Candidate Biomarkers for Genetic and Clinicopathological Diagnosis of Endometrial Cancer


Abstract
:1. Introduction
2. Genetic Abnormalities in Endometrial Cancer
3. Epigenetic Aberrant Methylation in Endometrial Cancer
4. microRNA Abnormalities in Endometrial Cancer
5. Diagnosis of Endometrial Cancer as a Familial Tumor
6. Biomarkers in Endometrial Cancer
7. Conclusions
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin 2010, 60, 277–300. [Google Scholar]
- Ushijima, K. Current status of gynecologic cancer in Japan. J. Gynecol. Oncol 2009, 20, 67–71. [Google Scholar]
- Honda, T.; Urabe, R.; Kurita, T.; Kagami, S.; Kawagoe, T.; Toki, N.; Matsuura, Y.; Hachisuga, T. Trends in the demographic and clinicopathological characteristics in Japanese patients with endometrial cancer, 1990–2010. Int. J. Womens Health 2012, 4, 207–212. [Google Scholar]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol 1983, 15, 10–17. [Google Scholar]
- Deligdisch, L.; Cohen, C.J. Histologic correlates and virulence implications of endometrial carcinoma associated with adenomatous hyperplasia. Cancer 1985, 56, 1452–1455. [Google Scholar]
- Oehler, M.K.; Brand, A.; Wain, G.V. Molecular genetics and endometrial cancer. J. Br. Menopause Soc 2003, 9, 27–31. [Google Scholar]
- Bansal, N.; Yendluri, V.; Wenham, R.M. The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control 2009, 1, 8–13. [Google Scholar]
- Gadducci, A.; Cosio, S.; Genazzani, A.R. Tissue and serum biomarkers as prognostic variables in endometrioid-type endometrial cancer. Crit. Rev. Oncol. Hematol 2011, 80, 181–192. [Google Scholar]
- Doll, A.; Abal, M.; Rigau, M.; Monge, M.; Gonzalez, M.; Demajo, S.; Colás, E.; Llauradó, M.; Alazzouzi, H.; Planagumá, J.; et al. Novel molecular profiles of endometrial cancer-new light through old windows. J. Steroid Biochem. Mol. Biol 2008, 108, 221–229. [Google Scholar]
