Construction of the Coding Sequence of the Transcription Variant 2 of the Human Renalase Gene and Its Expression in the Prokaryotic System

Abstract
:1. Introduction
2. Results
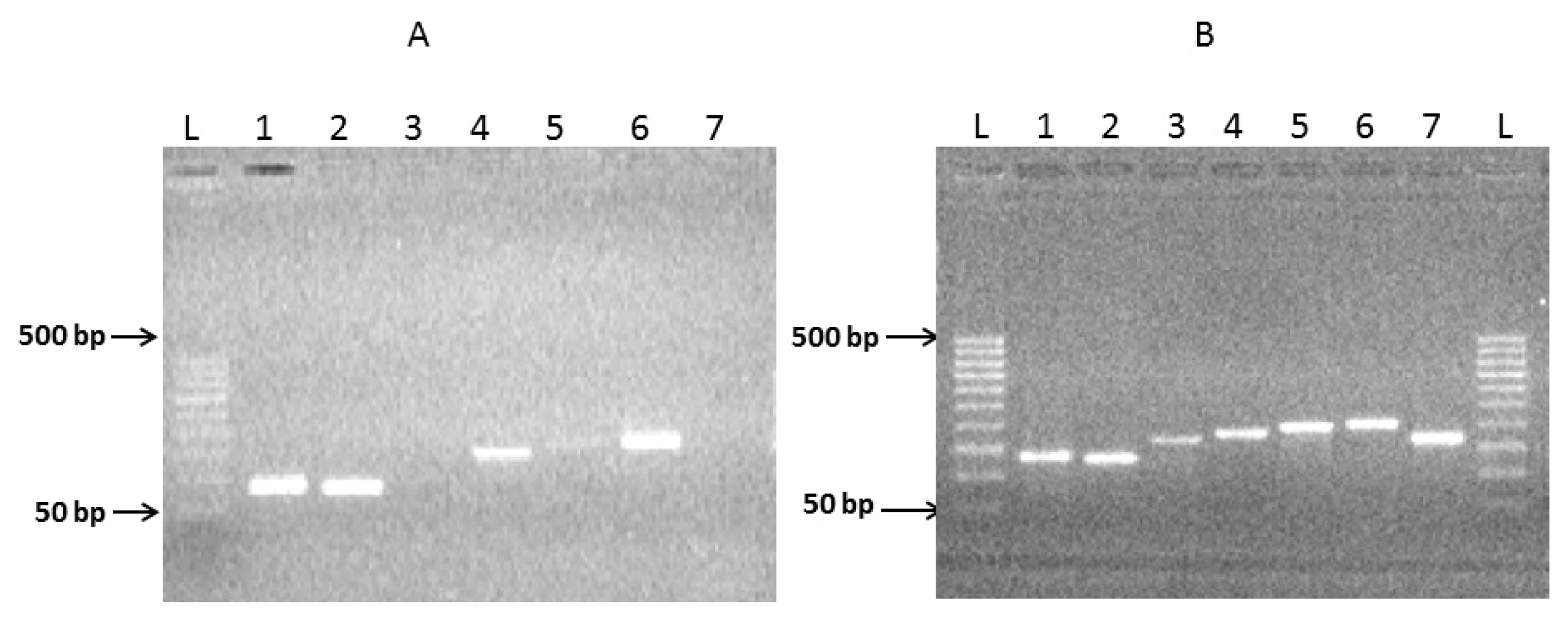
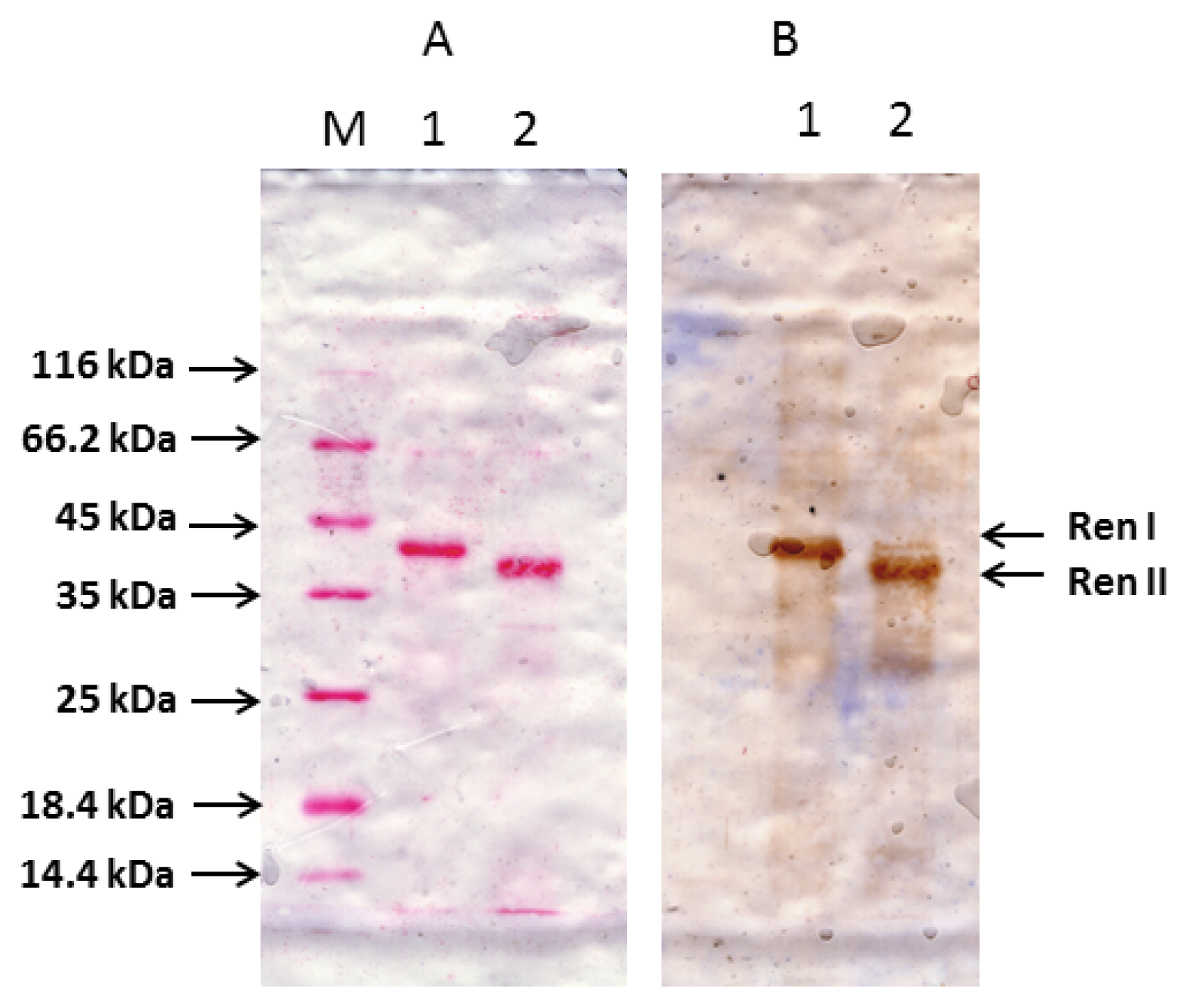
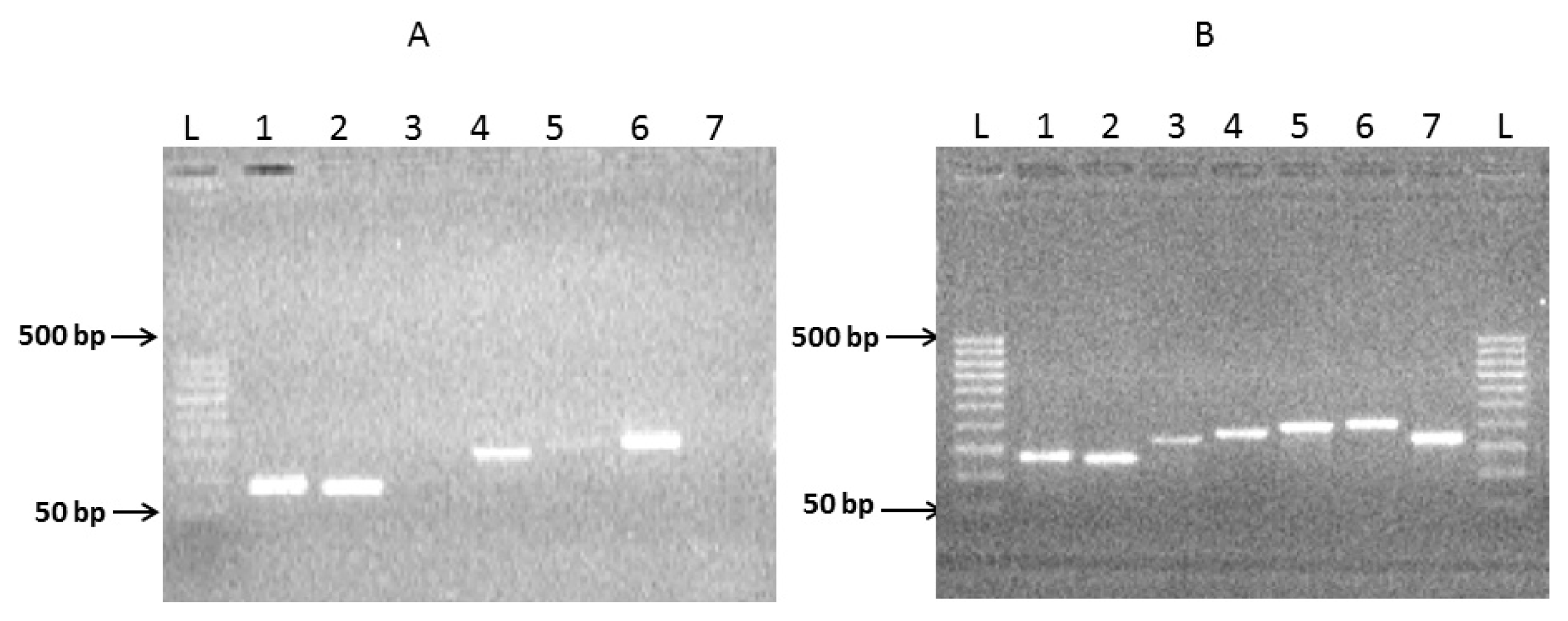
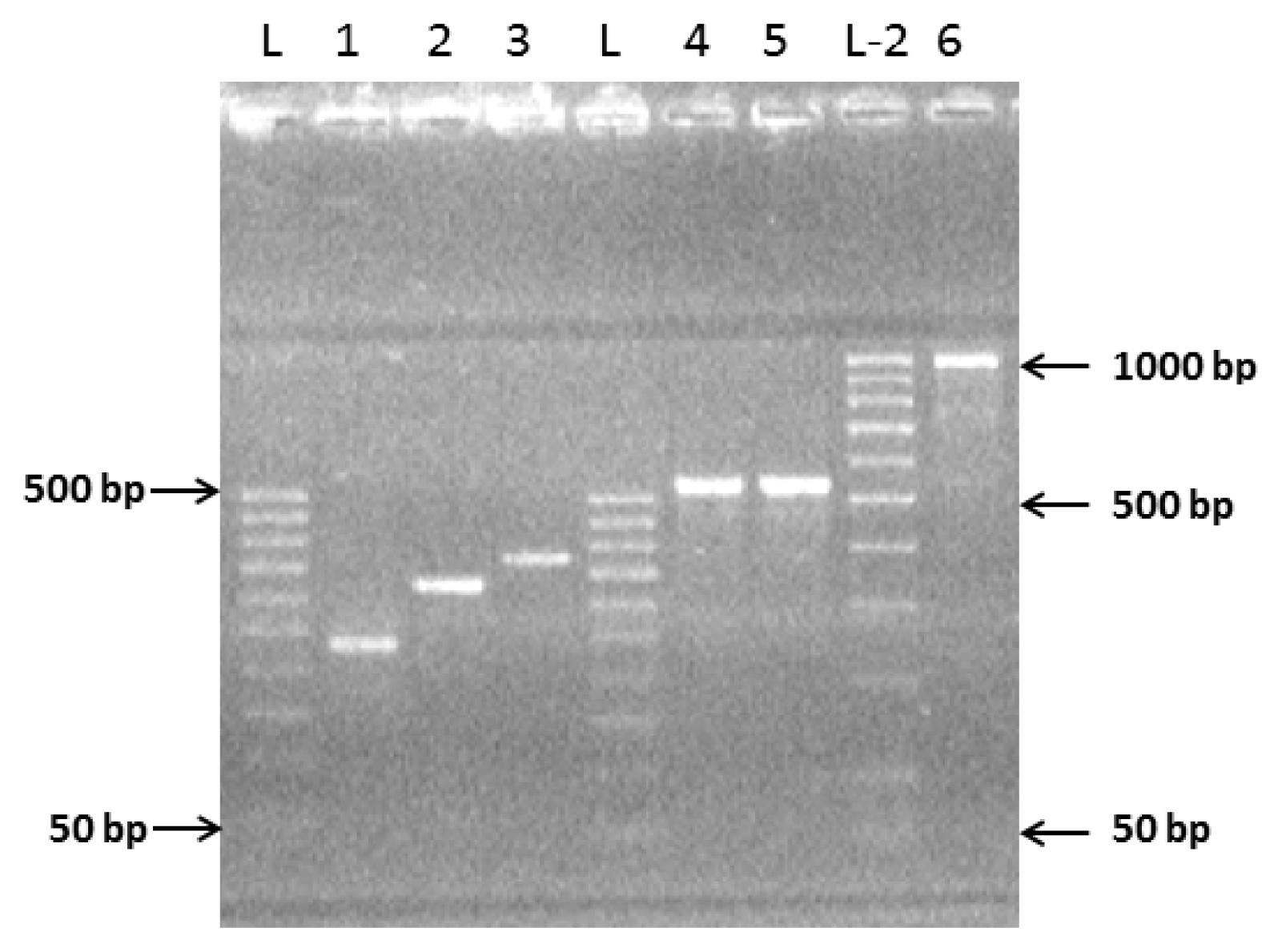
2.1. Preparation of Complete Coding Sequence of hRenalase1 and Its Expression in the Prokaryotic System
2.2. Preparation of the Complete Coding Sequence of hRenalase2 and Its Expression in the Prokaryotic System
