Posttranslational Modification of the Androgen Receptor in Prostate Cancer

Abstract
:1. Introduction
2. Phosphorylation of the AR
2.1. Serine and Threonine Phosphorylation of the AR
2.2. Tyrosine Phosphorylation of the AR
2.3. Phosphatases That Affect the AR
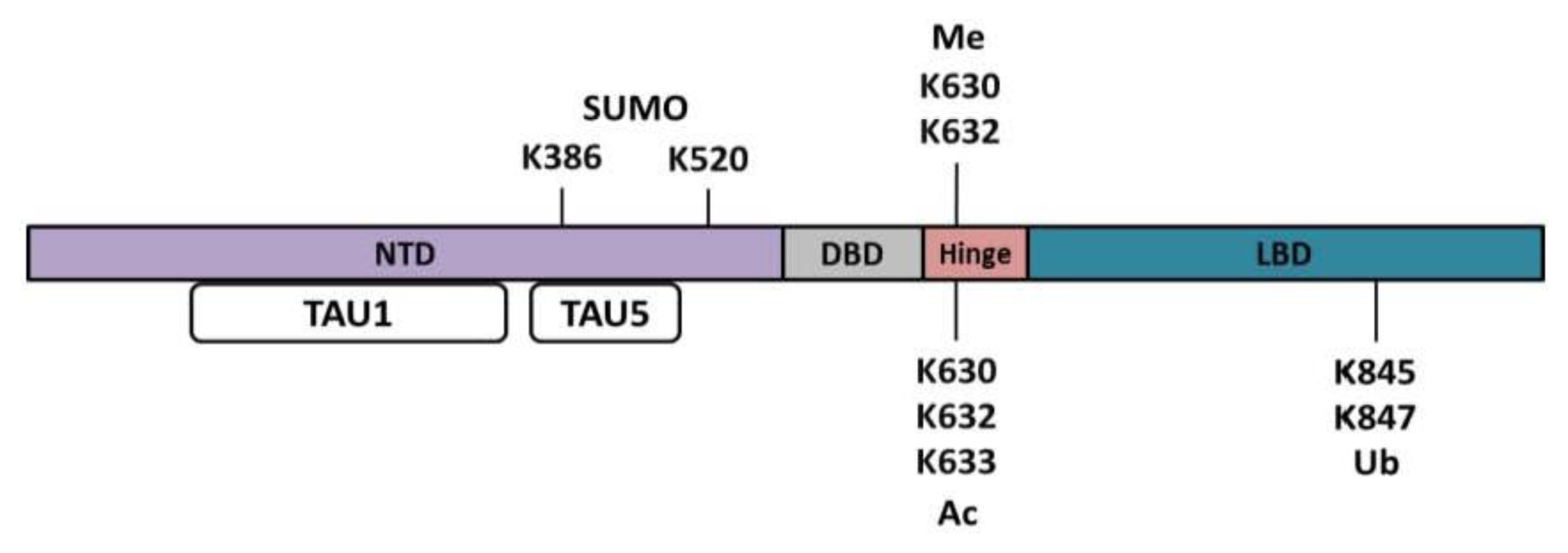
3. Acetylation of the AR
4. Methylation of the AR
5. Ubiquitination of the AR
6. SUMOylation of the AR
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci 2000, 21, 381–388. [Google Scholar]
- Dehm, S.M.; Tindall, D.J. Molecular regulation of androgen action in prostate cancer. J. Cell. Biochem 2006, 99, 333–344. [Google Scholar]
- Zhang, L.; Johnson, M.; Le, K.H.; Sato, M.; Ilagan, R.; Iyer, M.; Gambhir, S.S.; Wu, L.; Carey, M. Interrogating androgen receptor function in recurrent prostate cancer. Cancer Res 2003, 63, 4552–4560. [Google Scholar]
- Hamy, F.; Brondani, V.; Spoerri, R.; Rigo, S.; Stamm, C.; Klimkait, T. Specific block of androgen receptor activity by antisense oligonucleotides. Prostate Cancer Prostatic Dis 2003, 6, 27–33. [Google Scholar]
- Yuan, X.; Li, T.; Wang, H.; Zhang, T.; Barua, M.; Borgesi, R.A.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Androgen receptor remains critical for cell-cycle progression in androgen-independent cwr22 prostate cancer cells. Am. J. Pathol 2006, 169, 682–696. [Google Scholar]
- Garcia, J.A.; Rini, B.I. Castration-resistant prostate cancer: Many treatments, many options, many challenges ahead. Cancer 2012, 118, 2583–2593. [Google Scholar]
- Lin, Y.; Kokontis, J.; Tang, F.; Godfrey, B.; Liao, S.; Lin, A.; Chen, Y.; Xiang, J. Androgen and its receptor promote bax-mediated apoptosis. Mol. Cell. Biol 2006, 26, 1908–1916. [Google Scholar]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol 2002, 20, 3001–3015. [Google Scholar]
- Ding, D.; Xu, L.; Menon, M.; Reddy, G.P.; Barrack, E.R. Effect of ggc (glycine) repeat length polymorphism in the human androgen receptor on androgen action. Prostate 2005, 62, 133–139. [Google Scholar]
- Ding, D.; Xu, L.; Menon, M.; Reddy, G.P.; Barrack, E.R. Effect of a short cag (glutamine) repeat on human androgen receptor function. Prostate 2004, 58, 23–32. [Google Scholar]
- Ferro, P.; Catalano, M.G.; Dell’Eva, R.; Fortunati, N.; Pfeffer, U. The androgen receptor cag repeat: A modifier of carcinogenesis? Mol. Cell. Endocrinol 2002, 193, 109–120. [Google Scholar]
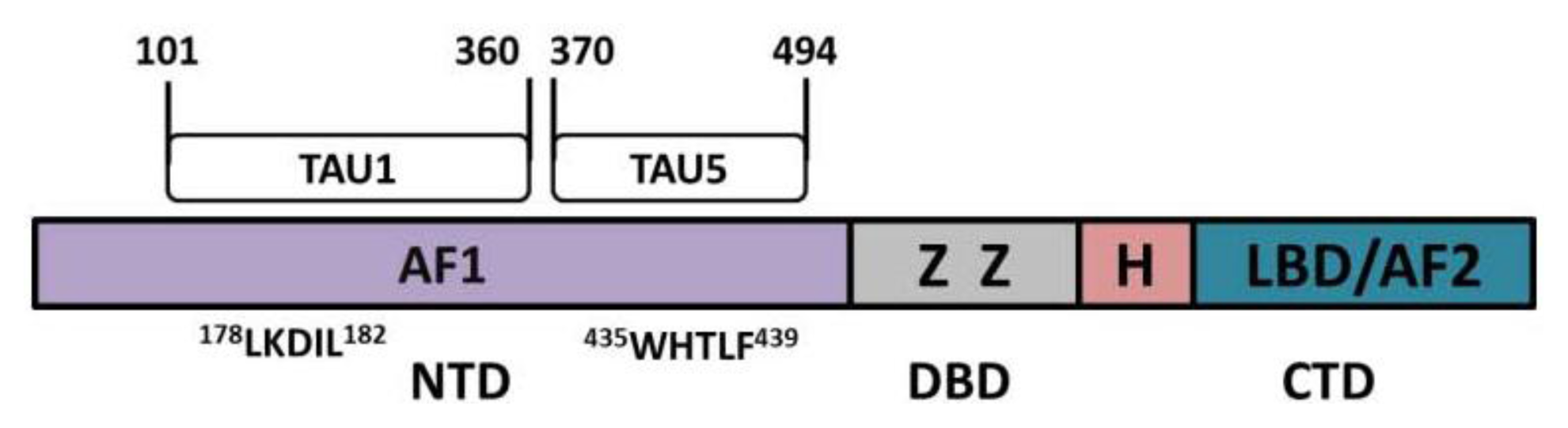
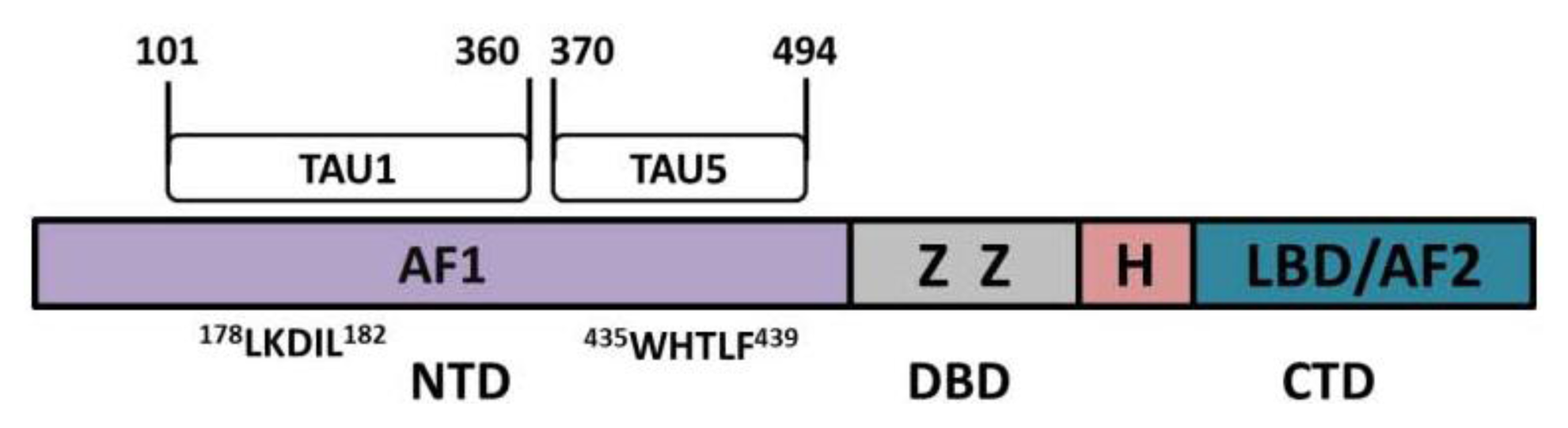
- Jenster, G.; van der Korput, H.A.; Trapman, J.; Brinkmann, A.O. Identification of two transcription activation units in the n-terminal domain of the human androgen receptor. J. Biol. Chem 1995, 270, 7341–7346. [Google Scholar]
- Wong, C.I.; Zhou, Z.X.; Sar, M.; Wilson, E.M. Steroid requirement for androgen receptor dimerization and DNA binding. Modulation by intramolecular interactions between the nh2-terminal and steroid-binding domains. J. Biol. Chem 1993, 268, 19004–19012. [Google Scholar]
- Kasper, S.; Rennie, P.S.; Bruchovsky, N.; Sheppard, P.C.; Cheng, H.; Lin, L.; Shiu, R.P.; Snoek, R.; Matusik, R.J. Cooperative binding of androgen receptors to two dna sequences is required for androgen induction of the probasin gene. J. Biol. Chem 1994, 269, 31763–31769. [Google Scholar]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar]
- He, B.; Gampe, R.T., Jr; Kole, A.J.; Hnat, A.T.; Stanley, T.B.; An, G.; Stewart, E.L.; Kalman, R.I.; Minges, J.T.; Wilson, E.M. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 2004, 16, 425–438. [Google Scholar]
