Genome-Wide Investigation of Multifocal and Unifocal Prostate Cancer — Are They Genetically Different?

Abstract
:1. Introduction
2. Results and Discussion
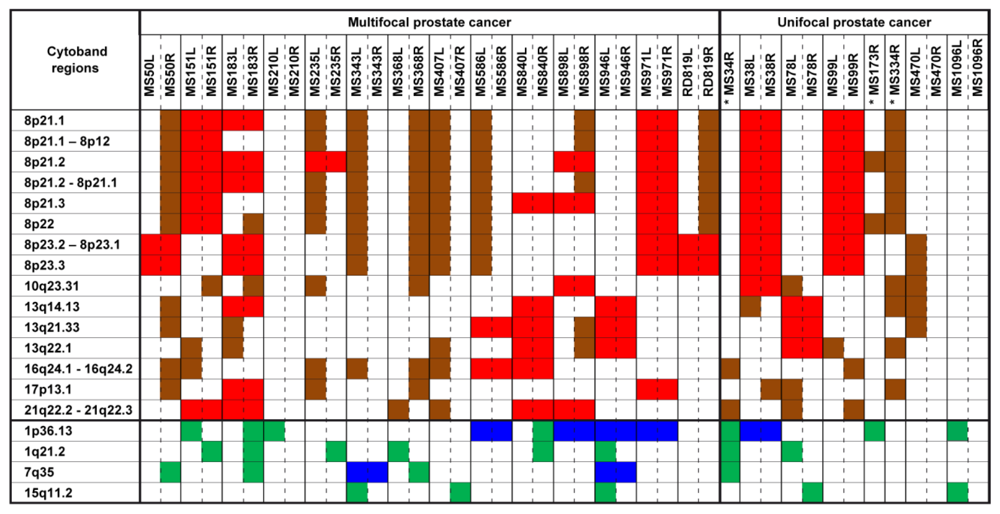
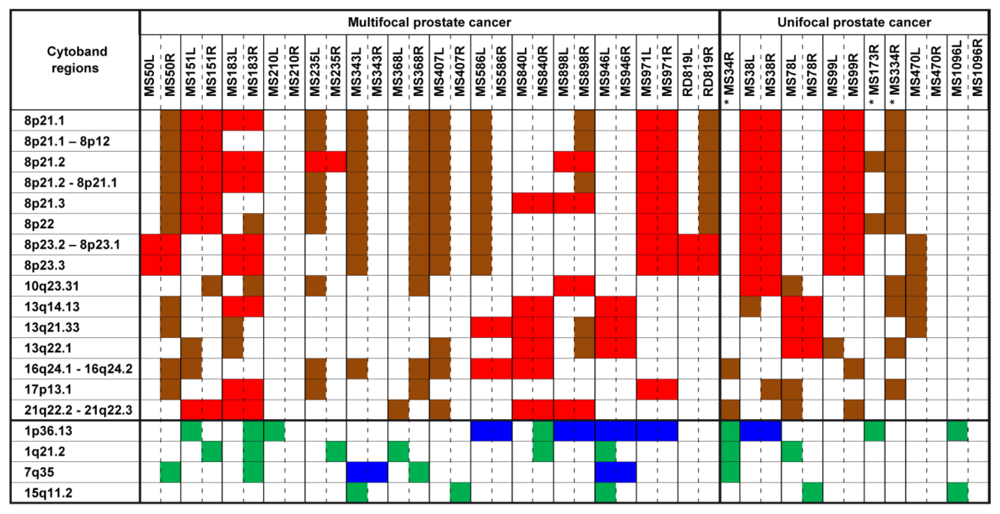
