Facile Synthesis of 5-Arylidene Thiohydantoin by Sequential Sulfonylation/Desulfination Reaction


Abstract
:1. Introduction
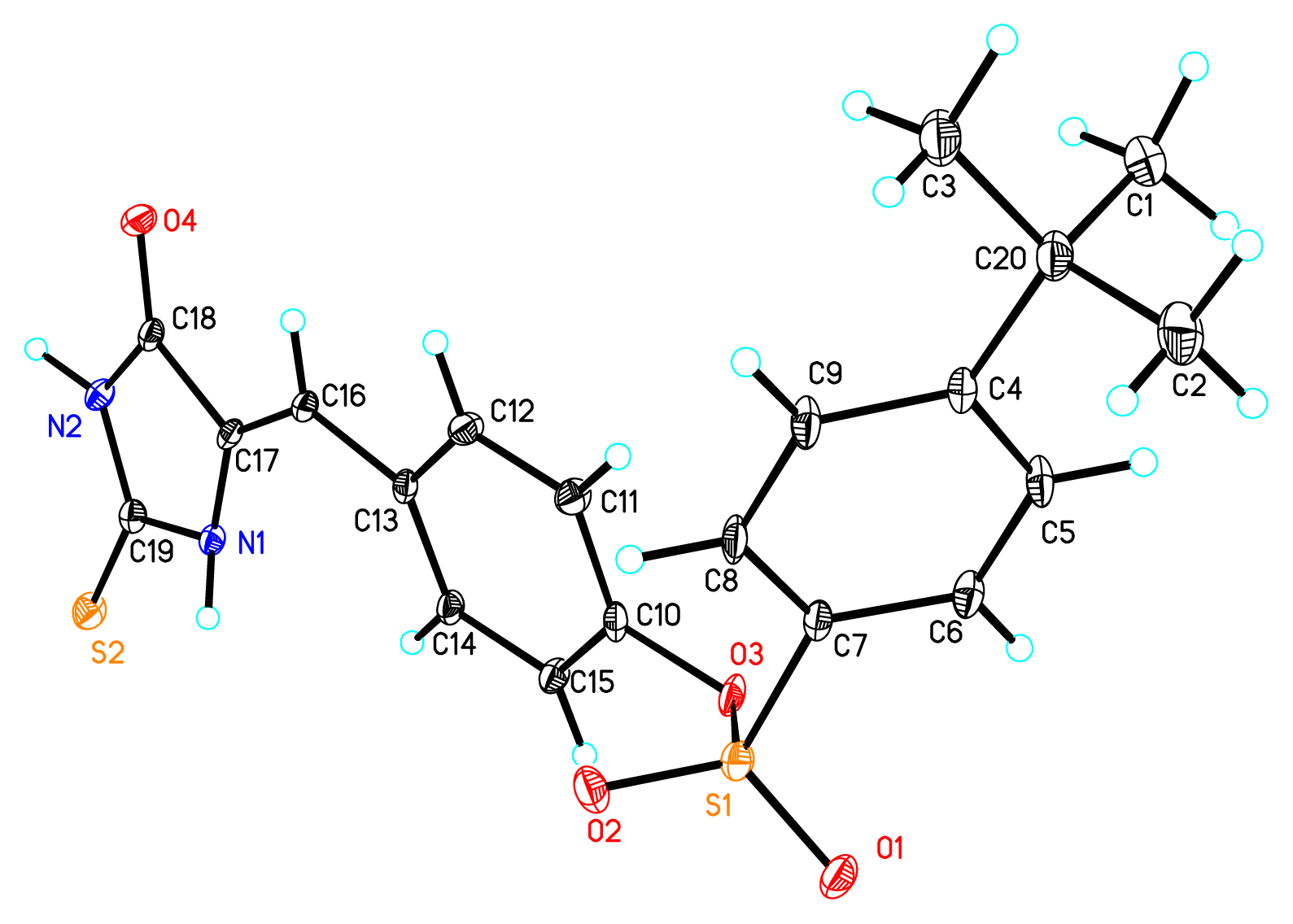
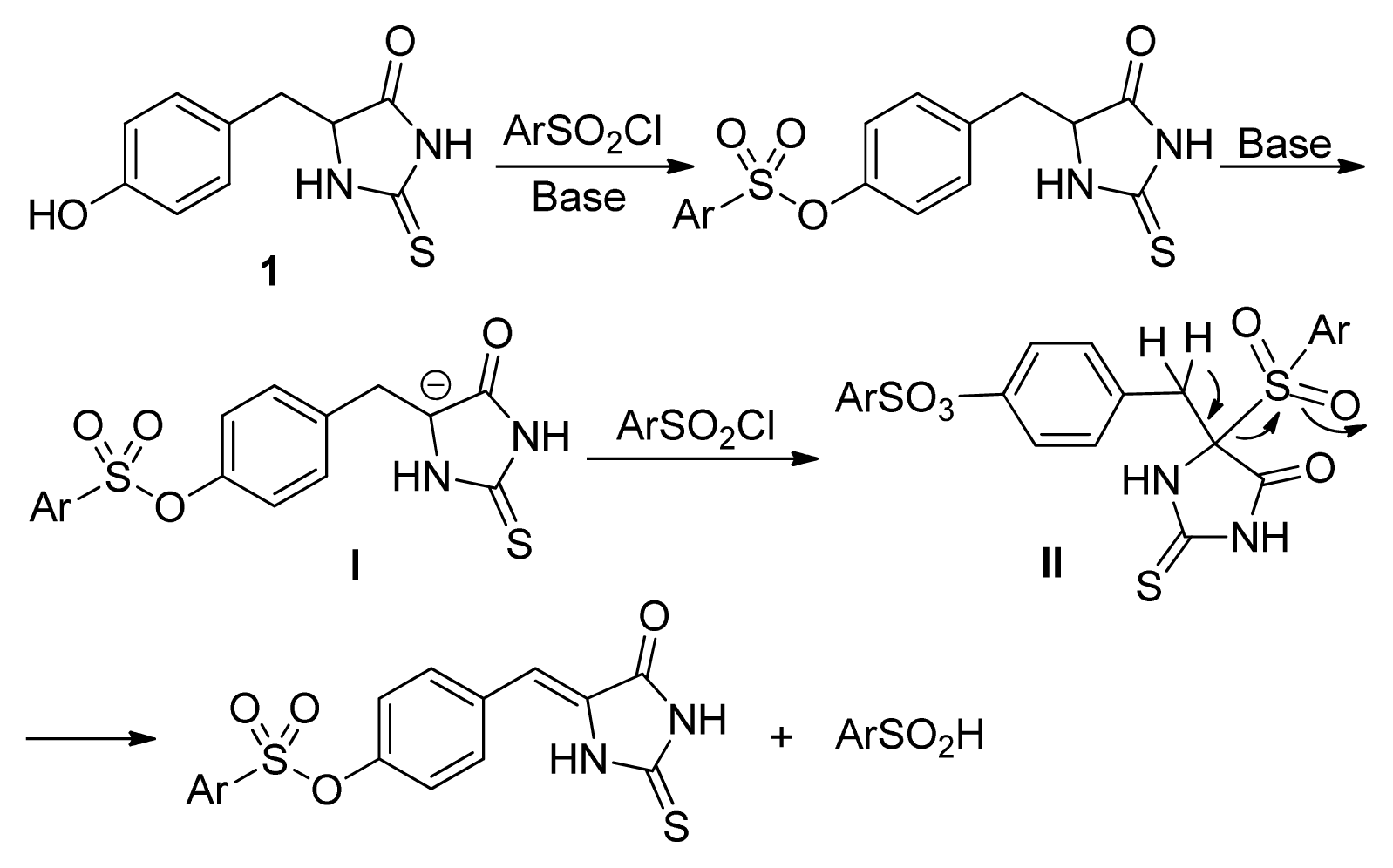
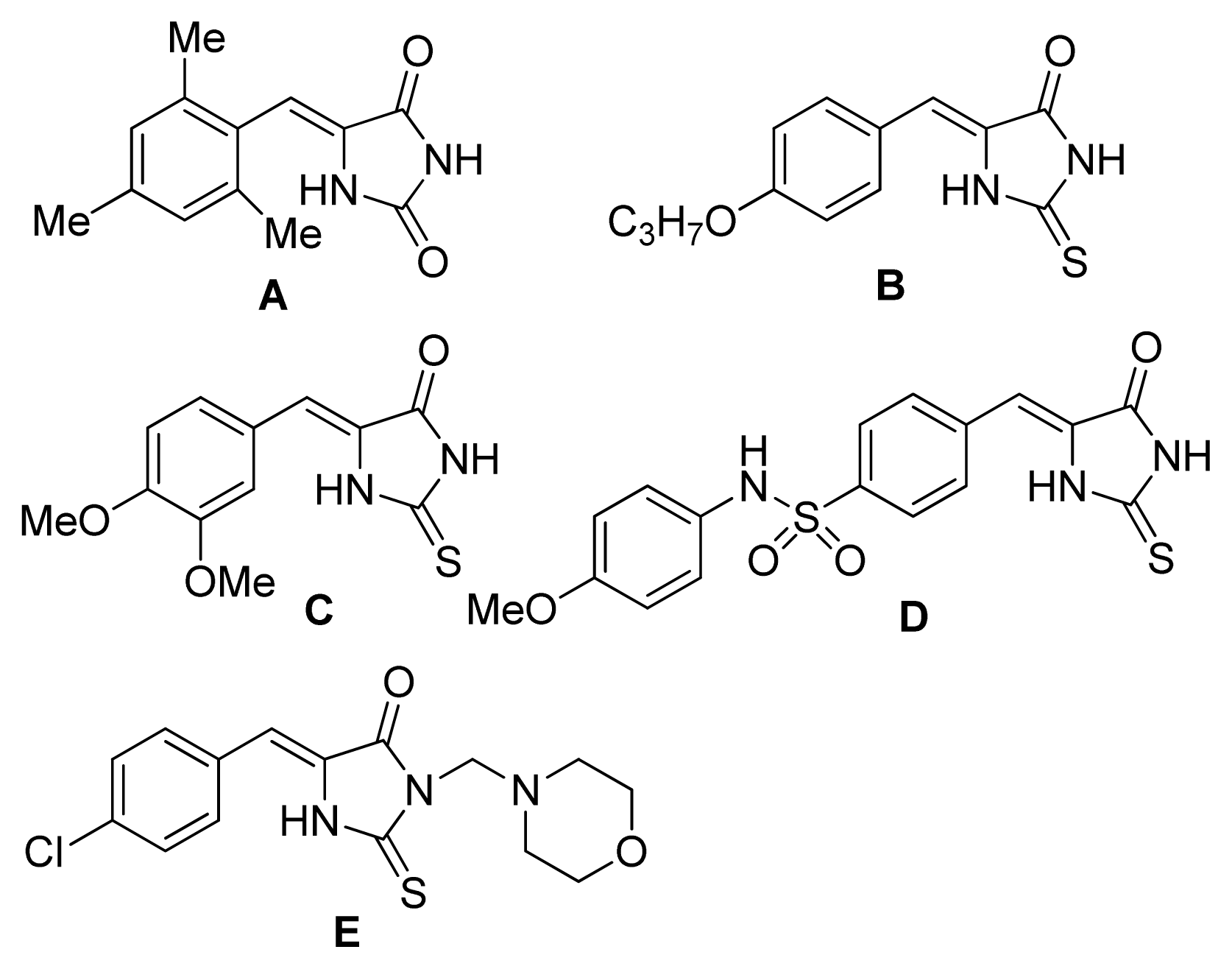
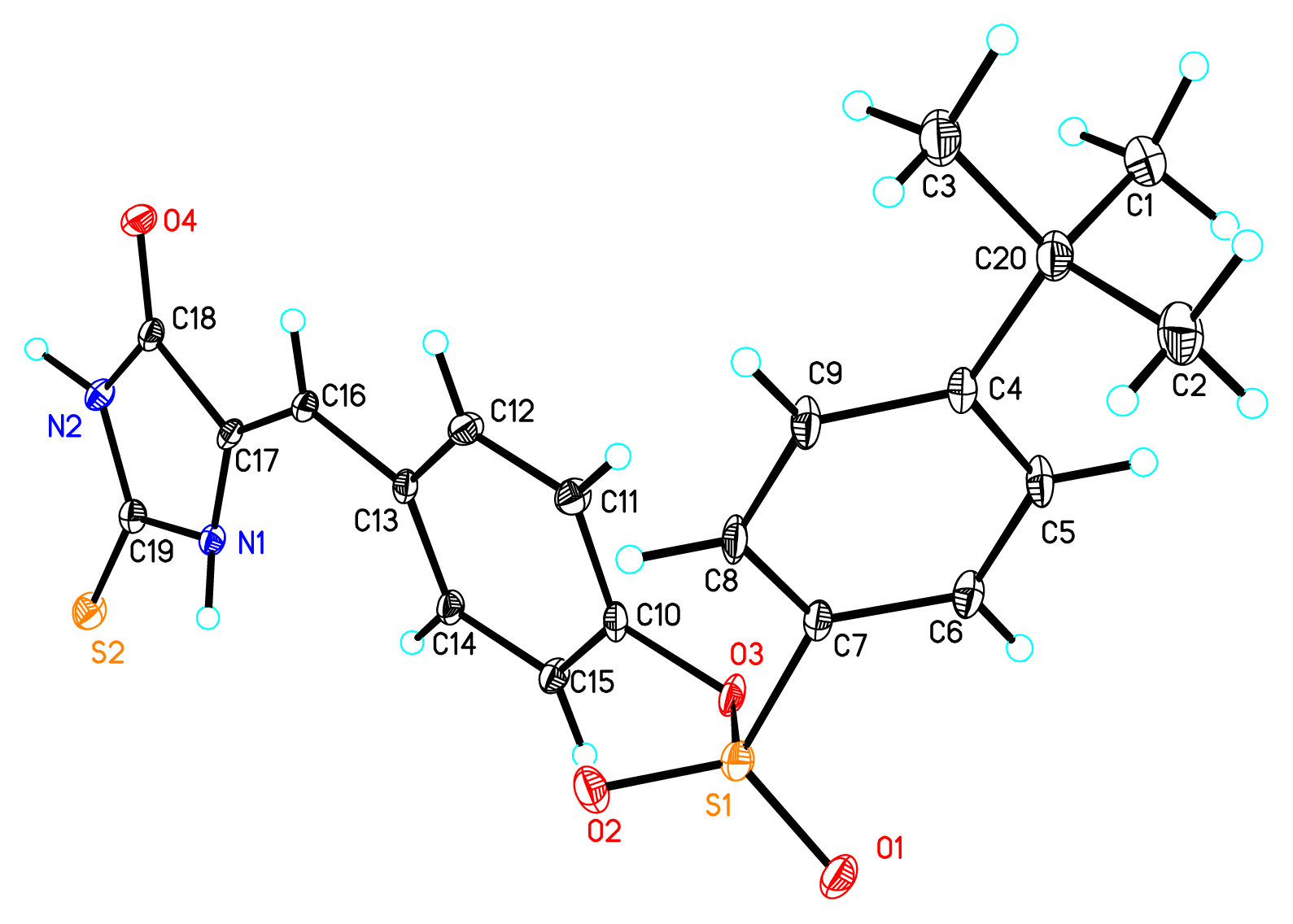
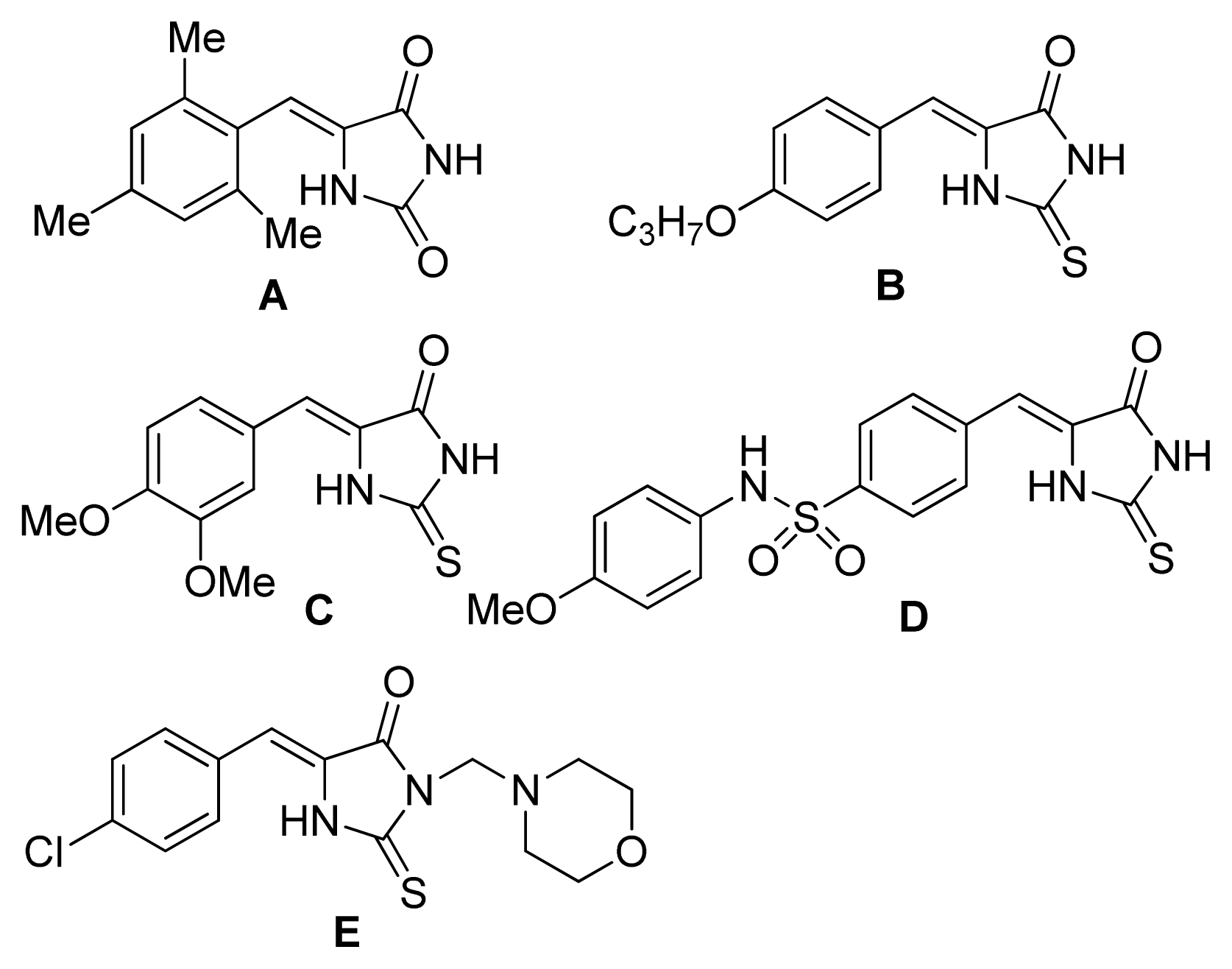
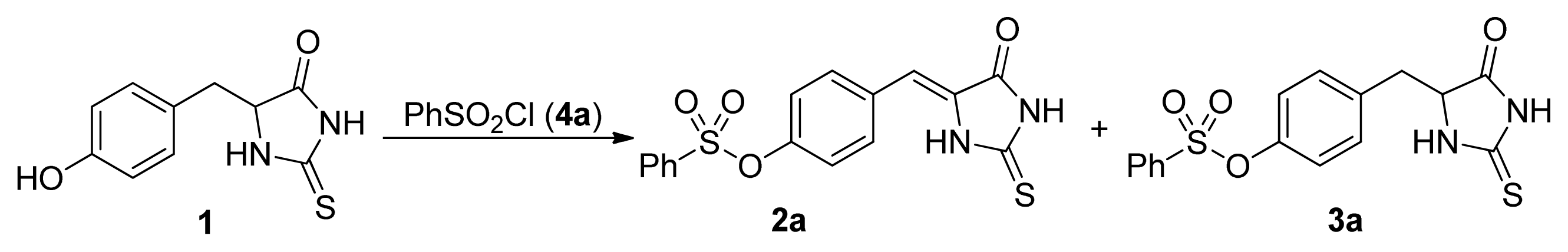
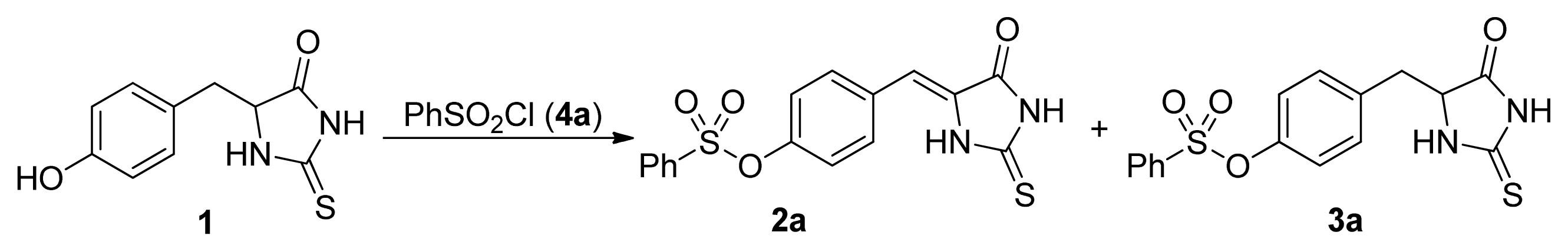
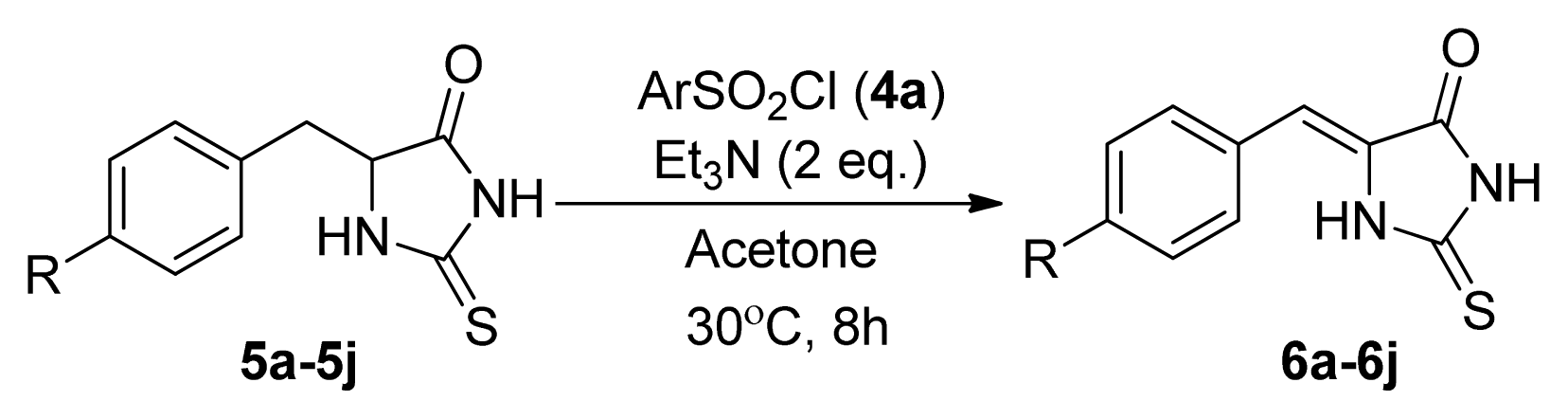
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. The Synthesis of 5-(4-Hydroxybenzyl)-Thiohydantoin (1)
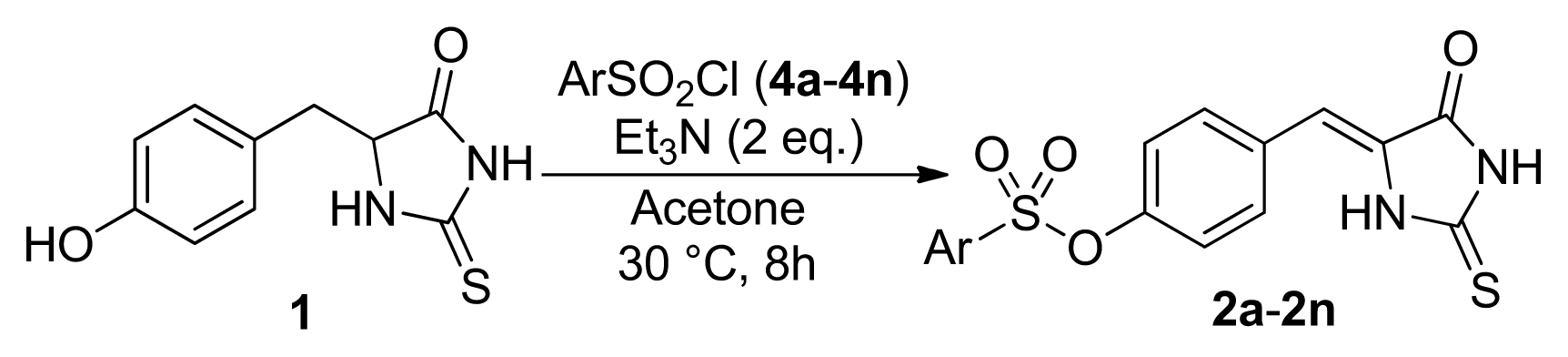
3.2.2. General Procedure for the Synthesis of Compounds 2 and 6
3.3. Bioassay of Fungicidal Activity
4. Conclusions
Supplementary Information
ijms-14-12484-s001.pdfAcknowledgments
Conflict of Interest
References and Notes
- Thenmozhiyal, J.C.; Wong, P.T.-H.; Chui, W.K. Anticonvulsant activity of phenylmethylene-hydantoins: A structure-activity relationship study. J. Med. Chem 2004, 47, 1527–1535. [Google Scholar]