- Hur, E.; Pfaff, S.J.; Payne, E.S.; Gron, H.; Buehrer, B.M.; Fletterick, R.J. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol 2004, 2, e274. [Google Scholar]
- Estebanez-Perpina, E.; Moore, J.M.; Mar, E.; Delgado-Rodrigues, E.; Nguyen, P.; Baxter, J.D.; Buehrer, B.M.; Webb, P.; Fletterick, R.J.; Guy, R.K. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J. Biol. Chem 2005, 280, 8060–8068. [Google Scholar]
- Dehm, S.M.; Regan, K.M.; Schmidt, L.J.; Tindall, D.J. Selective role of an nh2-terminal wxxlf motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res 2007, 67, 10067–10077. [Google Scholar]
- Dehm, S.M.; Tindall, D.J. Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J. Biol. Chem 2006, 281, 27882–27893. [Google Scholar]
- Debes, J.D.; Comuzzi, B.; Schmidt, L.J.; Dehm, S.M.; Culig, Z.; Tindall, D.J. P300 regulates androgen receptor-independent expression of prostate-specific antigen in prostate cancer cells treated chronically with interleukin-6. Cancer Res 2005, 65, 5965–5973. [Google Scholar]
- Chamberlain, N.L.; Whitacre, D.C.; Miesfeld, R.L. Delineation of two distinct type 1 activation functions in the androgen receptor amino-terminal domain. J. Biol. Chem 1996, 271, 26772–26778. [Google Scholar]
- Veldscholte, J.; Berrevoets, C.A.; Zegers, N.D.; van der Kwast, T.H.; Grootegoed, J.A.; Mulder, E. Hormone-induced dissociation of the androgen receptor-heat-shock protein complex: Use of a new monoclonal antibody to distinguish transformed from nontransformed receptors. Biochemistry 1992, 31, 7422–7430. [Google Scholar]
- Zoubeidi, A.; Zardan, A.; Beraldi, E.; Fazli, L.; Sowery, R.; Rennie, P.; Nelson, C.; Gleave, M. Cooperative interactions between androgen receptor (ar) and heat-shock protein 27 facilitate ar transcriptional activity. Cancer Res 2007, 67, 10455–10465. [Google Scholar]
- Pratt, W.B. Interaction of hsp90 with steroid receptors: Organizing some diverse observations and presenting the newest concepts. Mol. Cell. Endocrinol 1990, 74, C69–C76. [Google Scholar]
- Smith, D.F.; Sullivan, W.P.; Marion, T.N.; Zaitsu, K.; Madden, B.; McCormick, D.J.; Toft, D.O. Identification of a 60-kilodalton stress-related protein, p60, which interacts with hsp90 and hsp70. Mol. Cell. Biol 1993, 13, 869–876. [Google Scholar]
- Smith, D.F.; Toft, D.O. Steroid receptors and their associated proteins. Mol. Endocrinol 1993, 7, 4–11. [Google Scholar]
- Kuil, C.W.; Berrevoets, C.A.; Mulder, E. Ligand-induced conformational alterations of the androgen receptor analyzed by limited trypsinization. Studies on the mechanism of antiandrogen action. J. Biol. Chem 1995, 270, 27569–27576. [Google Scholar]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev 2004, 25, 276–308. [Google Scholar]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin 1972, 22, 232–240. [Google Scholar]
- Pienta, K.J.; Bradley, D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res 2006, 12, 1665–1671. [Google Scholar]
- Shamash, J.; Dancey, G.; Barlow, C.; Wilson, P.; Ansell, W.; Oliver, R.T. Chlorambucil and lomustine (cl56) in absolute hormone refractory prostate cancer: Re-induction of endocrine sensitivity an unexpected finding. Br. J. Cancer 2005, 92, 36–40. [Google Scholar]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar]
- Buchanan, G.; Irvine, R.A.; Coetzee, G.A.; Tilley, W.D. Contribution of the androgen receptor to prostate cancer predisposition and progression. Cancer Metastasis Rev 2001, 20, 207–223. [Google Scholar]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (ar) coregulators: A diversity of functions converging on and regulating the ar transcriptional complex. Endocr. Rev 2007, 28, 778–808. [Google Scholar]