3. Discussion
4. Experimental Section
4.1. Materials
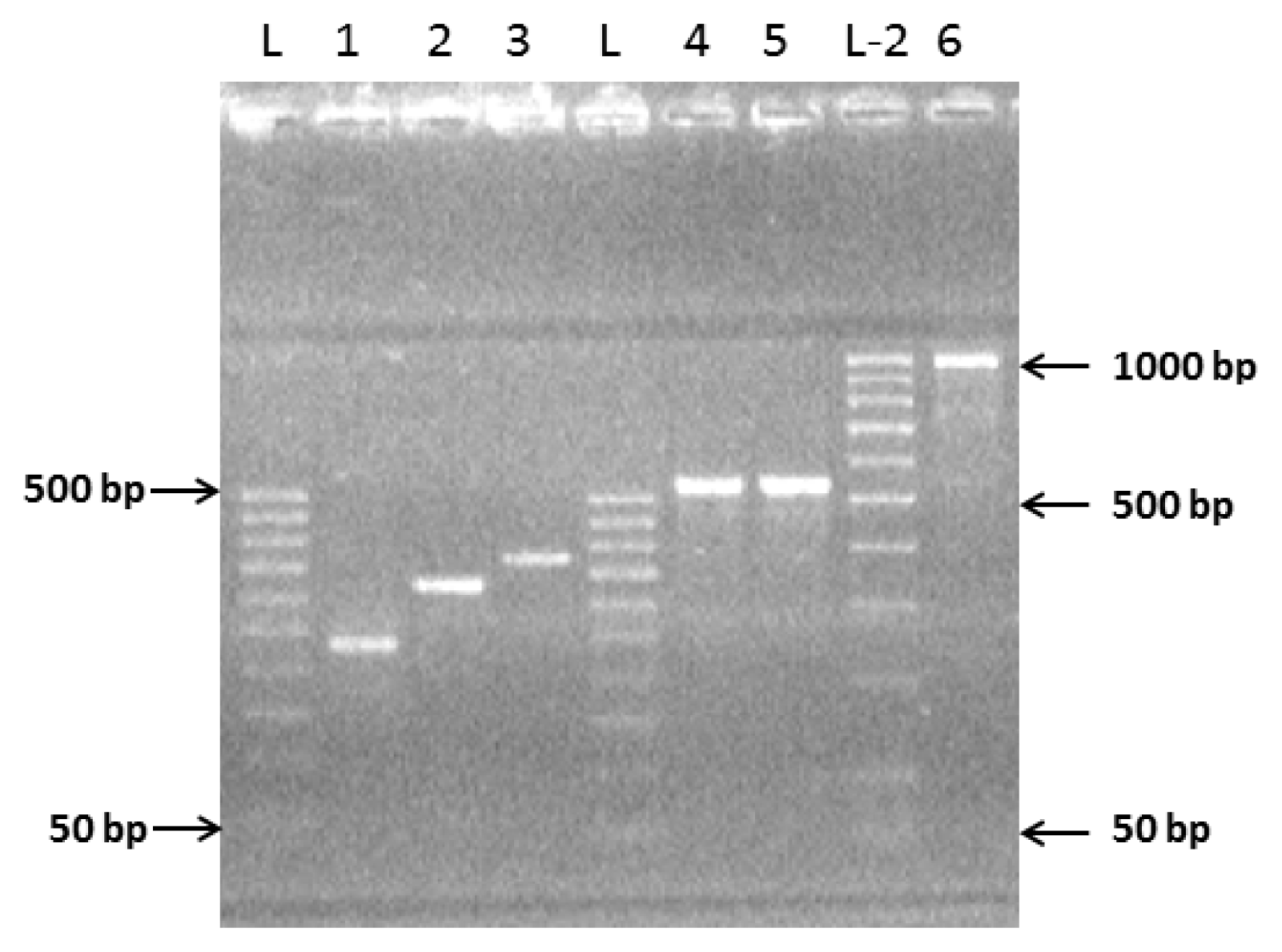
4.2. Experimental Design: Construction of a Full-Length Renalase Coding Sequence
4.3. Preparation of the Genomic DNA Template
4.4. Exon Amplification and Assembly
4.5. Plasmid Construction Cloning and Sequencing
4.6. Renalase Expression in Prokaryotic Cells and Purification
4.7. Western Blotting
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Baroni, S.; Milani, M.; Pandini, V.; Pavesi, G.; Horner, D.; Aliverti, A. Is renalase a novel