- Kiec-Kononowicz, K.; Szymanska, E. Antimycobacterial activity of 5-arylidene derivatives of hydantoin. II Farmaco 2002, 57, 909–916. [Google Scholar]
- Marton, J.; Enisz, J.; Hosztafi, S.; Timar, T. Preparation and fungicidal activity of 5-substituted hydantoins and their 2-thio analogs. J. Agric. Food Chem 1993, 41, 148–152. [Google Scholar]
- Kiec-Kononowicz, K.; Karolak-Wojicechowska, J.; Muller, C.E.; Schumacher, B.; Pekela, E.; Szymanska, E. Imidazo-thiazine, -diazinone and -diazepinone derivatives. Synthesis, structure and benzodiazepine receptor binding. Eur. J. Med. Chem 2001, 36, 407–419. [Google Scholar]
- Szymanska, E.; Kiec-Kononowicz, K.; Bialecka, E.; Kasprowicz, A. Antimicrobial activity of 5-arylidene aromatic derivatives of hydantoin. II Farmaco 2002, 57, 39–44. [Google Scholar]
- Szymanska, E.; Kiec-Kononowicz, K. Antimycobacterial activity of 5-arylidene aromatic derivatives of hydantoin. II Farmaco 2002, 57, 355–362. [Google Scholar]
