Lymphocytes of Patients with Alzheimer’s Disease Display Different DNA Damage Repair Kinetics and Expression Profiles of DNA Repair and Stress Response Genes

Abstract
:1. Introduction
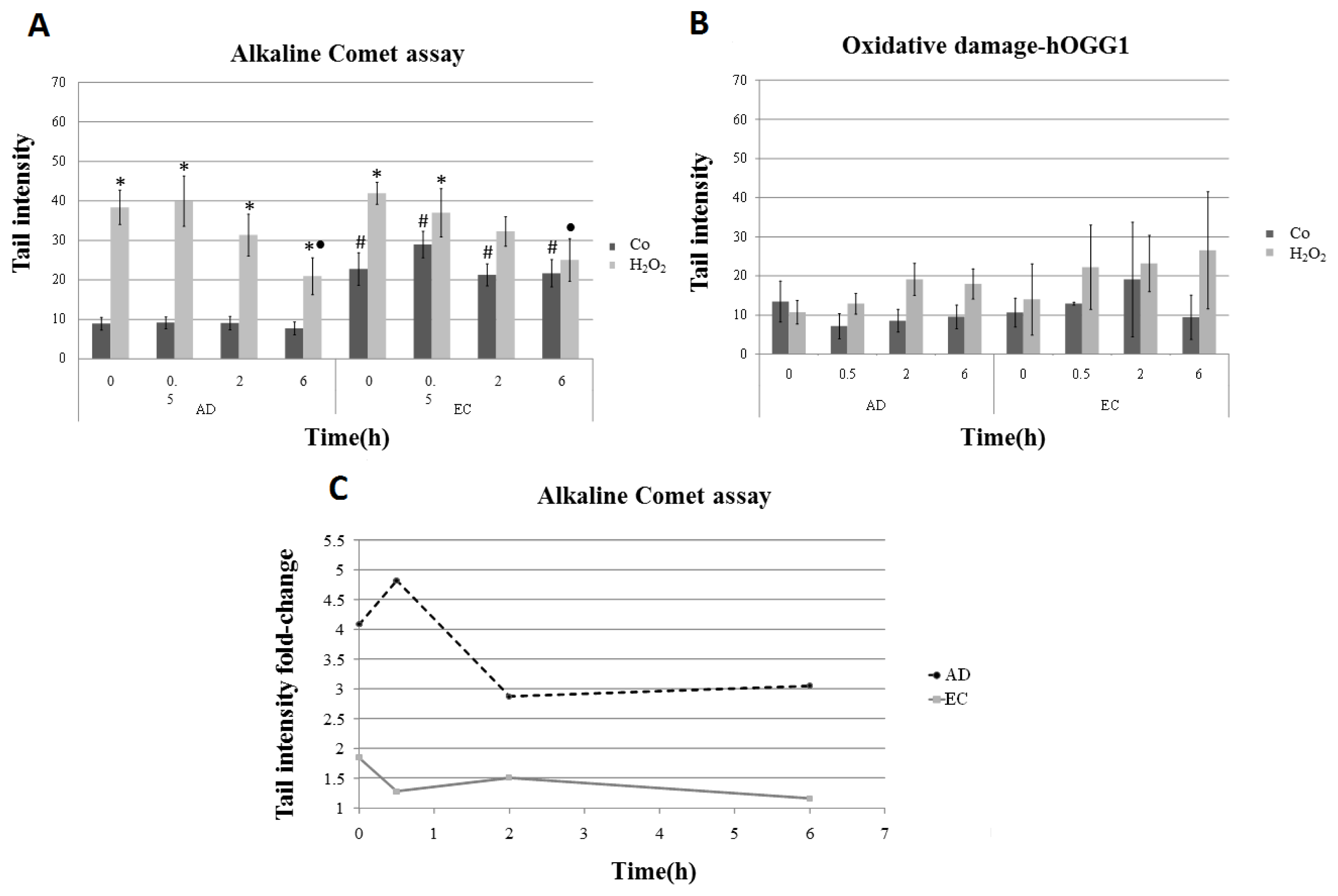
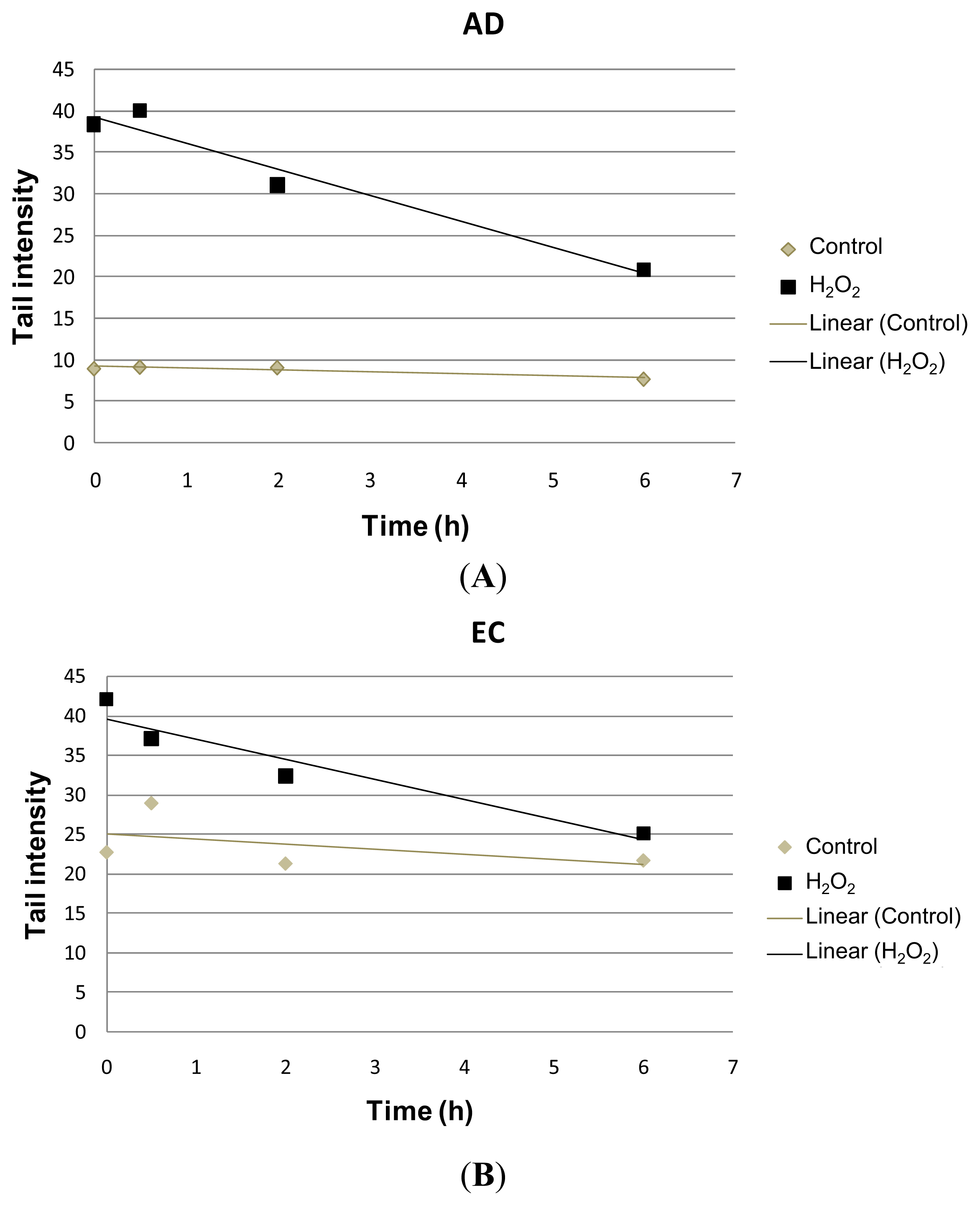
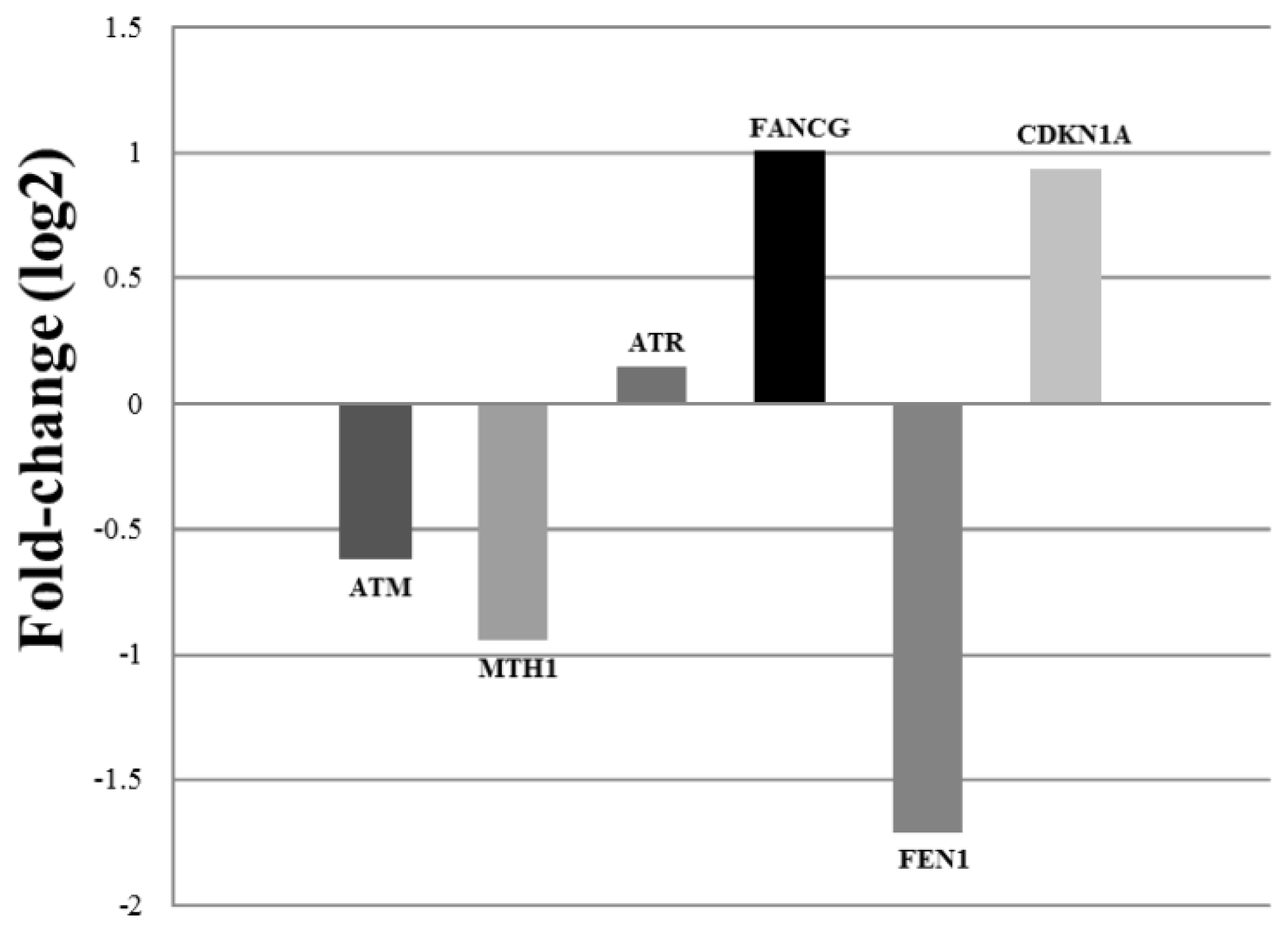
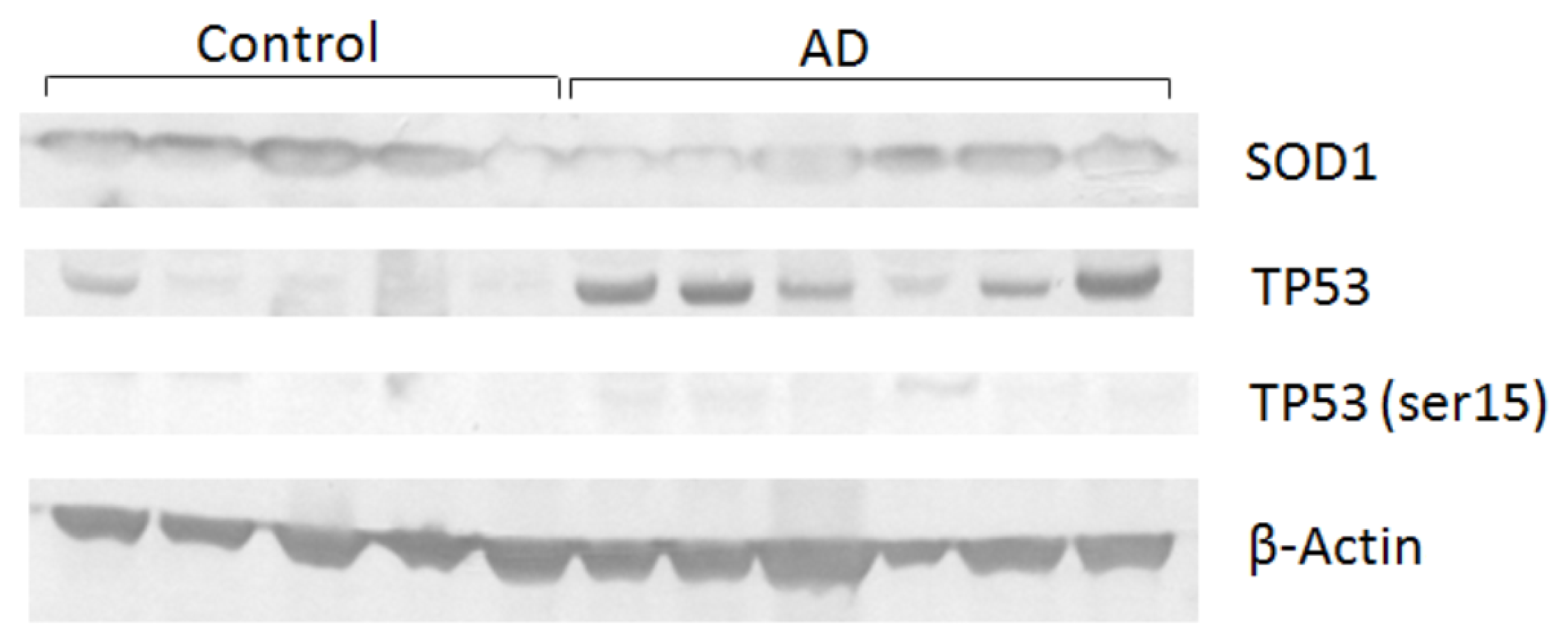
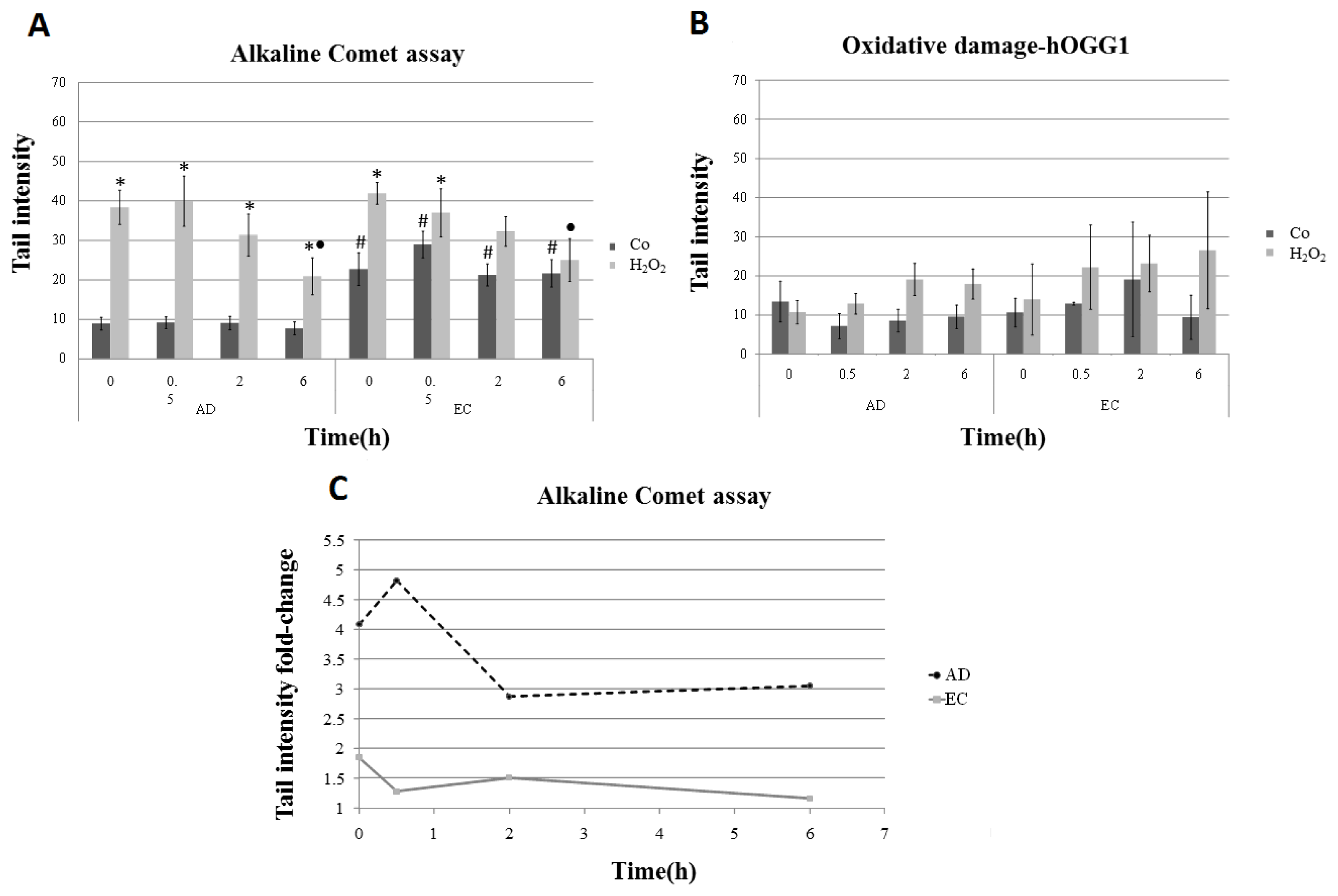
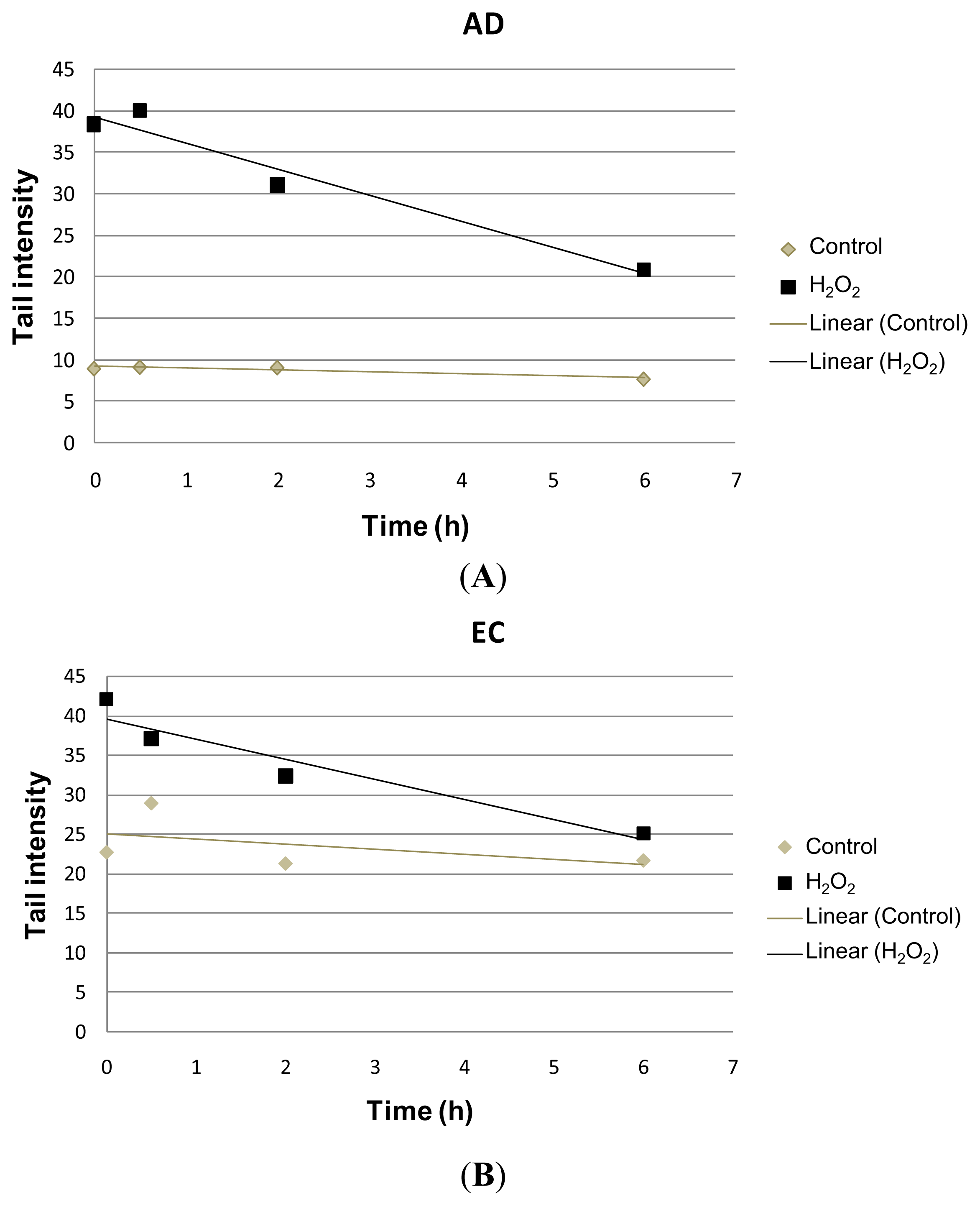
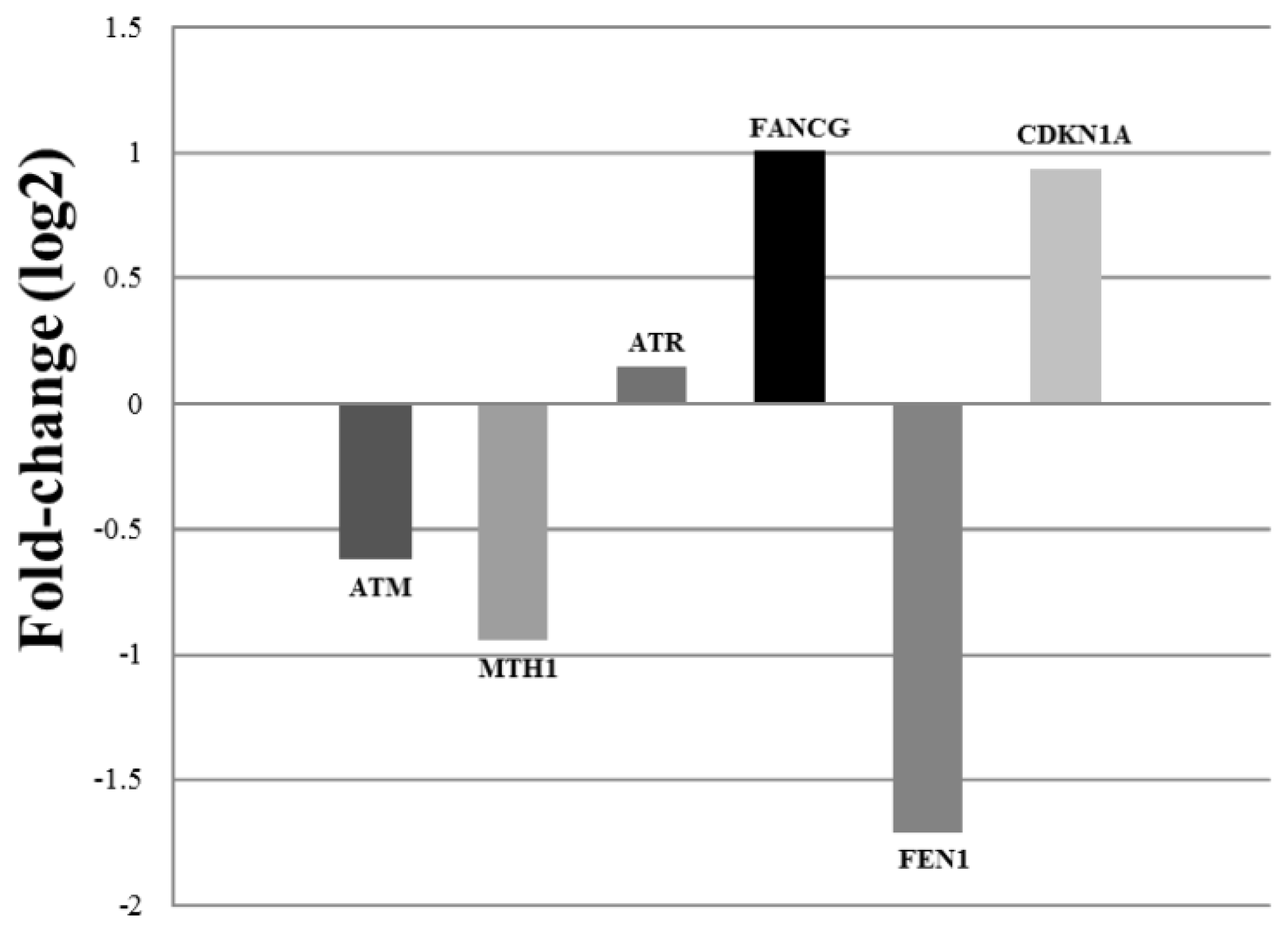
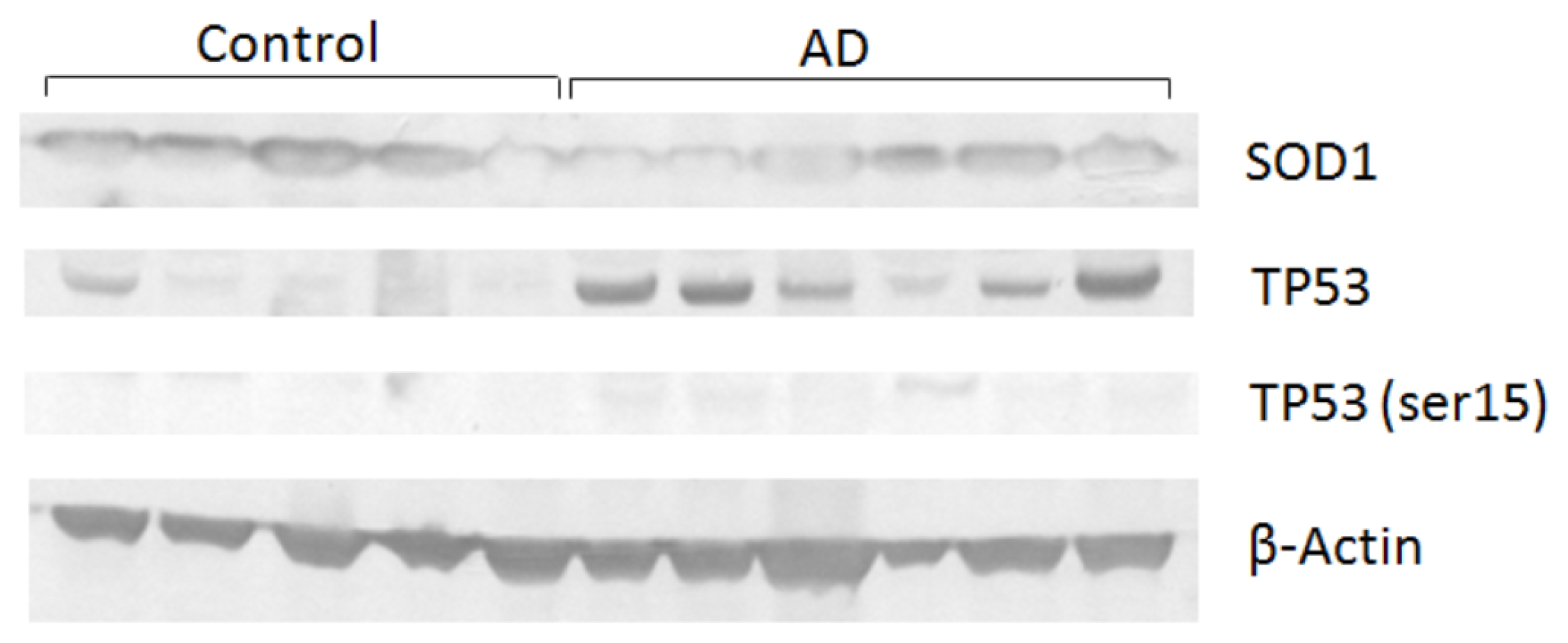
2. Results and Discussion
3. Experimental Section
3.1. Subjects
3.2. Blood Collection
3.3. Cell Culture and Comet Assay
3.4. Total RNA and Protein Extraction
3.5. Quantitative Real-Time PCR (qPCR)
3.6. Western Blot
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The public health impact of Alzheimer’s disease, 2000–2050: Potential implication of treatment advances. Annu. Rev. Public Health 2002, 23, 213–231. [Google Scholar]
- Avramopoulos, D. Genetics of Alzheimer’s disease: Recent advances. Genome Med 2009, 1, 34. [Google Scholar]
- Perry, G.; Nunomura, A.; Raina, A.K.; Aliev, G.; Siedlak, S.L.; Harris, P.L.; Casadesus, G.; Petersen, R.B.; Bligh-Glover, W.; Balraj, E.; et al. A metabolic basis for Alzheimer disease. Neurochem. Res 2003, 28, 1549–1552. [Google Scholar]
- Smith, M.A.; Richey, H.P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci 1997, 17, 2653–2657. [Google Scholar]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar]
- Haines, J.L. The genetics of Alzheimer disease--a teasing problem. Am. J. Hum. Genet 1991, 48, 1021–1025. [Google Scholar]
- Levy-Lahad, E.; Wijsman, E.M.; Nemens, E.; Anderson, L.; Goddard, K.A.; Weber, J.L.; Bird, T.D.; Schellenberg, G.D. A familial Alzheimer’s disease locus on chromosome 1. Science 1995, 269, 970–973. [Google Scholar]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar]
- Tanzi, R.; Gaston, S.; Bush, A.; Romano, D.; Pettingell, W.; Peppercorn, J.; Paradis, M.; Gurubhagavatula, S.; Jenkins, B.; Wasco, W. Genetic heterogeneity of gene defects responsible for familial Alzheimer disease. Genetica 1993, 91, 255–263. [Google Scholar]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar]