- Esteller, M. Dormant hypermethylated tumour suppressor genes: Questions and answers. J. Pathol 2005, 205, 172–180. [Google Scholar]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol 2005, 23, 3971–3993. [Google Scholar]
- Krusche, C.A.; Vloet, A.J.; Classen-Linke, I.; von Rango, U.; Beier, H.M.; Alfer, J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum. Reprod 2007, 22, 2956–2966. [Google Scholar]
- Weichert, W.; Denkert, C.; Noske, A.; Darb-Esfahani, S.; Dietel, M.; Kalloger, S.E.; Huntsam, D.G.; Köbel, M. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 2008, 10, 1021–1027. [Google Scholar]
- Esteller, M.; Levine, R.; Baylin, S.B.; Ellenson, L.H.; Herman, J.G. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 1998, 17, 2413–2417. [Google Scholar]
- Hecht, J.L.; Mutter, G.L. Molecular and pathologic aspects of endometrial carcinogenesis. J. Clin. Oncol 2006, 24, 4783–4791. [Google Scholar]
- Bilbao, C.; Lara, P.C.; Ramírez, R.; Henríquez-Hernández, L.A.; Rodríguez, G.; Falcón, O.; León, L.; Perucho, M.; Díaz-Chico, B.N.; Díaz-Chico, J.C. Microsatellite instability predicts clinical outcome in radiation-treated endometrioid endometrial cancer. Int. J. Radiat. Oncol. Biol. Phys 2010, 76, 9–13. [Google Scholar]
- Costello, J.F.; Plass, C. Methylation matters. J. Med. Genet 2001, 38, 285–303. [Google Scholar]
- Di Domenico, M.; Santoro, A.; Ricciardi, C.; Iaccarino, M.; Iaccarino, S.; Freda, M.; Feola, A.; Sanguedolce, F.; Losito, S.; Pasquali, D.; et al. Epigenetic fingerprint in endometrial carcinogenesis: The hypothesis of a uterine field cancerization. Cancer Biol. Ther 2011, 12, 447–457. [Google Scholar]
- Scolnick, D.M.; Halazonetis, T.D. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 2000, 27, 430–435. [Google Scholar]
- Chin, C.F.; Yeong, F.M. Safeguarding entry into mitosis the antephase checkpoint. Mol. Cell Biol 2010, 30, 22–23. [Google Scholar]