- Kumar, R.; Chauhan, P.M.S. A one-pot chemoselective S-alkylation and acetylation of thiohydantoins using the alkyl orthoformate-ZnCl2-Ac2O reagent system. Tetrahedron Lett 2008, 49, 5475–5479. [Google Scholar]
- Bourahla, K.; Paquin, L.; Lozach, O.; Meijer, L.; Carreaux, F.; Bazureau, J.P. A practical approach to new (5Z) 2-alkylthio-5-arylmethylene-1-methyl-1,5-dihydro-4H-imidazol-4-one derivatives. Molecules 2011, 16, 7377–7390. [Google Scholar]
- Shalaby, A.F.A.; Daboun, H.A.; Moghdadi, S.S.M. Reactions with 1-phenyl-4-thiohydantoin. Preparation of 5-arylazo-1-phenyl-4-thiohydantoin, their reactions towards alkylating agents and Mannich reaction. Z. Naturforsch 1976, B31, 865–869. [Google Scholar]
- Porwal, S.; Kumar, R.; Maulik, P.R.; Chauhan, P.M.S. A multicomponent reaction efficiently producing arylmethylene 2-thiohydantoins. Tetrahedron Lett 2006, 47, 5863–5866. [Google Scholar]
- Tripathy, P.K. Microwave assisted rapid condensation of 2-thiohydantoin/hydantoin with Schiff bases. Asian J. Chem 2005, 17, 1207–1210. [Google Scholar]
- Meanwell, N.A.; Herbert, R.R.; Edward, C.R. 1,3-Dihydro-2H-imidazo[4,5-b]quinolin-2-onesinhibitors of blood platelet cAMP phosphodiesterase and induced aggregation. J. Med. Chem 1991, 34, 2906–2916. [Google Scholar]
- Zhu, Z.J.; Lippa, B.S.; Townsend, L.B. A novel photo-assisted annulation reaction for the synthesis of 6,7-dichloroimidazo[4,5-b]quinolin-2-one. Tetrahedron Lett 1996, 37, 1937–1940. [Google Scholar]
- Pedro, M.; Alberto, T.; David, C. Synthesis of imidazo[1,5-c][1,3]benzodiazepines via an aza-Wittig/carbodiimide-mediated annulation process. Tetrahedron 1997, 53, 15895–15902. [Google Scholar]
- Kuninobu, Y.; Kikuchi, K.; Takai, K. Manganese-catalyzed synthesis of hydantoin derivatives from terminal alkynes and isocyanates. Chem. Lett 2008, 37, 740–741. [Google Scholar]