player in catecholaminergic signaling? The mystery of the catalytic activity of an intriguing new flavoenzyme. Curr. Pharm. Des 2013, 19, 2540–2551. [Google Scholar]
- Desir, G.V.; Wang, L.; Peixoto, A.J. Human renalase: A review of its biology, function, and implications for hypertension. J. Am. Soc. Hypertens 2012, 6, 417–426. [Google Scholar]
- Malyszko, J.; Malyszko, J.S.; Rysz, J.; Mysliwiec, M.; Tesar, V.; Levin-Iaina, N.; Banach, M. Renalase, Hypertension, and Kidney—The Discussion Continues. Angiology 2013, 64, 181–187. [Google Scholar]
- Medvedev, A.E.; Veselovsky, A.V.; Fedchenko, V.I. Renalase, a new secretory enzyme responsible for selective degradation of catecholamines: Achievements and unsolved problems. Biochemistry 2010, 75, 951–958. [Google Scholar]
- Xu, J.; Li, G.; Wang, P.; Velazquez, H.; Yao, X.; Li, Y.; Wu, Y.; Peixoto, A.; Crowley, S.; Desir, G.V. Renalase is a novel, soluble monoamine oxidase that regulates cardiac function and blood pressure. J. Clin. Invest 2005, 115, 1275–1280. [Google Scholar]
- Pandini, V.; Ciriello, F.; Tedeschi, G.; Rossoni, G.; Zanetti, G.; Aliverti, A. Synthesis of human renalase1 in Escherichia coli and its purification as a FAD-containing holoprotein. Protein Expr. Purif 2010, 72, 244–253. [Google Scholar]
- Milani, M.; Ciriello, F.; Baroni, S.; Pandini, V.; Canevari, G.; Bolognesi, M.; Aliverti, A. FAD-Binding site and NADP reactivity in human renalase: A new enzyme involved in blood pressure regulation. J. Mol. Biol 2011, 411, 463–473. [Google Scholar]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=full_report&list_uids=55328 (accessed on 9 June 2013).