- Yanokura, M.; Banno, K.; Kawaguchi, M.; Hirao, N.; Hirasawa, A.; Susumu, N.; Tsukazaki, K.; Aoki, D. Relationship of aberrant DNA hypermethylation of CHFR with sensitivity to taxanes in endometrial cancer. Oncol. Rep 2007, 17, 41–48. [Google Scholar]
- Bogen, K.T. Efficient tumorigenesis by mutation-induced failure to terminate microRNA-mediated adaptive hyperplasia. Med. Hypotheses 2013, 80, 83–90. [Google Scholar]
- Zhi, F.; Wang, S.; Wang, R.; Xia, X.; Yang, Y. From small to big: microRNAs as new players in medulloblastomas. Tumor Biol 2013, 34, 9–15. [Google Scholar]
- Yanokura, M.; Banno, K.; Kobayashi, Y.; Kisu, I.; Ueki, A.; Ono, A.; Masuda, K.; Nomura, H.; Hirasawa, A.; Susumu, N.; et al. MicroRNA and endometrial cancer: Roles of small RNAs in human tumors and clinical applications. Oncol. Lett 2010, 1, 935–940. [Google Scholar]
- Devor, E.J.; Hovey, A.M.; Goodheart, M.J.; Ramachandran, S.; Leslie, K.K. microRNA expression profiling of endometrial endometrioid adenocarcinomas and serous adenocarcinomas reveals profiles containing shared, unique and differentiating groups of microRNAs. Oncol. Rep 2011, 26, 995–1002. [Google Scholar]
- Snowdon, J.; Zhang, X.; Childs, T.; Tron, V.A.; Feilotter, H. The microRNA-200 family is upregulated in endometrial carcinoma. PLoS One 2011, 6, e22828. [Google Scholar]
- Lee, J.W.; Park, Y.A.; Choi, J.J.; Lee, Y.Y.; Kim, C.J.; Choi, C.; Kim, T.J.; Lee, N.W.; Kim, B.G.; Bae, D.S. The expression of the miRNA-200 family in endometrial endometrioid carcinoma. Gynecol. Oncol 2011, 120, 56–62. [Google Scholar]
- Torres, A.; Torres, K.; Pesci, A.; Ceccaroni, M.; Paszkowski, T.; Cassandrini, P.; Zamboni, G.; Maciejewski, R. Diagnostic and prognostic significance of miRNA signatures in tissues and plasma of endometrioid endometrial carcinoma patients. Int. J. Cancer 2013, 132, 1633–1645. [Google Scholar]