- Han, J.T.; Wang, J.M.; Dong, H.B.; Xu, Z.H.; Liu, B.; Wang, M.A. Synthesis and biological activity of novel phosphoramidate with hydantoin. Chin. J. Org. Chem 2013, 33, 596–601. [Google Scholar]
- Xu, Z.H.; Wang, J.M.; Han, J.T.; Liu, B.; Wang, M.A. Synthesis and biological activity of novel (thio)phosphates with hydantoin. Chin. J. Org. Chem 2012, 32, 2134–2140. [Google Scholar]
- Han, J.T.; Wang, J.M.; Dong, H.B.; Lei, J.P.; Wang, M.A.; Fang, J.X. Synthesis and herbicidal activity of 5-(4-hydroxybenzyl)-2-thioxoimidazolidin-4-one esters. Molecules 2011, 16, 2833–2845. [Google Scholar]
- Walters, E.W.; Lee, S.F.; Niderman, T.; Bemasconi, P.; Subramanian, M.V.; Siehl, D.L. Adenylosuccinate synthetase from maize. Purification, properties, and mechanism of inhibition by 5′-phosphohydantocidin. Plant Physiol 1997, 114, 549–555. [Google Scholar]
- Cseke, C.; Gerwick, B.C.; Crouse, G.D.; Murdoch, M.G.; Green, S.B.; Heim, D.R. 2-Phosphohydantocidin: The in vivo adenylosuccinate synthetase inhibitor responsible for hydantocidin phytotoxicity. Pestic. Biochem. Physiol 1996, 55, 210–217. [Google Scholar]
- Fonne-Pfister, R.; Chemla, P.; Ward, E.; Girardet, M.; Kreuz, K.E.; Hinzatko, R.B.; Fromm, H.; Schar, H.P.; Grutter, M.G.; Cowan-Jacob, S.W. The mode of action and the structure of a herbicide in complex with its target: binding of activated hydantocidin to the feedback regulation site of adenylosuccinate synthetase. Proc. Natl. Acad. Sci. USA 1996, 93, 9431–9436. [Google Scholar]
- Brady, S.F.; Bauer, J.D.; Clarke-Pearson, M.F.; Daniels, R. Natural products from isnA-containing biosynthetic gene clusters recovered from the genomes of cultured and uncultured bacteria. J. Am. Chem. Soc 2007, 129, 12102–12103. [Google Scholar]
- The quantum chemical calculation results showed that the molecular energy for Z-isomers are lower 6–10 Kcal/mol than that of E-isomers, which indicated the Z-isomers are much stable.