- Li, J.C.; Kaminskas, E. Deficient repair of DNA lesions in Alzheimer’s disease fibroblasts. Biochem. Biophys. Res. Commun 1985, 129, 733–738. [Google Scholar]
- Migliore, L.; Fontana, I.; Trippi, F.; Colognato, R.; Coppede, F.; Tognoni, G.; Nucciarone, B.; Siciliano, G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol. Ag 2005, 26, 567–573. [Google Scholar]
- Robison, S.H.; Munzer, J.S.; Tandan, R.; Bradley, W.G. Alzheimer’s disease cells exhibit defective repair of alkylating agent-induced DNA damage. Ann. Neurol 1987, 21, 250–258. [Google Scholar]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem 2006, 96, 825–832. [Google Scholar]
- Kadioglu, E.; Sardas, S.; Aslan, S.; Isik, E.; Karakaya, A.E. Detection of oxidative DNA damage in lymphocytes of patients with Alzheimer’s disease. Biomarkers 2004, 9, 203–209. [Google Scholar]
- Chinnery, P.F.; Taylor, G.A.; Howell, N.; Brown, D.T.; Parsons, T.J.; Turnbull, D.M. Point mutations of the mtDNA control region in normal and neurodegenerative human brains. Am. J. Hum. Genet 2001, 68, 529–532. [Google Scholar]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar]
- Melov, S. Modeling mitochondrial function in aging neurons. Trends Neurosci 2004, 27, 601–606. [Google Scholar]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci 1999, 19, 1959–1964. [Google Scholar]
- Gasparini, L.; Racchi, M.; Binetti, G.; Trabucchi, M.; Solerte, S.B.; Alkon, D.; Etcheberrigaray, R.; Gibson, G.; Blass, J.; Paoletti, R.; et al. Peripheral markers in testing pathophysiological hypotheses and diagnosing Alzheimer’s disease. FASEB J 1998, 12, 17–34. [Google Scholar]
- Yoon, S.C.; Kwon, Y.A.; Kim, H.; Kim, S.; Ahn-Jo, S.; Kim, D.K. Altered cell viability and proliferation activity of peripheral lymphocytes in patients with Alzheimer’s disease. Psychiatry Investig 2010, 7, 68–71. [Google Scholar]