- Hennebry, S.C.; Eikelis, N.; Socratous, F.; Desir, G.; Lambert, G.; Schlaich, M. Renalase, a novel soluble FAD-dependent protein, is synthesized in the brain and peripheral nerves. Mol. Psychiatry 2010, 15, 234–236. [Google Scholar]
- Malyszko, J.; Zbroch, E.; Malyszko, J.S.; Koc-Zorawska, E.; Mysliwiec, M. Renalase, a novel regulator of blood pressure, is predicted by kidney function in renal transplant Recipients. Transplant. Proc. 2011, 43, 3004–3007. [Google Scholar]
- Wang, F.; Zhao, Q.; Xing, T.; Li, J.; Wang, N. Renalase-specific polyclonal antibody and its application in the detection of Renalase’s expression. Hybridoma 2012, 31, 378–381. [Google Scholar]
- Wang, F.; Xing, T.; Li, J.; Bai, M.; Hu, R.; Zhao, Z.; Tian, S.; Zhang, Z.; Wang, N. Renalase’s expression and distribution in renal tissue and cells. PLoS One 2012, 7, e46442. [Google Scholar]
- Fedchenko, V.I.; Buneeva, O.A.; Kopylov, A.T.; Kaloshin, A.A.; Aksenova, L.N.; Zgoda, V.G.; Medvdev, A.E. Mass spectrometry detection of monomeric renalase in human urine. Biomed. Khim 2012, 58, 599–607. [Google Scholar]
- Przybylowski, P.; Malyszko, J.; Kozlowska, S.; Malyszko, J.; Koc-Zorawska, E.; Mysliwiec, M. Serum renalase depends on kidney function but not on blood pressure in heart transplant recipients. Transplant. Proc 2011, 43, 3888–3891. [Google Scholar]
- Zbroch, E.; Malyszko, J.; Malyszko, J.S.; Koc-Zorawska, E.; Mysliwiec, M. Renalase, a novel enzyme involved in blood pressure regulation, is related to kidney function but not to blood pressure in hemodialysis patients. Kidney Blood Press Res 2012, 35, 395–399. [Google Scholar]
- Pelling, A.L.; Thorne, A.W.; Crane-Robinson, C. A human genomic library enriched in transcriptionally active sequences (aDNA library). Genome Res 2000, 10, 874–886. [Google Scholar]
- Wang, J.; Qi, S.; Cheng, W.; Li, L.; Wang, F.; Li, Y.Z.; Zhang, S.P. Identification, expression and tissue distribution of a renalase homologue from mouse. Mol. Biol. Rep 2008, 35, 613–620. [Google Scholar]
- An, X.; Lu, J.; Huang, J.D.; Zhang, X.; Chen, J.; Zhou, Y.; Tong, Y. Rapid assembly of multiple-exon cDNA directly from genomic DNA. PLoS One 2007, 2, e1179. [Google Scholar]
- Davies, W.L.; Carvalho, L.S.; Hunt, D.M. SPLICE: A technique for generation in vitro spliced coding sequeces from genomic DNA. BioTechniques 2007, 43, 785–789. [Google Scholar]
- Jayakumar, A.; Huang, W.-Y.; Raetz, B.; Chirala, S.S.; Wakil, S.J. Cloning and expression of the multifunctional human fatty acid synthase and its subdomains in Escherichia coli. Proc. Natl. Acad. Sci. USA 1996, 93, 14509–14514. [Google Scholar]
- Booth, P.M.; Buchman, G.W.; Rashtchian, A. Assembly and cloning of coding sequences for neurotrophic factors directly from genomic DNA using polymerase chain reaction and uracil DNA glycosylase. Gene 1994, 146, 303–308. [Google Scholar]
- Grimberg, J.; Nawoschik, S.; Belluscio, L.; McKee, R.; Turck, A.; Eisenberg, A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 1989, 17, 8390. [Google Scholar]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988, 16, 1215. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar]
- OMIGA, version 2.0. Available online: http://www.highbeam.com/doc/1G1-61963309.html (accessed on 21 April 2000).