- Ramón, L.A.; Braza-Boïls, A.; Gilabert, J.; Chirivella, M.; España, F.; Estellés, A.; Gilabert-Estellés, J. microRNAs related to angiogenesis are dysregulated in endometrioid endometrial cancer. Hum. Reprod 2012, 27, 3036–3045. [Google Scholar]
- Hiroki, E.; Suzuki, F.; Akahira, J.; Nagase, S.; Ito, K.; Sugawara, J.; Miki, Y.; Suzuki, T.; Sasano, H.; Yaegashi, N. MicroRNA-34b functions as a potential tumor suppressor in endometrial serous adenocarcinoma. Int J. Cancer 2012, 131, E395–404. [Google Scholar]
- Tsuruta, T.; Kozaki, K.; Uesugi, A.; Furuta, M.; Hirasawa, A.; Imoto, I.; Susumu, N.; Aoki, D.; Inazawa, J. miR-152 is a tumor suppressor microRNA that is silenced by DNA hypermethylation in endometrial cancer. Cancer Res 2011, 71, 6450–6462. [Google Scholar]
- Ketabi, Z.; Mosgaard, B.J.; Gerdes, A.M.; Ladelund, S.; Bernstein, I.T. Awareness of endometrial cancer risk and compliance with screening in hereditary nonpolyposis colorectal cancer. Obstet. Gynecol 2012, 120, 1005–1012. [Google Scholar]
- Kalady, M.F.; Lipman, J.; McGannon, E.; Church, J.M. Risk of colonic neoplasia after proctectomy for rectal cancer in hereditary nonpolyposis colorectal cancer. Ann. Surg 2012, 255, 1121–1125. [Google Scholar]
- Banno, K.; Yanokura, M.; Kobayashi, Y.; Kawaguchi, M.; Nomura, H.; Hirasawa, A.; Susumu, N.; Aoki, D. Endometrial cancer as a familial tumor: Pathology and molecular carcinogenesis (review). Curr. Genomics 2009, 10, 127–132. [Google Scholar]
- Banno, K.; Susumu, N.; Hirao, T.; Yanokura, M.; Hirasawa, A.; Aoki, D.; Udagawa, Y.; Sugano, K.; Nozawa, S. Two Japanese kindreds occurring endometrial cancer meeting new clinical criteria for hereditary non-polyposis colorectal cancer (HNPCC), Amsterdam Criteria II. J. Obstet. Gynaecol. Res 2004, 30, 287–292. [Google Scholar]