- Collins, A.R.; Oscoz, A.A.; Brunborg, G.; Gaivao, I.; Giovannelli, L.; Kruszewski, M.; Smith, C.C.; Stetina, R. The comet assay: Topical issues. Mutagenesis 2008, 23, 143–151. [Google Scholar]
- Otten, U.; Ehrhard, P.; Peck, R. Nerve growth factor induces growth and differentiation of human B lymphocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 10059–10063. [Google Scholar]
- Wick, G.; Grubeck-Loebenstein, B. Immunity and aging. Dev. Comp. Immunol 1997, 21, 455–460. [Google Scholar]
- Richartz-Salzburger, E.; Batra, A.; Stransky, E.; Laske, C.; Kohler, N.; Bartels, M.; Buchkremer, G.; Schott, K. Altered lymphocyte distribution in Alzheimer’s disease. J. Psychiatr Res 2007, 41, 174–178. [Google Scholar]
- Leutner, S.; Schindowski, K.; Frolich, L.; Maurer, K.; Kratzsch, T.; Eckert, A.; Muller, W.E. Enhanced ROS-generation in lymphocytes from Alzheimer’s patients. Pharmacopsychiatry 2005, 38, 312–315. [Google Scholar]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res 2008, 5, 525–532. [Google Scholar]
- Torbergsen, A.C.; Collins, A.R. Recovery of human lymphocytes from oxidative DNA damage; the apparent enhancement of DNA repair by carotenoids is probably simply an antioxidant effect. Eur. J. Nutr 2000, 39, 80–85. [Google Scholar]
- Dezor, M.; Dorszewska, J.; Florczak, J.; Kempisty, B.; Jaroszewska-Kolecka, J.; Rozycka, A.; Polrolniczak, A.; Bugaj, R.; Jagodzinski, P.P.; Kozubski, W. Expression of 8-oxoguanine DNA glycosylase 1 (OGG1) and the level of p53 and TNF-alphalpha proteins in peripheral lymphocytes of patients with Alzheimer’s disease. Folia Neuropathol 2011, 49, 123–131. [Google Scholar]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol 2001, 60, 759–767. [Google Scholar]
- Dorszewska, J.; Florczak, J.; Rozycka, A.; Jaroszewska-Kolecka, J.; Trzeciak, W.H.; Kozubski, W. Polymorphisms of the CHRNA4 gene encoding the alpha4 subunit of nicotinic acetylcholine receptor as related to the oxidative DNA damage and the level of apoptotic proteins in lymphocytes of the patients with Alzheimer’s disease. DNA Cell Biol 2005, 24, 786–794. [Google Scholar]