- Debes, J.D.; Tindall, D.J. Mechanisms of androgen-refractory prostate cancer. N. Engl. J. Med 2004, 351, 1488–1490. [Google Scholar]
- Askew, E.B.; Gampe, R.T., Jr; Stanley, T.B.; Faggart, J.L.; Wilson, E.M. Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J. Biol. Chem. 2007, 282, 25801–25816. [Google Scholar]
- Bergerat, J.P.; Ceraline, J. Pleiotropic functional properties of androgen receptor mutants in prostate cancer. Hum. Mutat 2009, 30, 145–157. [Google Scholar]
- Zhu, M.L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 2008, 15, 841–849. [Google Scholar]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res 2008, 68, 6407–6415. [Google Scholar]
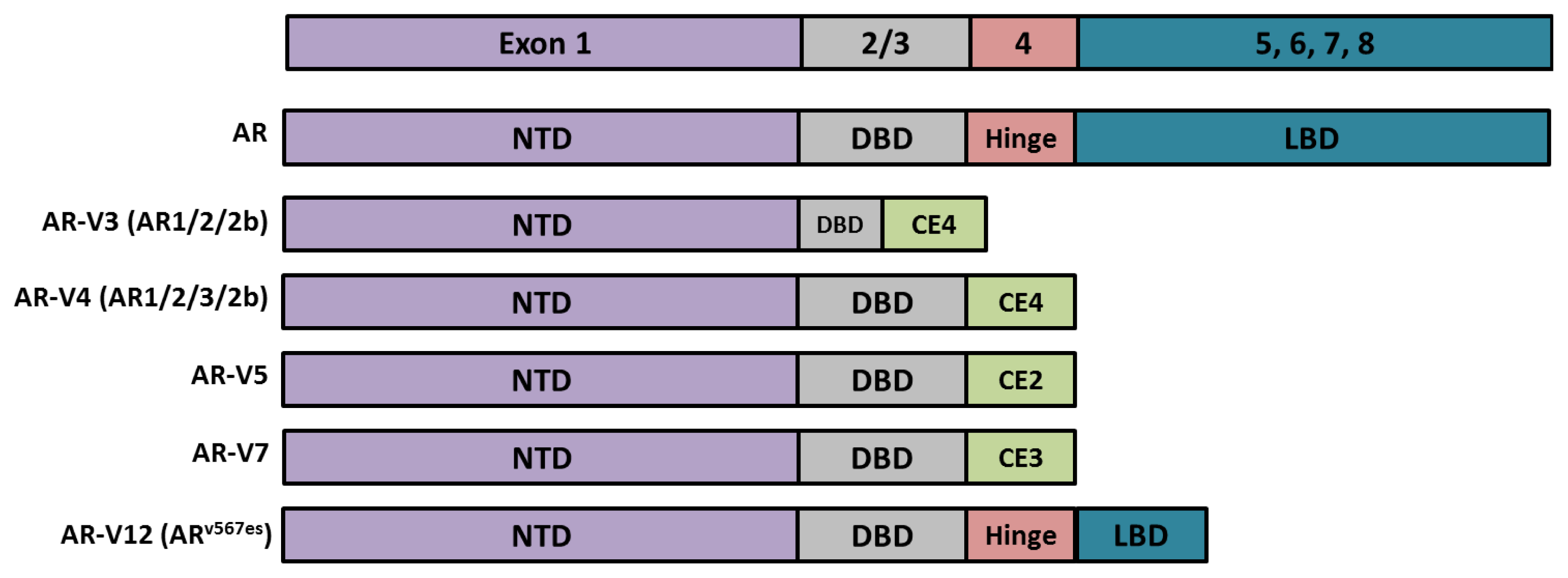
- Haile, S.; Sadar, M.D. Androgen receptor and its splice variants in prostate cancer. Cell Mol. Life Sci 2011, 68, 3971–3981. [Google Scholar]
- Ceraline, J.; Cruchant, M.D.; Erdmann, E.; Erbs, P.; Kurtz, J.E.; Duclos, B.; Jacqmin, D.; Chopin, D.; Bergerat, J.P. Constitutive activation of the androgen receptor by a point mutation in the hinge region: A new mechanism for androgen-independent growth in prostate cancer. Int. J. Cancer 2004, 108, 152–157. [Google Scholar]
- Libertini, S.J.; Tepper, C.G.; Rodriguez, V.; Asmuth, D.M.; Kung, H.J.; Mudryj, M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res 2007, 67, 9001–9005. [Google Scholar]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 2011, 6, e19059. [Google Scholar]
- Harada, N.; Inoue, K.; Yamaji, R.; Nakano, Y.; Inui, H. Androgen deprivation causes truncation of the C-terminal region of androgen receptor in human prostate cancer lncap cells. Cancer Sci 2012, 103, 1022–1027. [Google Scholar]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008, 68, 5469–5477. [Google Scholar]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest 2010, 120, 2715–2730. [Google Scholar]
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One 2011, 6, e27970. [Google Scholar]
- Carrión-Salip, D.; Panosa, C.; Menendez, J.A.; Puig, T.; Oliveras, G.; Pandiella, A.; De Llorens, R.; Massaguer, A. Androgen-independent prostate cancer cells circumvent egfr inhibition by overexpression of alternative her receptors and ligands. Int. J. Oncol 2012, 41, 1128–1138. [Google Scholar]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar]
- Jenster, G.; van der Korput, H.A.; van Vroonhoven, C.; van der Kwast, T.H.; Trapman, J.; Brinkmann, A.O. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol 1991, 5, 1396–1404. [Google Scholar]
- Slagsvold, T.; Kraus, I.; Bentzen, T.; Palvimo, J.; Saatcioglu, F. Mutational analysis of the androgen receptor af-2 (activation function 2) core domain reveals functional and mechanistic differences of conserved residues compared with other nuclear receptors. Mol. Endocrinol 2000, 14, 1603–1617. [Google Scholar]
- MacLean, H.E.; Warne, G.L.; Zajac, J.D. Localization of functional domains in the androgen receptor. J. Steroid. Biochem. Mol. Biol 1997, 62, 233–242. [Google Scholar]
- Gioeli, D.; Paschal, B.M. Post-translational modification of the androgen receptor. Mol. Cell. Endocrinol 2012, 352, 70–78. [Google Scholar]
- Coffey, K.; Robson, C.N. Regulation of the androgen receptor by post-translational modifications. J. Endocrinol 2012, 215, 221–237. [Google Scholar]
- Lavery, D.N.; Bevan, C.L. Androgen receptor signalling in prostate cancer: The functional consequences of acetylation. J. Biomed. Biotechnol 2011, 2011, 862125. [Google Scholar]