- Hirai, Y.; Banno, K.; Suzuki, M.; Ichikawa, Y.; Udagawa, Y.; Sugano, K.; Miki, Y. Molecular epidemiological and mutational analysis of DNA mismatch repair (MMR) genes in endometrial cancer patients with HNPCC-associated familial predisposition to cancer. Cancer Sci 2008, 99, 1715–1719. [Google Scholar]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and cancer: A new carcinogenic mechanism of Lynch syndrome. Int. J. Oncol 2012, 41, 793–797. [Google Scholar]
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 2002, 62, 3925–3928. [Google Scholar]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet 2009, 41, 12–17. [Google Scholar]
- Nelen, M.R.; Kremer, H.; Konings, I.B.; Schoute, F.; van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN mutations in patients with Cowden disease: Absence of clear genotype-phenotype correlations. Eur. J. Hum. Genet 1999, 7, 267–273. [Google Scholar]
- Nelen, M.R.; Padberg, G.W.; Peeters, E.A.; Lin, A.Y.; van den Helm, B.; Frants, R.R.; Coulon, V.; Goldstein, A.M.; van Reen, M.M.; Easton, D.F.; et al. Localization of the gene for Cowden disease to chromosome 10q22–23. Nat. Genet 1996, 13, 114–116. [Google Scholar]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet 1997, 16, 64–67. [Google Scholar]
- Salem, O.S.; Steck, W.D. Cowden’s disease (multiple hamartoma and neoplasia syndrome). A case report and review of the English literature. J. Am. Acad. Dermatol 1983, 8, 686–696. [Google Scholar]
- Higuchi, M.; Onishi, K.; Kikuchi, C.; Gotoh, Y. Scaffolding function of PAK in the PDK1-Akt pathway. Nat. Cell. Biol 2008, 10, 1356–1364. [Google Scholar]
- Beggs, A.D.; Latchford, A.R.; Vasen, H.F.; Moslein, G.; Alonso, A.; Aretz, S.; Bertario, L.; Blanco, I.; Bulow, S.; Burn, J.; et al. Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut 2010, 59, 975–986. [Google Scholar]
- Scully, R.E. Sex cord tumor with annular tubules a distinctive ovarian tumor of the Peutz-Jeghers syndrome. Cancer 1970, 25, 1107–1121. [Google Scholar]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz-Correa, M.; Offerhaus, J.A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Biomarkers in endometrial cancer: Possible clinical applications. Oncol. Lett 2012, 3, 1175–1180. [Google Scholar]