- Dorszewska, J.; Florczak, J.; Rozycka, A.; Kempisty, B.; Jaroszewska-Kolecka, J.; Chojnacka, K.; Trzeciak, W.H.; Kozubski, W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson’s diseases. Acta Neurobiol. Exp 2007, 67, 113–129. [Google Scholar]
- Dorszewska, J.; Kempisty, B.; Jaroszewska-Kolecka, J.; Rozycka, A.; Florczak, J.; Lianeri, M.; Jagodzinski, P.P.; Kozubski, W. Expression and polymorphisms of gene 8-oxoguanine glycosylase 1 and the level of oxidative DNA damage in peripheral blood lymphocytes of patients with Alzheimer’s disease. DNA Cell Biol 2009, 28, 579–588. [Google Scholar]
- Dumont, M.; Beal, M.F. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic. Biol. Med 2011, 51, 1014–1026. [Google Scholar]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem 1998, 71, 2034–2040. [Google Scholar]
- Morocz, M.; Kalman, J.; Juhasz, A.; Sinko, I.; McGlynn, A.P.; Downes, C.S.; Janka, Z.; Rasko, I. Elevated levels of oxidative DNA damage in lymphocytes from patients with Alzheimer’s disease. Neurobiol. Ag 2002, 23, 47–53. [Google Scholar]
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alzheimer’s Disease. Biochim. Biophys. Acta 2000, 1502, 139–144. [Google Scholar]
- Iida, T.; Furuta, A.; Nishioka, K.; Nakabeppu, Y.; Iwaki, T. Expression of 8-oxoguanine DNA glycosylase is reduced and associated with neurofibrillary tangles in Alzheimer’s disease brain. Acta Neuropathol 2002, 103, 20–25. [Google Scholar]
- Bialopiotrowicz, E.; Kuzniewska, B.; Kachamakova-Trojanowska, N.; Barcikowska, M.; Kuznicki, J.; Wojda, U. Cell cycle regulation distinguishes lymphocytes from sporadic and familial Alzheimer’s disease patients. Neurobiol. Ag 2011, 32, 2319, e13–2319.e26.. [Google Scholar]
- Stieler, J.T.; Lederer, C.; Bruckner, M.K.; Wolf, H.; Holzer, M.; Gertz, H.J.; Arendt, T. Impairment of mitogenic activation of peripheral blood lymphocytes in Alzheimer’s disease. Neuroreport 2001, 12, 3969–3972. [Google Scholar]