- Clinckemalie, L.; Vanderschueren, D.; Boonen, S.; Claessens, F. The hinge region in androgen receptor control. Mol. Cell. Endocrinol 2012, 358, 1–8. [Google Scholar]
- Hsu, F.N.; Chen, M.C.; Chiang, M.C.; Lin, E.; Lee, Y.T.; Huang, P.H.; Lee, G.S.; Lin, H. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J. Biol. Chem 2011, 286, 33141–33149. [Google Scholar]
- Yang, C.S.; Vitto, M.J.; Busby, S.A.; Garcia, B.A.; Kesler, C.T.; Gioeli, D.; Shabanowitz, J.; Hunt, D.F.; Rundell, K.; Brautigan, D.L.; et al. Simian virus 40 small t antigen mediates conformation-dependent transfer of protein phosphatase 2a onto the androgen receptor. Mol. Cell. Biol 2005, 25, 1298–1308. [Google Scholar]
- Lin, H.K.; Yeh, S.; Kang, H.Y.; Chang, C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7200–7205. [Google Scholar]
- Palazzolo, I.; Burnett, B.G.; Young, J.E.; Brenne, P.L.; La Spada, A.R.; Fischbeck, K.H.; Howell, B.W.; Pennuto, M. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet 2007, 16, 1593–1603. [Google Scholar]
- Lin, H.K.; Hu, Y.C.; Yang, L.; Altuwaijri, S.; Chen, Y.T.; Kang, H.Y.; Chang, C. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/akt pathway in prostate cancer lncap cells with different passage numbers. J. Biol. Chem 2003, 278, 50902–50907. [Google Scholar]
- Chen, S.; Gulla, S.; Cai, C.; Balk, S.P. Androgen receptor serine 81 phosphorylation mediates chromatin binding and transcriptional activation. J. Biol. Chem 2012, 287, 8571–8583. [Google Scholar]
- Mahajan, K.; Challa, S.; Coppola, D.; Lawrence, H.; Luo, Y.; Gevariya, H.; Zhu, W.; Chen, Y.A.; Lawrence, N.J.; Mahajan, N.P. Effect of ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate 2010, 70, 1274–1285. [Google Scholar]
- Mahajan, N.P.; Liu, Y.; Majumder, S.; Warren, M.R.; Parker, C.E.; Mohler, J.L.; Earp, H.S.; Whang, Y.E. Activated cdc42-associated kinase ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl. Acad. Sci. USA 2007, 104, 8438–8443. [Google Scholar]
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated tyrosine kinase ack1 promotes prostate tumorigenesis: Role of ack1 in polyubiquitination of tumor suppressor wwox. Cancer Res 2005, 65, 10514–10523. [Google Scholar]
- Liu, Y.; Karaca, M.; Zhang, Z.; Gioeli, D.; Earp, H.S.; Whang, Y.E. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by ack1 and src kinases. Oncogene 2010, 29, 3208–3216. [Google Scholar]
- Shu, S.K.; Liu, Q.; Coppola, D.; Cheng, J.Q. Phosphorylation and activation of androgen receptor by aurora-a. J. Biol. Chem 2010, 285, 33045–33053. [Google Scholar]
- Chymkowitch, P.; Le May, N.; Charneau, P.; Compe, E.; Egly, J.M. The phosphorylation of the androgen receptor by tfiih directs the ubiquitin/proteasome process. EMBO J 2011, 30, 468–479. [Google Scholar]
- Ponguta, L.A.; Gregory, C.W.; French, F.S.; Wilson, E.M. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J. Biol. Chem 2008, 283, 20989–21001. [Google Scholar]
- Guo, Z.; Dai, B.; Jiang, T.; Xu, K.; Xie, Y.; Kim, O.; Nesheiwat, I.; Kong, X.; Melamed, J.; Handratta, V.D.; et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006, 10, 309–319. [Google Scholar]
- Gioeli, D.; Black, B.E.; Gordon, V.; Spencer, A.; Kesler, C.T.; Eblen, S.T.; Paschal, B.M.; Weber, M.J. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol. Endocrinol 2006, 20, 503–515. [Google Scholar]
- Zhou, Z.X.; Kemppainen, J.A.; Wilson, E.M. Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol. Endocrinol 1995, 9, 605–615. [Google Scholar]