- Lee, E.J.; Kim, T.J.; Kim, D.S.; Choi, C.H.; Lee, J.W.; Lee, J.H.; Bae, D.S.; Kim, B.G. p53 alteration independently predicts poor outcomes in patients with endometrial cancer: A clinicopathologic study of 131 cases and literature review. Gynecol. Oncol 2010, 116, 533–538. [Google Scholar]
- Risinger, J.I.; Hayes, K.; Maxwell, G.L.; Carney, M.E.; Dodge, R.K.; Barrett, J.C.; Berchuck, A. PTEN mutation in endometrial cancers is associated with favorable clinical and pathologic characteristics. Clin. Cancer Res 1998, 4, 3005–3010. [Google Scholar]
- Mackay, H.J.; Gallinger, S.; Tsao, M.S.; McLachlin, C.M.; Tu, D.; Keiser, K.; Eisenhauer, E.A.; Oza, A.M. Prognostic value of microsatellite instability (MSI) and PTEN expression in women with endometrial cancer: Results from studies of the NCIC Clinical Trials Group (NCIC CTG). Eur. J. Cancer 2010, 46, 1365–1373. [Google Scholar]
- Saegusa, M.; Hashimura, M.; Yoshida, T.; Okayasu, I. Beta-catenin mutations and aberrant nuclear expression during endometrial tumorigenesis. Br. J. Cancer 2001, 84, 209–217. [Google Scholar]
- Mizuuchi, H.; Nasim, S.; Kudo, R.; Silverberg, S.G.; Greenhouse, S.; Garrett, C.T. Clinical implications of K-ras mutations in malignant epithelial tumors of the endometrium. Cancer Res 1992, 52, 2777–2781. [Google Scholar]
- Jo, H.; Kim, J.W.; Kang, G.H.; Park, N.H.; Song, Y.S.; Kang, S.B.; Lee, H.P. Association of promoter hypermethylation of the RASSF1A gene with prognostic parameters in endometrial cancer. Oncol. Res 2006, 16, 205–209. [Google Scholar]
- Mizumoto, Y.; Kyo, S.; Mori, N.; Sakaguchi, J.; Ohno, S.; Maida, Y.; Hashimoto, M.; Takakura, M.; Inoue, M. Activation of ERK1/2 occurs independently of KRAS or BRAF status in endometrial cancer and is associated with favorable prognosis. Cancer Sci 2007, 98, 652–658. [Google Scholar]