- Horvathova, E.; Slamenova, D.; Hlincikova, L.; Mandal, T.K.; Gabelova, A.; Collins, A.R. The nature and origin of DNA single-strand breaks determined with the comet assay. Mutat. Res 1998, 409, 163–171. [Google Scholar]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem 2005, 280, 11648–11655. [Google Scholar]
- Mecocci, P.; Polidori, M.C.; Cherubini, A.; Ingegni, T.; Mattioli, P.; Catani, M.; Rinaldi, P.; Cecchetti, R.; Stahl, W.; Senin, U. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch. Neurol 2002, 59, 794–798. [Google Scholar]
- Coppede, F.; Migliore, L. DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res 2009, 6, 36–47. [Google Scholar]
- Dizdaroglu, M.; Rao, G.; Halliwell, B.; Gajewski, E. Damage to the DNA bases in mammalian chromatin by hydrogen peroxide in the presence of ferric and cupric ions. Arch. Biochem. Biophys 1991, 285, 317–324. [Google Scholar]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 1988, 240, 640–642. [Google Scholar]
- Joenje, H. Genetic toxicology of oxygen. Mutat. Res 1989, 219, 193–208. [Google Scholar]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar]
- Degan, P.; Bonassi, S.; de Caterina, M.; Korkina, L.G.; Pinto, L.; Scopacasa, F.; Zatterale, A.; Calzone, R.; Pagano, G. In vivo accumulation of 8-hydroxy-2′-deoxyguanosine in DNA correlates with release of reactive oxygen species in Fanconi’s anaemia families. Carcinogenesis 1995, 16, 735–741. [Google Scholar]
- Futaki, M.; Igarashi, T.; Watanabe, S.; Kajigaya, S.; Tatsuguchi, A.; Wang, J.; Liu, J.M. The FANCG Fanconi anemia protein interacts with CYP2E1: Possible role in protection against oxidative DNA damage. Carcinogenesis 2002, 23, 67–72. [Google Scholar]
- Pagano, G.; Youssoufian, H. Fanconi anaemia proteins: Major roles in cell protection against oxidative damage. Bioessays 2003, 25, 589–595. [Google Scholar]
- Taniguchi, T.; Garcia-Higuera, I.; Xu, B.; Andreassen, P.R.; Gregory, R.C.; Kim, S.T.; Lane, W.S.; Kastan, M.B.; D’Andrea, A.D. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002, 109, 459–472. [Google Scholar]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 2004, 18, 1958–1963. [Google Scholar]