- Gordon, V.; Bhadel, S.; Wunderlich, W.; Zhang, J.; Ficarro, S.B.; Mollah, S.A.; Shabanowitz, J.; Hunt, D.F.; Xenarios, I.; Hahn, W.C.; et al. Cdk9 regulates ar promoter selectivity and cell growth through serine 81 phosphorylation. Mol. Endocrinol 2010, 24, 2267–2280. [Google Scholar]
- Linn, D.E.; Yang, X.; Xie, Y.; Alfano, A.; Deshmukh, D.; Wang, X.; Shimelis, H.; Chen, H.; Li, W.; Xu, K.; et al. Differential regulation of androgen receptor by pim-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin e3 ligases. J. Biol. Chem 2012, 287, 22959–22968. [Google Scholar]
- Gioeli, D.; Ficarro, S.B.; Kwiek, J.J.; Aaronson, D.; Hancock, M.; Catling, A.D.; White, F.M.; Christian, R.E.; Settlage, R.E.; Shabanowitz, J.; et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J. Biol. Chem 2002, 277, 29304–29314. [Google Scholar]
- Yang, C.S.; Xin, H.W.; Kelley, J.B.; Spencer, A.; Brautigan, D.L.; Paschal, B.M. Ligand binding to the androgen receptor induces conformational changes that regulate phosphatase interactions. Mol. Cell. Biol 2007, 27, 3390–3404. [Google Scholar]
- Langley, E.; Zhou, Z.X.; Wilson, E.M. Evidence for an anti-parallel orientation of the ligand-activated human androgen receptor dimer. J. Biol. Chem 1995, 270, 29983–29990. [Google Scholar]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006, 66, 2815–2825. [Google Scholar]
- Chen, S.; Xu, Y.; Yuan, X.; Bubley, G.J.; Balk, S.P. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc. Natl. Acad. Sci. USA 2006, 103, 15969–15974. [Google Scholar]
- Ngan, E.S.; Hashimoto, Y.; Ma, Z.Q.; Tsai, M.J.; Tsai, S.Y. Overexpression of cdc25b, an androgen receptor coactivator, in prostate cancer. Oncogene 2003, 22, 734–739. [Google Scholar]
- Maddison, L.A.; Huss, W.J.; Barrios, R.M.; Greenberg, N.M. Differential expression of cell cycle regulatory molecules and evidence for a “Cyclin switch” During progression of prostate cancer. Prostate 2004, 58, 335–344. [Google Scholar]
- Ozen, M.; Ittmann, M. Increased expression and activity of cdc25c phosphatase and an alternatively spliced variant in prostate cancer. Clin. Cancer Res 2005, 11, 4701–4706. [Google Scholar]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar]
- Tarricone, C.; Dhavan, R.; Peng, J.; Areces, L.B.; Tsai, L.H.; Musacchio, A. Structure and regulation of the cdk5-p25(nck5a) complex. Mol. Cell 2001, 8, 657–669. [Google Scholar]
- Wong, H.Y.; Burghoorn, J.A.; van Leeuwen, M.; de Ruiter, P.E.; Schippers, E.; Blok, L.J.; Li, K.W.; Dekker, H.L.; de Jong, L.; Trapman, J.; et al. Phosphorylation of androgen receptor isoforms. Biochem. J 2004, 383, 267–276. [Google Scholar]
- La Montagna, R.; Caligiuri, I.; Maranta, P.; Lucchetti, C.; Esposito, L.; Paggi, M.G.; Toffoli, G.; Rizzolio, F.; Giordano, A. Androgen receptor serine 81 mediates pin1 interaction and activity. Cell Cycle 2012, 11, 3415–3420. [Google Scholar]
- Ha, S.; Iqbal, N.J.; Mita, P.; Ruoff, R.; Gerald, W.L.; Lepor, H.; Taneja, S.S.; Lee, P.; Melamed, J.; Garabedian, M.J.; et al. Phosphorylation of the androgen receptor by pim1 in hormone refractory prostate cancer. Oncogene 2012. [Google Scholar] [CrossRef]
- Deep, G.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin b causes androgen receptor degradation in human prostate carcinoma cells via pi3k-akt-mdm2-mediated pathway. Oncogene 2008, 27, 3986–3998. [Google Scholar]
- Chen, S.; Kesler, C.T.; Paschal, B.M.; Balk, S.P. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1. J. Biol. Chem 2009, 284, 25576–25584. [Google Scholar]
