN 406 | VAL 407 | TRP 439 | MET 443 |
| TcAChE | GLY 437 | VAL 438 | ILE 439 | HIS 440 | GLY 441 | TYR 442 | GLU 443 | ILE 444 | - | ||||||
| EeAChE | GLY 444 | VAL 445 | PRO 446 | HIS 447 | GLY 448 | TYR 449 | GLU 450 | ILE 451 | |||||||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sepsova, V.; Karasova, J.Z.; Korabecny, J.; Dolezal, R.; Zemek, F.; Bennion, B.J.; Kuca, K. Oximes: Inhibitors of Human Recombinant Acetylcholinesterase. A Structure-Activity Relationship (SAR) Study. Int. J. Mol. Sci. 2013, 14, 16882-16900. https://doi.org/10.3390/ijms140816882
Sepsova V, Karasova JZ, Korabecny J, Dolezal R, Zemek F, Bennion BJ, Kuca K. Oximes: Inhibitors of Human Recombinant Acetylcholinesterase. A Structure-Activity Relationship (SAR) Study. International Journal of Molecular Sciences. 2013; 14(8):16882-16900. https://doi.org/10.3390/ijms140816882
Chicago/Turabian StyleSepsova, Vendula, Jana Zdarova Karasova, Jan Korabecny, Rafael Dolezal, Filip Zemek, Brian J. Bennion, and Kamil Kuca. 2013. "Oximes: Inhibitors of Human Recombinant Acetylcholinesterase. A Structure-Activity Relationship (SAR) Study" International Journal of Molecular Sciences 14, no. 8: 16882-16900. https://doi.org/10.3390/ijms140816882