- Pichierri, P.; Averbeck, D.; Rosselli, F. DNA cross-link-dependent RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C protein. Hum. Mol. Genet 2002, 11, 2531–2546. [Google Scholar]
- Gu, Y.; Turck, C.W.; Morgan, D.O. Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature 1993, 366, 707–710. [Google Scholar]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 2010, 22, 1003–1012. [Google Scholar]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 1995, 9, 935–944. [Google Scholar]
- Nakabeppu, Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res 2001, 477, 59–70. [Google Scholar]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G—T and A—C substitutions. J. Biol. Chem 1992, 267, 166–172. [Google Scholar]
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355, 273–275. [Google Scholar]
- Sekiguchi, M. MutT-related error avoidance mechanism for DNA synthesis. Genes Cells 1996, 1, 139–145. [Google Scholar]
- Mo, J.Y.; Maki, H.; Sekiguchi, M. Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: Sanitization of nucleotide pool. Proc. Natl. Acad. Sci. USA 1992, 89, 11021–11025. [Google Scholar]
- Taddei, F.; Hayakawa, H.; Bouton, M.; Cirinesi, A.; Matic, I.; Sekiguchi, M.; Radman, M. Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 1997, 278, 128–130. [Google Scholar]
- Oda, H.; Nakabeppu, Y.; Furuichi, M.; Sekiguchi, M. Regulation of expression of the human MTH1 gene encoding 8-oxo-dGTPase. Alternative splicing of transcription products. J. Biol. Chem 1997, 272, 17843–17850. [Google Scholar]
- Song, X.N.; Zhang, L.Q.; Liu, D.G.; Lin, J.; Zheng, J.D.; Dai, D.P.; Hei, A.L.; Hayakawa, H.; Sekiguchi, M.; Cai, J.P. Oxidative Damage to RNA and Expression Patterns of MTH1 in the Hippocampi of Senescence-Accelerated SAMP8 Mice and Alzheimer’s Disease Patients. Neurochem. Res 2011, 36, 1558–1565. [Google Scholar]
- Liu, Y.; Kao, H.I.; Bambara, R.A. Flap endonuclease 1: A central component of DNA metabolism. Annu. Rev. Biochem 2004, 73, 589–615. [Google Scholar]