- Sasayama, T.; Marumoto, T.; Kunitoku, N.; Zhang, D.; Tamaki, N.; Kohmura, E.; Saya, H.; Hirota, T. Over-expression of aurora-a targets cytoplasmic polyadenylation element binding protein and promotes mrna polyadenylation of cdk1 and cyclin b1. Genes Cells 2005, 10, 627–638. [Google Scholar]
- Zong, H.; Chi, Y.; Wang, Y.; Yang, Y.; Zhang, L.; Chen, H.; Jiang, J.; Li, Z.; Hong, Y.; Wang, H.; et al. Cyclin d3/cdk11p58 complex is involved in the repression of androgen receptor. Mol. Cell. Biol 2007, 27, 7125–7142. [Google Scholar]
- Zhou, Z.X.; Sar, M.; Simental, J.A.; Lane, M.V.; Wilson, E.M. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by nh2-terminal and carboxyl-terminal sequences. J. Biol. Chem 1994, 269, 13115–13123. [Google Scholar]
- Jenster, G.; van der Korput, J.A.; Trapman, J.; Brinkmann, A.O. Functional domains of the human androgen receptor. J. Steroid. Biochem. Mol. Biol 1992, 41, 671–675. [Google Scholar]
- Picard, D.; Yamamoto, K.R. Two signals mediate hormone-dependent nuclear localization of the glucocorticoid receptor. EMBO J 1987, 6, 3333–3340. [Google Scholar]
- Poukka, H.; Aarnisalo, P.; Santti, H.; Janne, O.A.; Palvimo, J.J. Coregulator small nuclear ring finger protein (snurf) enhances sp1- and steroid receptor-mediated transcription by different mechanisms. J. Biol. Chem 2000, 275, 571–579. [Google Scholar]
- Kraus, S.; Gioeli, D.; Vomastek, T.; Gordon, V.; Weber, M.J. Receptor for activated c kinase 1 (rack1) and src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res 2006, 66, 11047–11054. [Google Scholar]
- Lee, L.F.; Louie, M.C.; Desai, S.J.; Yang, J.; Chen, H.W.; Evans, C.P.; Kung, H.J. Interleukin-8 confers androgen-independent growth and migration of lncap: Differential effects of tyrosine kinases src and fak. Oncogene 2004, 23, 2197–2205. [Google Scholar]
- Lee, L.F.; Guan, J.; Qiu, Y.; Kung, H.J. Neuropeptide-induced androgen independence in prostate cancer cells: Roles of nonreceptor tyrosine kinases etk/bmx, src, and focal adhesion kinase. Mol. Cell. Biol 2001, 21, 8385–8397. [Google Scholar]
- Drake, J.M.; Graham, N.A.; Stoyanova, T.; Sedghi, A.; Goldstein, A.S.; Cai, H.; Smith, D.A.; Zhang, H.; Komisopoulou, E.; Huang, J.; et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, 1643–1648. [Google Scholar]
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the her-2/neu tyrosine kinase. Nat. Med 1999, 5, 280–285. [Google Scholar]
- Craft, N.; Chhor, C.; Tran, C.; Belldegrun, A.; DeKernion, J.; Witte, O.N.; Said, J.; Reiter, R.E.; Sawyers, C.L. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res 1999, 59, 5030–5036. [Google Scholar]
- Bedolla, R.; Prihoda, T.J.; Kreisberg, J.I.; Malik, S.N.; Krishnegowda, N.K.; Troyer, D.A.; Ghosh, P.M. Determining risk of biochemical recurrence in prostate cancer by immunohistochemical detection of pten expression and akt activation. Clin. Cancer Res 2007, 13, 3860–3867. [Google Scholar]
- Graff, J.R.; Konicek, B.W.; McNulty, A.M.; Wang, Z.; Houck, K.; Allen, S.; Paul, J.D.; Hbaiu, A.; Goode, R.G.; Sandusky, G.E.; et al. Increased akt activity contributes to prostate cancer progression by dramatically accelerating prostate tumor growth and diminishing p27kip1 expression. J. Biol. Chem 2000, 275, 24500–24505. [Google Scholar]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. P300 and p300/camp-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem 2000, 275, 20853–20860. [Google Scholar]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol. Cell. Biol 2003, 23, 8563–8575. [Google Scholar]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010, 70, 540–554. [Google Scholar]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem 2002, 277, 25904–25913. [Google Scholar]