- Gallagher, S.; Winston, S.E.; Fuller, S.A.; Hurrell, J.G.R. Immunoblotting and immunodetection. Curr. Protoc. Cell Biol 2011. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exons | Primers forward (5′-3′) | Primers reverse (5′-3′) |
|---|---|---|
| 1ExRe1 | 1Pr-f: cgtcgccatggcgcaggtgctgatcg | 1Pr-r: cctgagtcgtcagccttgtcc |
| 2ExRe1 | 2Pr-f: ggacaaggctgaCgactcagggggaagaatgactacagc | 2Pr-r: cgttggtgttttttggcataatg |
| 3ExRe1 | 3Pr-f: cattatgccaaaaaacaccaacgtttttatgatgaactgttagc | 3Pr-r: ctgattctttcaagtaatgc |
| 4ExRe1 | 4Pr-f: gcattacttgaaagaatcaggtgcagaagtctacttc | 4Pr-r: aggtggtgatgtcaccttg |
| 5ExRe1 | 5Pr-f: caaggtgacatcaccaccttaattagtgaatgccaaaggc | 5Pr-r: ctatattgcgcttcttattatc |
| 6ExRe1 | 6Pr-f: gataataagaacggcaatatagagtcatcagaaattgggc | 6Pr-r: cctgtgaatgtctccatttttgg |
| 7ExRe1 | 7Pr-f: ccaaaaatggagacattcacaggttacaaatgctgctgcc | 7Pr-r: gaggagctcgagaatataattctttaaagc |
| 1ExRe2 | 1PrRe2-r: cctgagtcctCagccttgtcc | |
| 2ExRe2 | 2PrRe2-f: ggacaaggctgaGgactcagggggaagaatgactacagc | |
| 7ExRe2 | 7PrRe2-f: ccaaaaatggagacattcacaggtaccaagtgctggtgtgattc | 7PrRe2-r: gaggagctcgaggatgggaaatccaatcgc |
| Protein form | Purification step | Volume (mL) | Protein (mg) | A280 nm |
|---|---|---|---|---|
| hRenalase1 | Bacterial culture | 2000 | 3500 | nd |
| Crude extract | 500 | 2750 | nd | |
| Ni-Sepharose affinity chromatography | 22.7 | 57.2 | 2.65 | |
| hRenalase2 | Bacterial culture | 2000 | 3350 | nd |
| Crude extract | 500 | 2350 | nd | |
| Ni-Sepharose affinity chromatography | 19.9 | 33.75 | 2.12 | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fedchenko, V.I.; Kaloshin, A.A.; Mezhevikina, L.M.; Buneeva, O.A.; Medvedev, A.E. Construction of the Coding Sequence of the Transcription Variant 2 of the Human Renalase Gene and Its Expression in the Prokaryotic System. Int. J. Mol. Sci. 2013, 14, 12764-12779. https://doi.org/10.3390/ijms140612764
Fedchenko VI, Kaloshin AA, Mezhevikina LM, Buneeva OA, Medvedev AE. Construction of the Coding Sequence of the Transcription Variant 2 of the Human Renalase Gene and Its Expression in the Prokaryotic System. International Journal of Molecular Sciences. 2013; 14(6):12764-12779. https://doi.org/10.3390/ijms140612764
Chicago/Turabian StyleFedchenko, Valerii I., Alexei A. Kaloshin, Lyudmila M. Mezhevikina, Olga A. Buneeva, and Alexei E. Medvedev. 2013. "Construction of the Coding Sequence of the Transcription Variant 2 of the Human Renalase Gene and Its Expression in the Prokaryotic System" International Journal of Molecular Sciences 14, no. 6: 12764-12779. https://doi.org/10.3390/ijms140612764