- Tsutakawa, S.E.; Classen, S.; Chapados, B.R.; Arvai, A.S.; Finger, L.D.; Guenther, G.; Tomlinson, C.G.; Thompson, P.; Sarker, A.H.; Shen, B.; et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 2011, 145, 198–211. [Google Scholar]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J 2009, 28, 3067–3073. [Google Scholar]
- Subba Rao, K. Mechanisms of disease: DNA repair defects and neurological disease. Nat. Clin. Pract. Neurol 2007, 3, 162–172. [Google Scholar]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 1999, 13, 152–157. [Google Scholar]
- Bieging, K.T.; Attardi, L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol 2012, 22, 97–106. [Google Scholar]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar]
- Efeyan, A.; Garcia-Cao, I.; Herranz, D.; Velasco-Miguel, S.; Serrano, M. Tumour biology: Policing of oncogene activity by p53. Nature 2006, 443, 159. [Google Scholar]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar]
- Eckert, A.; Oster, M.; Zerfass, R.; Hennerici, M.; Muller, W.E. Elevated levels of fragmented DNA nucleosomes in native and activated lymphocytes indicate an enhanced sensitivity to apoptosis in sporadic Alzheimer’s disease. Specific differences to vascular dementia. Dement Geriatr. Cogn. Disord 2001, 12, 98–105. [Google Scholar]
- Culmsee, C.; Mattson, M.P. p53 in neuronal apoptosis. Biochem. Biophys. Res. Commun 2005, 331, 761–777. [Google Scholar]
- Uberti, D.; Lanni, C.; Racchi, M.; Govoni, S.; Memo, M. Conformationally altered p53: A putative peripheral marker for Alzheimer’s disease. Neurodegener Dis 2008, 5, 209–211. [Google Scholar]
- Fridovich, I. Superoxide dismutases. Annu. Rev. Biochem 1975, 44, 147–159. [Google Scholar]
- De las Cuevas, N.; Munoz, U.; Hermida, O.G.; Martin-Requero, A. Altered transcriptional regulators in response to serum in immortalized lymphocytes from Alzheimer’s disease patients. Neurobiol. Ag 2005, 26, 615–624. [Google Scholar]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000; p. 943. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. Alzheimers Dement 2011, 7, 263–269.