- Dai, Y.; Ngo, D.; Forman, L.W.; Qin, D.C.; Jacob, J.; Faller, D.V. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol. Endocrinol 2007, 21, 1807–1821. [Google Scholar]
- Montie, H.L.; Pestell, R.G.; Merry, D.E. Sirt1 modulates aggregation and toxicity through deacetylation of the androgen receptor in cell models of sbma. J. Neurosci 2011, 31, 17425–17436. [Google Scholar]
- Fu, M.; Liu, M.; Sauve, A.A.; Jiao, X.; Zhang, X.; Wu, X.; Powell, M.J.; Yang, T.; Gu, W.; Avantaggiati, M.L.; et al. Hormonal control of androgen receptor function through sirt1. Mol. Cell. Biol 2006, 26, 8122–8135. [Google Scholar]
- Kauffman, E.C.; Robinson, B.D.; Downes, M.J.; Powell, L.G.; Lee, M.M.; Scherr, D.S.; Gudas, L.J.; Mongan, N.P. Role of androgen receptor and associated lysine-demethylase coregulators, lsd1 and jmjd2a, in localized and advanced human bladder cancer. Mol. Carcinog 2011, 50, 931–944. [Google Scholar]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. Lsd1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, kdm4b, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res 2013, 41, 4433–4446. [Google Scholar]
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Kong, X.; et al. Regulation of androgen receptor transcriptional activity and specificity by rnf6-induced ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar]
- Rees, I.; Lee, S.; Kim, H.; Tsai, F.T. The e3 ubiquitin ligase chip binds the androgen receptor in a phosphorylation-dependent manner. Biochim. Biophys. Acta 2006, 1764, 1073–1079. [Google Scholar]
- Lin, H.K.; Wang, L.; Hu, Y.C.; Altuwaijri, S.; Chang, C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by akt require mdm2 e3 ligase. EMBO J 2002, 21, 4037–4048. [Google Scholar]
- He, B.; Bai, S.; Hnat, A.T.; Kalman, R.I.; Minges, J.T.; Patterson, C.; Wilson, E.M. An androgen receptor nh2-terminal conserved motif interacts with the cooh terminus of the hsp70-interacting protein (chip). J. Biol. Chem 2004, 279, 30643–30653. [Google Scholar]
- Saporita, A.J.; Zhang, Q.; Navai, N.; Dincer, Z.; Hahn, J.; Cai, X.; Wang, Z. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J. Biol. Chem 2003, 278, 41998–42005. [Google Scholar]
- Gong, Y.; Wang, D.; Dar, J.A.; Singh, P.; Graham, L.; Liu, W.; Ai, J.; Xin, Z.; Guo, Y.; Wang, Z. Nuclear export signal of androgen receptor (nesar) regulation of androgen receptor level in human prostate cell lines via ubiquitination and proteasome-dependent degradation. Endocrinology 2012, 153, 5716–5725. [Google Scholar]
- Felten, A.; Brinckmann, D.; Landsberg, G.; Scheidtmann, K.H. Zipper-interacting protein kinase is involved in regulation of ubiquitination of the androgen receptor, thereby contributing to dynamic transcription complex assembly. Oncogene 2012. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol 2007, 8, 947–956. [Google Scholar]
- Wang, Y.; Dasso, M. Sumoylation and desumoylation at a glance. J. Cell. Sci 2009, 122, 4249–4252. [Google Scholar]
- Nishida, T.; Yasuda, H. Pias1 and piasxalpha function as sumo-e3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem 2002, 277, 41311–41317. [Google Scholar]
- Kotaja, N.; Karvonen, U.; Janne, O.A.; Palvimo, J.J. Pias proteins modulate transcription factors by functioning as sumo-1 ligases. Mol. Cell. Biol 2002, 22, 5222–5234. [Google Scholar]