- Chen, C.A.; Cheng, W.F.; Lee, C.N.; Wei, L.H.; Chu, J.S.; Hsieh, F.J.; Hsieh, C.Y. Cytosol vascular endothelial growth factor in endometrial carcinoma: Correlation with disease-free survival. Gynecol. Oncol 2001, 80, 207–212. [Google Scholar]
- Zaino, R.J.; Davis, A.T.; Ohlsson-Wilhelm, B.M.; Brunetto, V.L. DNA content is an independent prognostic indicator in endometrial adenocarcinoma. A Gynecologic Oncology Group study. Int. J. Gynecol. Pathol 1998, 17, 312–319. [Google Scholar]
- Scambia, G.; Gadducci, A.; Panici, P.B.; Foti, E.; Ferdeghini, M.; Ferrandina, G.; Amoroso, M.; Castellani, C.; Facchini, V.; Mancuso, S. Combined use of CA 125 and CA 15–3 in patients with endometrial carcinoma. Gynecol. Oncol 1994, 54, 292–297. [Google Scholar]
- Sood, A.K.; Buller, R.E.; Burger, R.A.; Dawson, J.D.; Sorosky, J.I.; Berman, M. Value of preoperative CA 125 level in the management of uterine cancer and prediction of clinical outcome. Obstet. Gynecol 1997, 90, 441–447. [Google Scholar]
- Diefenbach, C.S.; Shah, Z.; Iasonos, A.; Barakat, R.R.; Levine, D.A.; Aghajanian, C.; Sabbatini, P.; Hensley, M.L.; Konner, J.; Tew, W.; et al. Preoperative serum YKL-40 is a marker for detection and prognosis of endometrial cancer. Gynecol. Oncol 2007, 104, 435–442. [Google Scholar]
- Moore, R.G.; Brown, A.K.; Miller, M.C.; Badgwell, D.; Lu, Z.; Allard, W.J.; Granai, C.O.; Bast, R.C., Jr; Lu, K. Utility of a novel serum tumor biomarker HE4 in patients with endometrioid adenocarcinoma of the uterus. Gynecol. Oncol 2008, 110, 196–201. [Google Scholar]
- Mutz-Dehbalaie, I.; Egle, D.; Fessler, S.; Hubalek, M.; Fiegl, H.; Marth, C.; Widschwendter, A. HE4 is an independent prognostic marker in endometrial cancer patients. Gynecol. Oncol 2012, 126, 186–191. [Google Scholar]
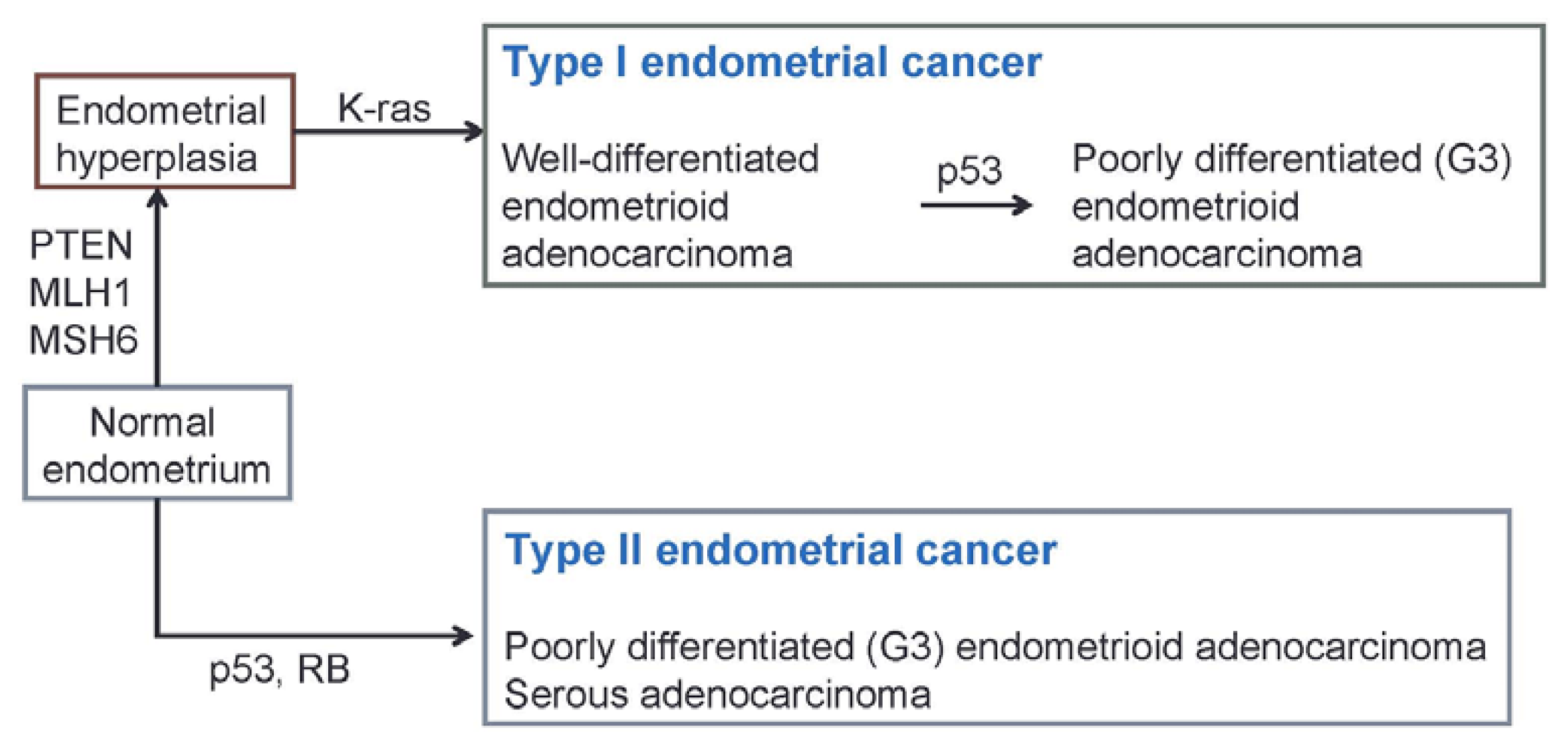

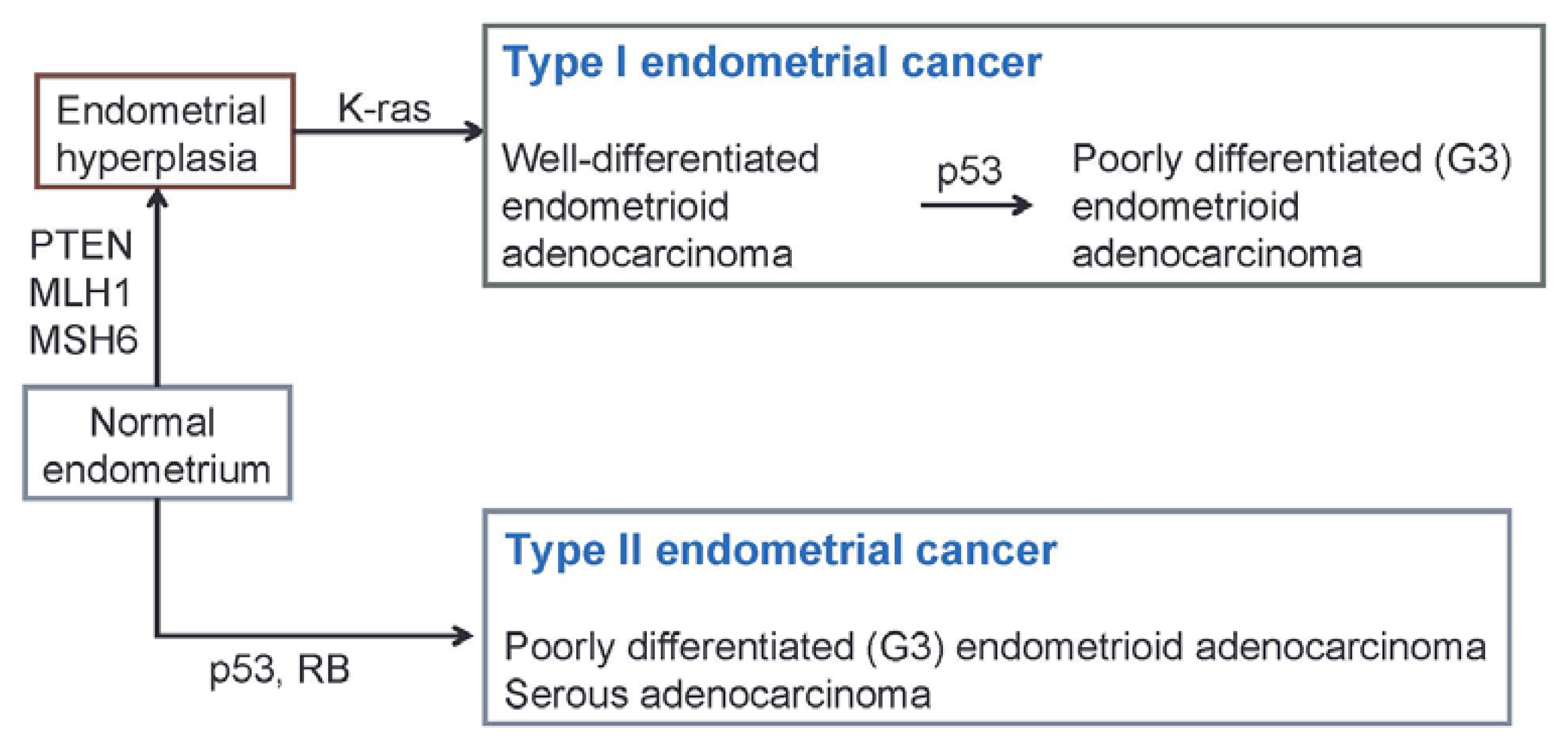{kind=link}
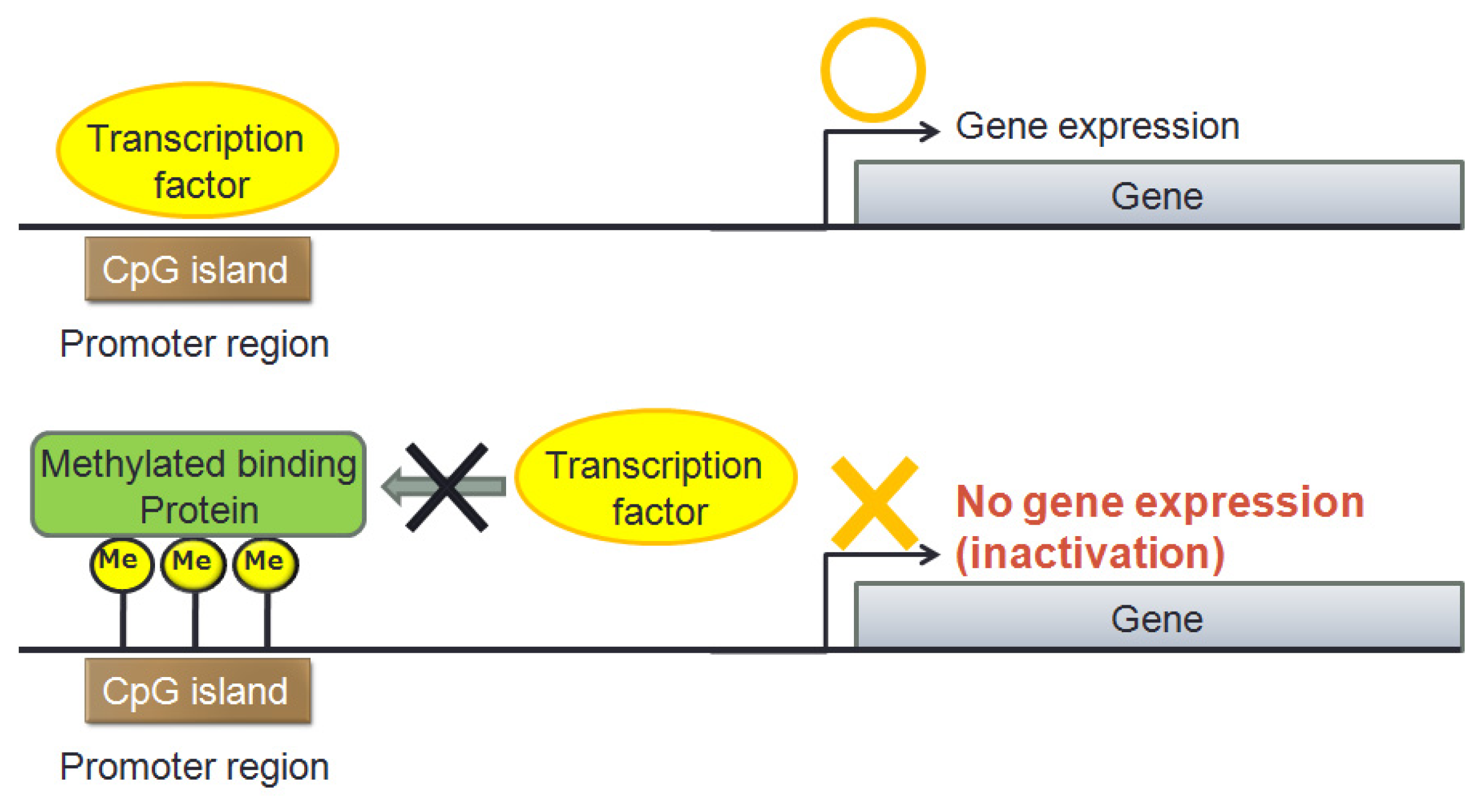