- The similar results were obtained with the other substituted phenylsulfonyl chloride such as 4c, 4d, and 4k. For example, the yields for 3k were 86%, 65%, 14%, <5%, the yields for 2k were 10%, 16%, 74%, 82% when 4k was used with 1, 1.2, 1.5, 2.0 equivalents, respectively.
- Cubbage, J.W.; Vos, B.W.; Jenks, W.S. Ei elimination: An unprecedented facet of sulfone chemistry. J. Am. Chem. Soc 2000, 122, 4968–4971. [Google Scholar]
- Cubbage, J.W.; Guo, Y.; McCulla, R.D.; Jenks, W.S. Thermolysis of alkyl sulfoxides and derivatives: A comparison of experiment and theory. J. Org. Chem 2001, 66, 8722–8736. [Google Scholar]
- Trost, B.M.; Seoane, P.; Mignani, S.; Acemoglu, M. Stereocontrolled cyclopentenone synthesis via cycloaddition. J. Am. Chem. Soc 1989, 111, 7487–7500. [Google Scholar]
- Baker-Glenn, C.A.G.; Barrett, A.G.M.; Gray, A.A.; Procopiou, P.A.; Ruston, M. Alkene synthesis: Elimination of arenesulfinic acid from alkyl aryl sulfones using potassium trimethylsilanolate as base. Tetrahedron Lett 2005, 46, 7427–7430. [Google Scholar]
- Barrett, A.G.M.; Cramp, S.M.; Hennessy, A.J.; Procopiou, P.A.; Roberts, R.S. Oxazole synthesis with minimal purification: Synthesis and application of a rompgel tosmic reagent. Org. Lett 2001, 3, 271–273. [Google Scholar]
- Schmidt-Winkel, P.; Wudl, F. A remarkably thermally stable, polar aliphatic polysulfone. Macromolecules 1998, 31, 2911–2917. [Google Scholar]
- Kiec-Kononowicz, K.; Szymanska, E.; Motyl, M.; Kasprowicz, A.; Bialecka, A.; Holzer, W. Synthesis, spectral, and antimicrobial properties of 5-chloroarylidene aromatic derivatives of imidazolin-4-one. Pharmazie 1998, 53, 680–684. [Google Scholar]
- Wang, Z.D.; Sheikh, S.O.; Zhang, Y.A. Simple synthesis of 2-thiohydantoins. Molecules 2006, 11, 739–750. [Google Scholar]
- Chen, N.C. The Bioassay Technologies for Pesticides; Beijing Agricultural University Press: Beijing, China, 1991; pp. 161–162. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ratio (1:4a) | Temp (°C) | Time (h) | Solvent | Base | 2a Yields (%) a | 3a yield (%) a |
| 1 | 1:1 | 30 | 8 | acetone | Et3N | 15 | 84 [24] |
| 2 | 1:1.2 | 30 | 8 | acetone | Et3N | 29 | 58 [24] |
| 3 | 1:1.5 | 30 | 8 | acetone | Et3N | 69 | 23 [24] |
| 4 | 1:2 | 30 | 8 | acetone | Et3N | 81 | <5 [24] |
| 5 | 1:3 | 30 | 8 | acetone | Et3N | 80 | - b |
| 6 | 1:2 | 30 | 6 | acetone | Et3N | 76 | - b |
| 7 | 1:2 | 30 | 4 | acetone | Et3N | 49 | - b |
| 8 | 1:2 | 30 | 2 | acetone | Et3N | 38 | - b |
| 9 | 1:2 | 0 | 8 | acetone | Et3N | 53 | - b |
| 10 | 1:2 | 60 | 8 | acetone | Et3N | 81 | - b |
| 11 | 1:2 | 30 | 8 | CH2Cl2 | Et3N | 41 | - b |
| 12 | 1:2 | 30 | 8 | CHCl3 | Et3N | 63 | - b |
| 13 | 1:2 | 30 | 8 | THF | Et3N | 66 | - b |