- Kaikkonen, S.; Jaaskelainen, T.; Karvonen, U.; Rytinki, M.M.; Makkonen, H.; Gioeli, D.; Paschal, B.M.; Palvimo, J.J. Sumo-specific protease 1 (senp1) reverses the hormone-augmented sumoylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol. Endocrinol 2009, 23, 292–307. [Google Scholar]
- Poukka, H.; Karvonen, U.; Janne, O.A.; Palvimo, J.J. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (sumo-1). Proc. Natl. Acad. Sci. USA 2000, 97, 14145–14150. [Google Scholar]
- Callewaert, L.; Verrijdt, G.; Haelens, A.; Claessens, F. Differential effect of small ubiquitin-like modifier (sumo)-ylation of the androgen receptor in the control of cooperativity on selective versus canonical response elements. Mol. Endocrinol 2004, 18, 1438–1449. [Google Scholar]
- Rytinki, M.; Kaikkonen, S.; Sutinen, P.; Paakinaho, V.; Rahkama, V.; Palvimo, J.J. Dynamic sumoylation is linked to the activity cycles of androgen receptor in the cell nucleus. Mol. Cell. Biol 2012, 32, 4195–4205. [Google Scholar]
- Krueckl, S.L.; Sikes, R.A.; Edlund, N.M.; Bell, R.H.; Hurtado-Coll, A.; Fazli, L.; Gleave, M.E.; Cox, M.E. Increased insulin-like growth factor i receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res 2004, 64, 8620–8629. [Google Scholar]
- Lorenzo, G.D.; Bianco, R.; Tortora, G.; Ciardiello, F. Involvement of growth factor receptors of the epidermal growth factor receptor family in prostate cancer development and progression to androgen independence. Clin. Prostate cancer 2003, 2, 50–57. [Google Scholar]
- Lee, D.K.; Duan, H.O.; Chang, C. Androgen receptor interacts with the positive elongation factor p-tefb and enhances the efficiency of transcriptional elongation. J. Biol. Chem 2001, 276, 9978–9984. [Google Scholar]



{kind=link}
{kind=link}
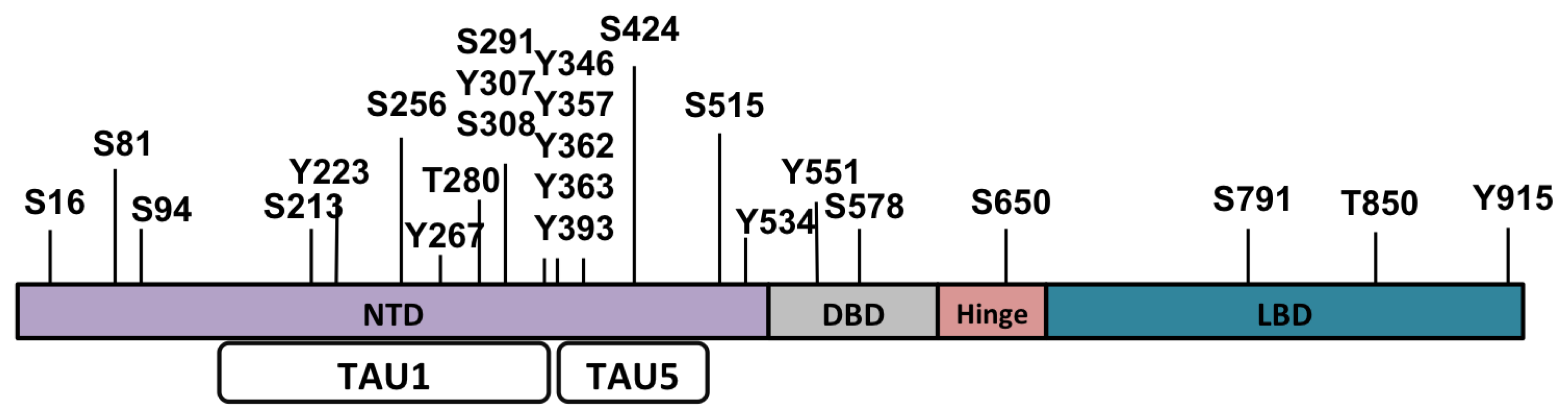{kind=link}
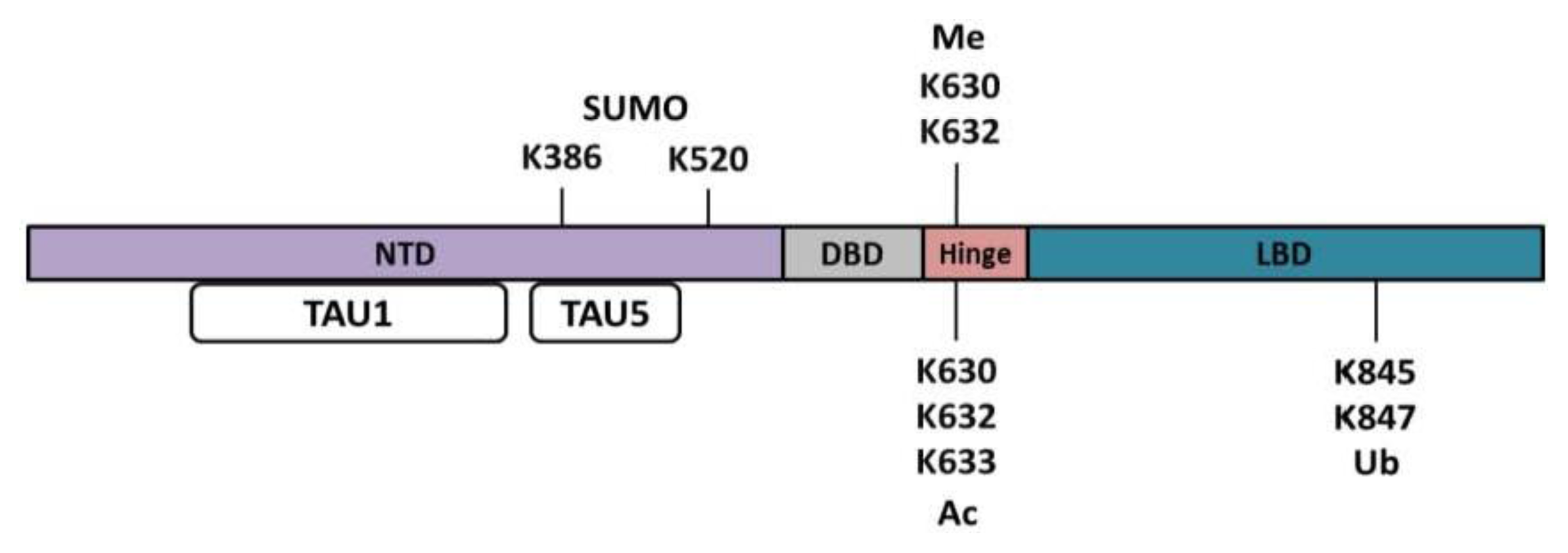{kind=link}
| Residue | Kinase/phosphatase | Function | References |
|---|---|---|---|
| S81 | CDK1, CDK5, CDK9 | Localization protein stability | [59] |
| PP2 | Cell growth transcription | [60] | |
| S94 | PP2 | Transcription | [60] |
| S213 | PI3K/AKT1 | Localization | [61–63] |
| PIM-1 | Stability | [59,64] | |
| Y267 | Ack | Cell growth transcription | [65–68] |
| Src | |||
| T280/S291 | AurA | Cell growth transcription | [69] |
| S308 | PP2 | Transcription | [60] |
| Y363 | Ack | Cell growth transcription | [66] |
| S424 | PP2 | Transcription stability | [60] |
| PP1 | |||
| S515 | MAPK | Transcription degradation | [70,71] |
| CDK7 | |||
| Y534 | Src | Localization cell cycle transcription | [68,72] |
| S578 | Localization transcription | [71] | |
| S650 | ERK1/JNK1/p38-alpha | Localization | [73] |
| Transcription | [74] | ||
| PP1 | Localization | [75] | |
| S791 | PI3K/AKT1 | Transcription apoptosis localization | [61–63] |
| T850 | PIM-1L | Stability | [76] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational Modification of the Androgen Receptor in Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 14833-14859. https://doi.org/10.3390/ijms140714833
Van der Steen T, Tindall DJ, Huang H. Posttranslational Modification of the Androgen Receptor in Prostate Cancer. International Journal of Molecular Sciences. 2013; 14(7):14833-14859. https://doi.org/10.3390/ijms140714833
Chicago/Turabian StyleVan der Steen, Travis, Donald J. Tindall, and Haojie Huang. 2013. "Posttranslational Modification of the Androgen Receptor in Prostate Cancer" International Journal of Molecular Sciences 14, no. 7: 14833-14859. https://doi.org/10.3390/ijms140714833