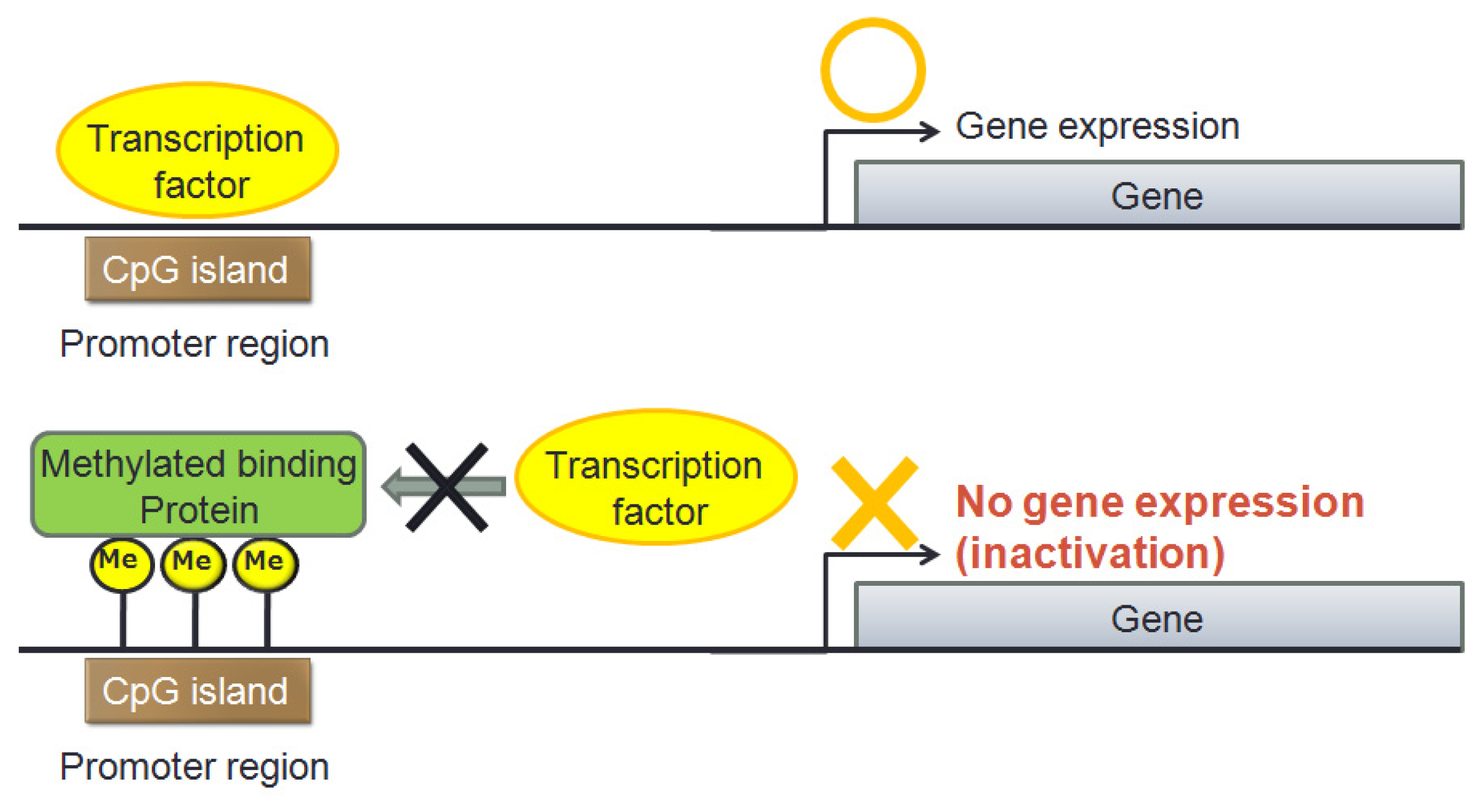{kind=link}
| Genetic alteration | Type 1 (%) | Type 2 (%) |
|---|---|---|
| PTEN inactivation | 50–80 | 10 |
| K-ras mutation | 15–30 | 0–5 |
| β-catenin mutation | 20–40 | 0–3 |
| Microsatellite instability | 20–40 | 0–5 |
| p53 mutation | 10–20 | 80–90 |
| HER-2/neu | 10–30 | 40–80 |
| p16 inactivation | 10 | 40 |
| E-cadherin | 10–20 | 60–90 |
| Upregulation | Downregulation |
|---|---|
| miR-200a | miR-410 |
| miR-200b | miR-15b |
| miR-200c | miR-17-5p |
| miR-429 | miR-20a |
| miR-203 | miR-34b |
| miR-205 | miR-152 |
| miR-210 | miR-125a |
| miR-214 | |
| miR-221 | |
| miR-222 | |
| miR-424 |
| Tissue biomarkers | Serum biomarkers |
|---|---|
| p53 | CA125 |
| PTEN-PIK3-mTOR signaling pathway (PTEN, PIK3, mTOR, 4E-BP1) | CA15-3 |
| MSI | YKL-40 |
| β-catenin | VEGF |
| Ras-MAPK-ERK signaling pathway (K-ras, RASSF1A, ERK) | HE-4 |
| VEGF | |
| DNA aneuploidy |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Banno, K.; Nogami, Y.; Kisu, I.; Yanokura, M.; Umene, K.; Masuda, K.; Kobayashi, Y.; Yamagami, W.; Susumu, N.; Aoki, D. Candidate Biomarkers for Genetic and Clinicopathological Diagnosis of Endometrial Cancer. Int. J. Mol. Sci. 2013, 14, 12123-12137. https://doi.org/10.3390/ijms140612123
Banno K, Nogami Y, Kisu I, Yanokura M, Umene K, Masuda K, Kobayashi Y, Yamagami W, Susumu N, Aoki D. Candidate Biomarkers for Genetic and Clinicopathological Diagnosis of Endometrial Cancer. International Journal of Molecular Sciences. 2013; 14(6):12123-12137. https://doi.org/10.3390/ijms140612123
Chicago/Turabian StyleBanno, Kouji, Yuya Nogami, Iori Kisu, Megumi Yanokura, Kiyoko Umene, Kenta Masuda, Yusuke Kobayashi, Wataru Yamagami, Nobuyuki Susumu, and Daisuke Aoki. 2013. "Candidate Biomarkers for Genetic and Clinicopathological Diagnosis of Endometrial Cancer" International Journal of Molecular Sciences 14, no. 6: 12123-12137. https://doi.org/10.3390/ijms140612123