| 14 | 1:2 | 30 | 8 | acetone | Pyridine | 34 | - b |
| 15 | 1:2 | 30 | 8 | acetone | Na2CO3 | 28 | - b |
| 16 c | 1:2 | 30 | 8 | acetone | Et3N | 63 | - b |
| 17 d | 1:2 | 30 | 8 | acetone | Et3N | 64 | - b |
| 18 e | 1:2 | 30 | 8 | acetone | Et3N | 65 | - b |
 | |||
|---|---|---|---|
| Entry | Ar | Product | Yield (%) |
| 1 | Ph (4a) | 2a | 81 |
| 2 | 4-FC6H4 (4b) | 2b | 78 |
| 3 | 4-ClC6H4 (4c) | 2c | 77 |
| 4 | 4-MeOC6H4 (4d) | 2d | 40 |
| 5 | 4-MeC6H4 (4e) | 2e | 46 |
| 6 | 4-NO2C6H4 (4f) | 2f | 84 |
| 7 | 4-BrC6H4 (4g) | 2g | 49 |
| 8 | 2-MeC6H4 (4h) | 2h | 42 |
| 9 | 3-CF3C6H4 (4i) | 2i | 80 |
| 10 | 4-IC6H4 (4j) | 2j | 49 |
| 11 | 4-t-ButC6H4 (4k) | 2k | 80 |
| 12 | 4-AcNHC6H4 (4l) | 2l | 48 |
| 13 | 2,5-Cl2C6H4 (4m) | 2m | 52 |
| 14 | 3,4-Cl2C6H4 (4n) | 2n | 54 |
 | |||
|---|---|---|---|
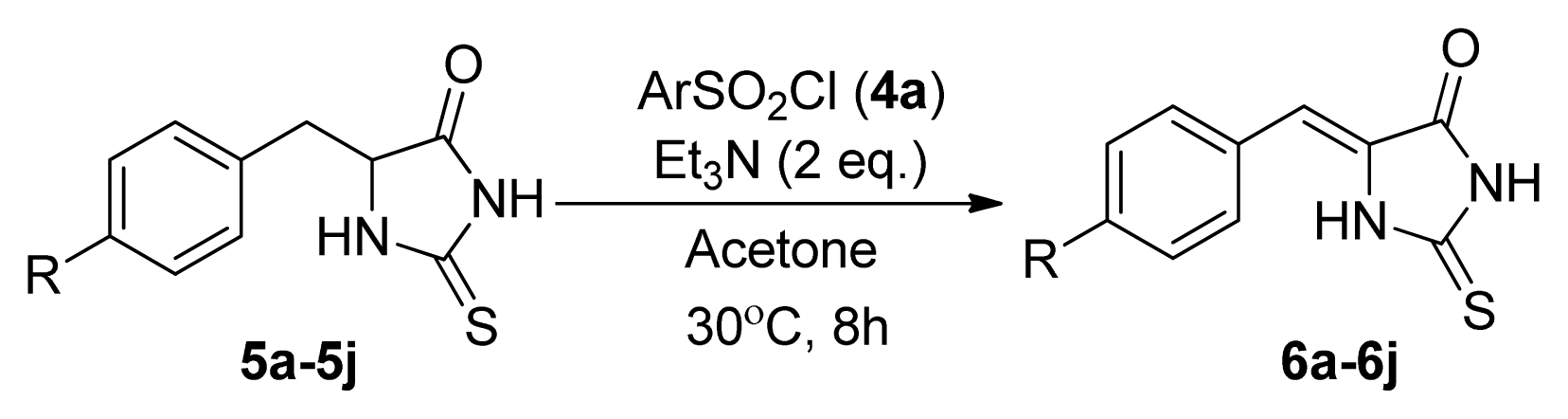
| Entry | R | Product a | Yield (%) |
| 1 | H (5a) | 6a | 38 |
| 2 | 4-F (5b) | 6b | 77 |
| 3 | 4-Cl (5c) | 6c | 74 |
| 4 | 4-Br (5d) | 6d | 65 |
| 5 | 4-Me (5e) | 6e | 48 |
| 6 | 4-NO2 (5f) | 6f | 76 |
| 7 | 4-MeO (5g) | 6g | 49 |
| 8 | 4-CF3 (5h) | 6h | 86 |
| 9 | 2,4-Cl2 (5i) | 6i | 75 |
| 10 | 4-NH2 (5j) | 6j | 0 |
| 11 | H (5a) | 6a | 52 |
| 12 | 4-NO2 (5f) | 6f | 84 |
| 13 | 4-CF3 (5h) | 6h | 88 |
| Compd. | Botrytis cinerea | Alternaria solani | Fusarium oxysporum | Dothiorella gregaria |
|---|---|---|---|---|
| 2a | 27.6 | 55.4 | 37.2 | −5.3 |
| 2b | 12.6 | 27.7 | 23.4 | −6.4 |
| 2c | 21.8 | 37.3 | 18.1 | −5.3 |
| 2d | 3.4 | 22.9 | 8.5 | −3.2 |
| 2e | 17.2 | 48.2 | 29.4 | 1.1 |
| 2f | 10.6 | 24.7 | 5.6 | −7.0 |
| 2g | 12.5 | 35.1 | 30.9 | 29.7 |
| 2h | 10.4 | 26.8 | 20.3 | 11.2 |
| 2i | 29.3 | 33.2 | 0 | 38.4 |
| 2j | 19.4 | 15.9 | 0 | 0 |
| 2k | 71.9 | 48.9 | 22.9 | 17.6 |
| 2l | 0 | 34.1 | 0 | 13.9 |
| 2m | 27.8 | 57.6 | 31.9 | 36.8 |
| 2n | 21.6 | 19.3 | 9.0 | 0 |
| Carbendazim | 8.4 | 10.9 | 100 | 97.6 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Han, J.; Dong, H.; Xu, Z.; Lei, J.; Wang, M. Facile Synthesis of 5-Arylidene Thiohydantoin by Sequential Sulfonylation/Desulfination Reaction. Int. J. Mol. Sci. 2013, 14, 12484-12495. https://doi.org/10.3390/ijms140612484
Han J, Dong H, Xu Z, Lei J, Wang M. Facile Synthesis of 5-Arylidene Thiohydantoin by Sequential Sulfonylation/Desulfination Reaction. International Journal of Molecular Sciences. 2013; 14(6):12484-12495. https://doi.org/10.3390/ijms140612484
Chicago/Turabian StyleHan, Jintao, Hongbo Dong, Zhihong Xu, Jianping Lei, and Mingan Wang. 2013. "Facile Synthesis of 5-Arylidene Thiohydantoin by Sequential Sulfonylation/Desulfination Reaction" International Journal of Molecular Sciences 14, no. 6: 12484-12495. https://doi.org/10.3390/ijms140612484