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res 1988, 175, 184–191. [Google Scholar]
- Collins, A.; Dusinska, M.; Franklin, M.; Somorovska, M.; Petrovska, H.; Duthie, S.; Fillion, L.; Panayiotidis, M.; Raslova, K.; Vaughan, N. Comet assay in human biomonitoring studies: Reliability, validation, and applications. Environ. Mol. Mutagen 1997, 30, 139–146. [Google Scholar]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002, 3, RESEARCH0034. [Google Scholar]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 2007, 35, W71–W74. [Google Scholar]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 2002, 30, e36. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection time (h) | Tail intensity (Alkaline comet assay) | |||
|---|---|---|---|---|
| AD | EC | |||
| Control | H2O2 | Control | H2O2 | |
| 0 | 8.96 (±4.57) | 38.40 (±12.35) | 22.73 (±11.73) | 41.99 (±7.37) |
| 0.5 | 9.14 (±4.13) | 39.99 (±18.04) | 28.97 (±9.60) | 37.04 (±17.26) |
| 2 | 9.08 (±4.79) | 31.08 (±15.07) | 21.30 (±7.88) | 32.30 (±10.44) |
| 6 | 7.74 (±4.54) | 20.93 (±13.16) | 21.70 (±9.09) | 25.08 (±14.32) |
| Collection time (h) | Tail intensity (Alkaline comet assay-hOGG1) | |||
| AD | EC | |||
| Control | H2O2 | Control | H2O2 | |
| 0 | 17.32 (±10.50) | 44.47 (±10.35) | 24.10 (±11.12) | 46.72 (±18.98) |
| 0.5 | 13.47 (±5.94) | 44.31 (±11.81) | 28.64 (±7.88) | 51.65 (± 20.33) |
| 2 | 12.98 (±7.08) | 43.90 (±11.89) | 34.65 (±21.20) | 53.26 (±21.99) |
| 6 | 15.70 (±8.19) | 31.59 (±11.57) | 27.85 (±17.29) | 46.84 (±20.66) |
| AD | ||||
|---|---|---|---|---|
| Sample | Age | MMSE | CDR | GENDER |
| AD 01 | 79 | 3 | 2 | M |
| AD 02 | 83 | 3 | 3 | M |
| AD 03 | 77 | 18 | 1 | F |
| AD 04 | 90 | 14 | 1 | F |
| AD 05 | 81 | 2 | 3 | F |
| AD 06 | 79 | 23 | – | F |
| AD 07 | 72 | 17 | 1 | M |
| AD 08 | 76 | 9 | 3 | F |
| AD 09 | 86 | 20 | 2 | F |
| AD 10 | 72 | 14 | 2 | F |
| AD 11 | 80 | 15 | 2 | F |
| AD 12 | 84 | – | 1 | F |
| AD 13 | 80 | 14 | 1 | F |
| EC | ||||
| Sample | Age | MMSE | CDR | GENDER |
| EC 01 | 86 | – | – | F |
| EC 02 | 69 | – | – | F |
| EC 03 | 72 | – | – | M |
| EC 04 | 70 | – | – | F |
| EC 05 | 72 | – | – | F |
| EC 06 | 69 | – | – | F |
| EC 07 | 76 | – | – | F |
| EC 08 | 74 | – | – | F |
| EC 09 | 78 | – | – | F |
| EC 10 | 70 | – | – | F |
| EC 11 | 74 | – | – | F |
| EC 12 | 68 | – | – | F |
| EC 13 | 83 | – | – | F |
| EC 14 | 74 | – | – | M |
| Primers | Sequence | Product size (pb) |
|---|---|---|
| ATM–forward | 5′–GACGTTACATGAGCCAGCAA–3′ | 100 |
| ATM–reverse | 5′–CACATGCGATGGAAAATGAG–3′ | |
| ATR–forward | 5′–GTGAGTGGAAGCCATGAGG–3′ | 109 |
| ATR–reverse | 5′–ACAAATGACAGGAGGGAGTTG–3′ | |
| FEN1–forward | 5′–ATTCCCATGGCAACACAGAG–3′ | 112 |
| FEN1–reverse | 5′–AGGGAGAGCGAGCTTAGGAC–3′ | |
| MTH1–forward | 5′–CGTGGAGAGCGACGAAAT–3′ | 103 |
| MTH1–reverse | 5′–CTGAAGCAGGAGTGGAAACC–3′ | |
| P21–forward | 5′–CTTCCTGTGGGCGGATTAG–3′ | 105 |
| P21–reverse | 5′–GACTCTCAGGGTCGAAAACG–3′ | |
| FANCG–forward | 5′–GACAGCAGTTGGCTCAGGAT–3′ | 102 |
| FANCG–reverse | 5′–CAGTCAGCTCCAAGGGAAGA–3′ | |
| B2M–forward | 5′–AGGCTATCCAGCGTACTCCA–3′ | 112 |
| B2M–reverse | 5′–TCAATGTCGGATGGATGAAA–3′ | |
| GUSB–forward | 5′–CACCAGGATCCACCTCTGAT–3′ | 115 |
| GUSB–reverse | 5′–TCCAAATGAGCTCTCCAACC–3′ | |
| HPRT1–forward | 5′–TCATTATGCTGAGGATTTGGA–3′ | 104 |
| HPRT1–reverse | 5′–GATGGCCTCCCATCTCCTT–3′ | |
| TBP–forward | 5′–AGGAGCCAAGAGTGAAGAACAG–3′ | 117 |
| TBP–reverse | 5′–CTCCCCACCATGTTCTGAAT–3′ |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leandro, G.S.; Lobo, R.R.; Oliveira, D.V.N.P.; Moriguti, J.C.; Sakamoto-Hojo, E.T. Lymphocytes of Patients with Alzheimer’s Disease Display Different DNA Damage Repair Kinetics and Expression Profiles of DNA Repair and Stress Response Genes. Int. J. Mol. Sci. 2013, 14, 12380-12400. https://doi.org/10.3390/ijms140612380
Leandro GS, Lobo RR, Oliveira DVNP, Moriguti JC, Sakamoto-Hojo ET. Lymphocytes of Patients with Alzheimer’s Disease Display Different DNA Damage Repair Kinetics and Expression Profiles of DNA Repair and Stress Response Genes. International Journal of Molecular Sciences. 2013; 14(6):12380-12400. https://doi.org/10.3390/ijms140612380
Chicago/Turabian StyleLeandro, Giovana S., Romulo R. Lobo, Douglas V. N. P. Oliveira, Julio C. Moriguti, and Elza T. Sakamoto-Hojo. 2013. "Lymphocytes of Patients with Alzheimer’s Disease Display Different DNA Damage Repair Kinetics and Expression Profiles of DNA Repair and Stress Response Genes" International Journal of Molecular Sciences 14, no. 6: 12380-12400. https://doi.org/10.3390/ijms140612380
APA StyleLeandro, G. S., Lobo, R. R., Oliveira, D. V. N. P., Moriguti, J. C., & Sakamoto-Hojo, E. T. (2013). Lymphocytes of Patients with Alzheimer’s Disease Display Different DNA Damage Repair Kinetics and Expression Profiles of DNA Repair and Stress Response Genes. International Journal of Molecular Sciences, 14(6), 12380-12400. https://doi.org/10.3390/ijms140612380