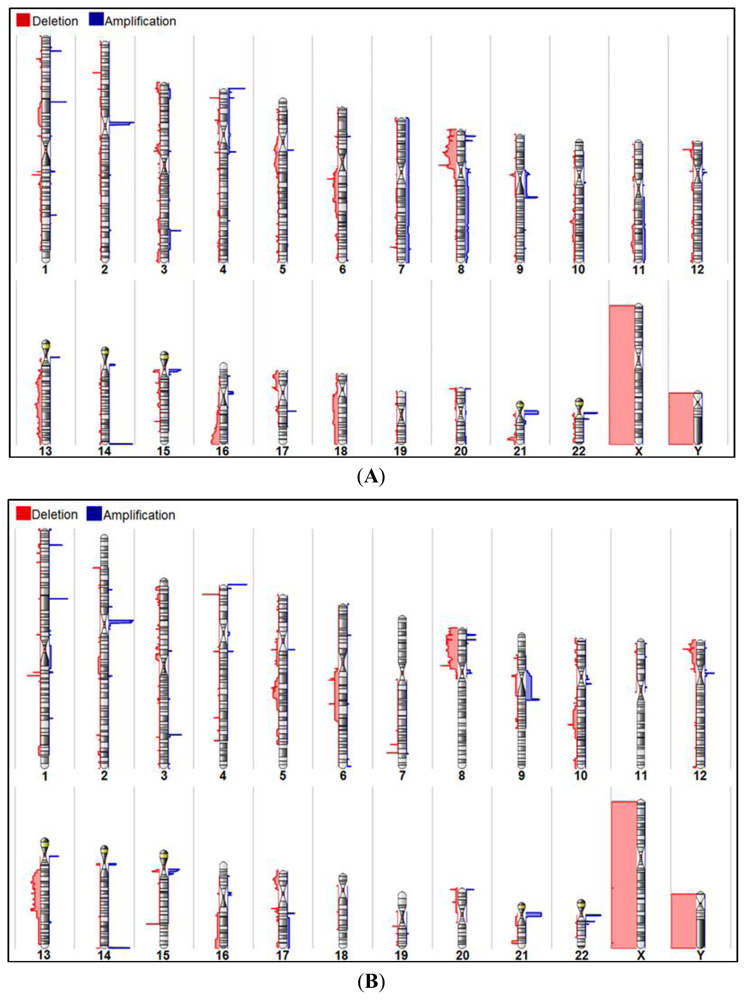
2.1. Chromosomal CNV Events
2.2. Comparison of Copy Number Profile Observed in Multifocal and Unifocal Prostate Cancers
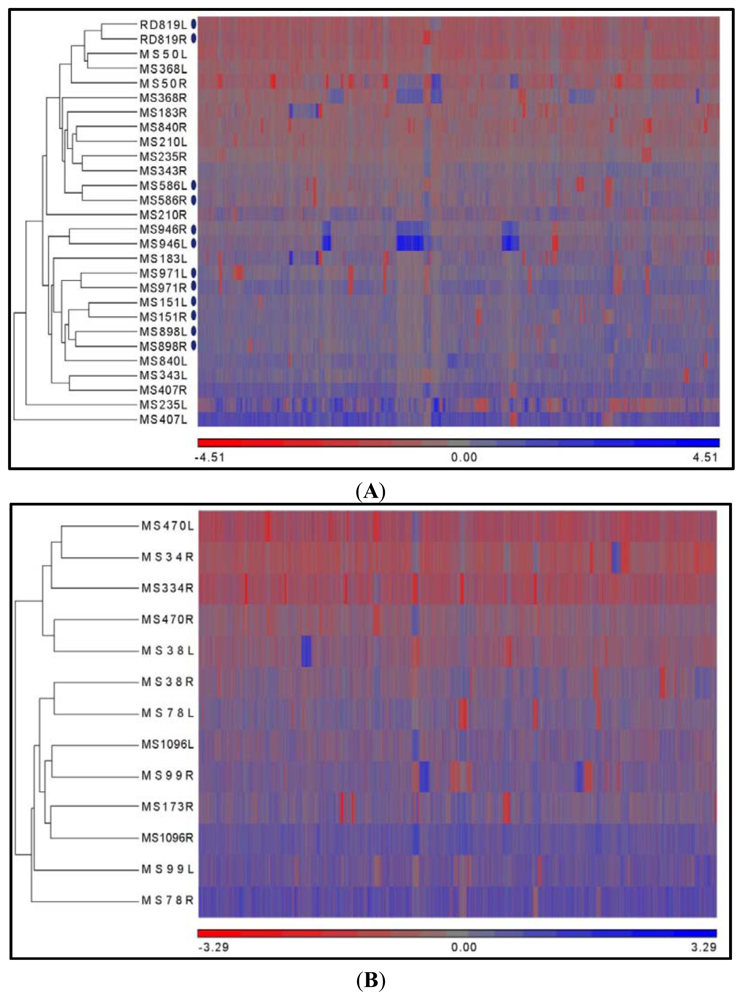
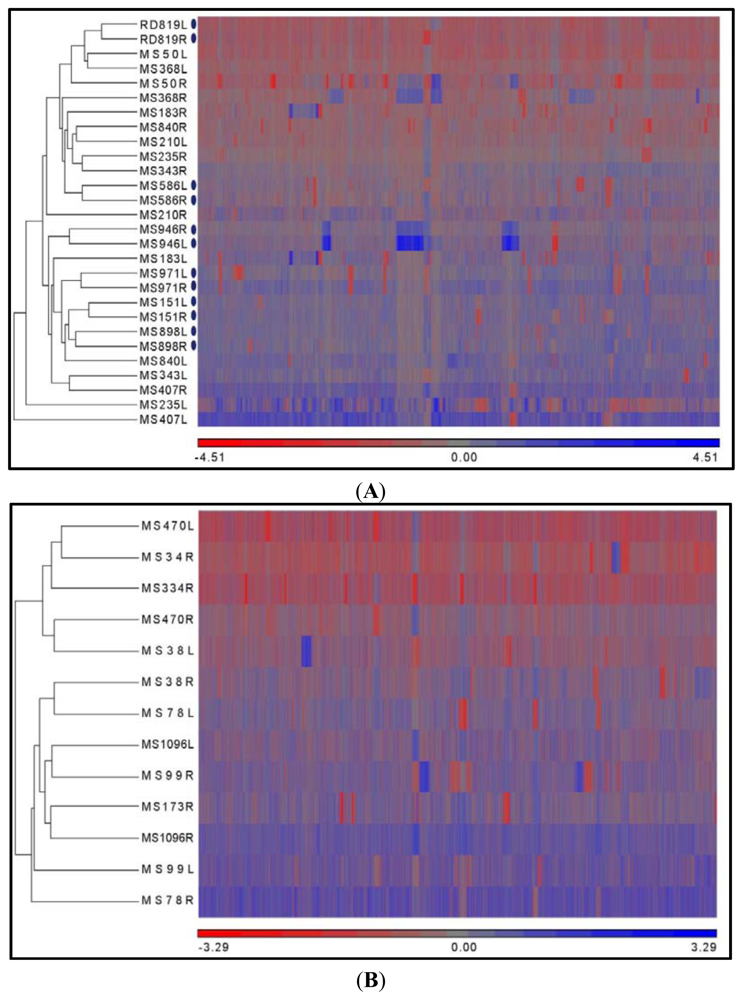
2.3. Hierarchical Clustering
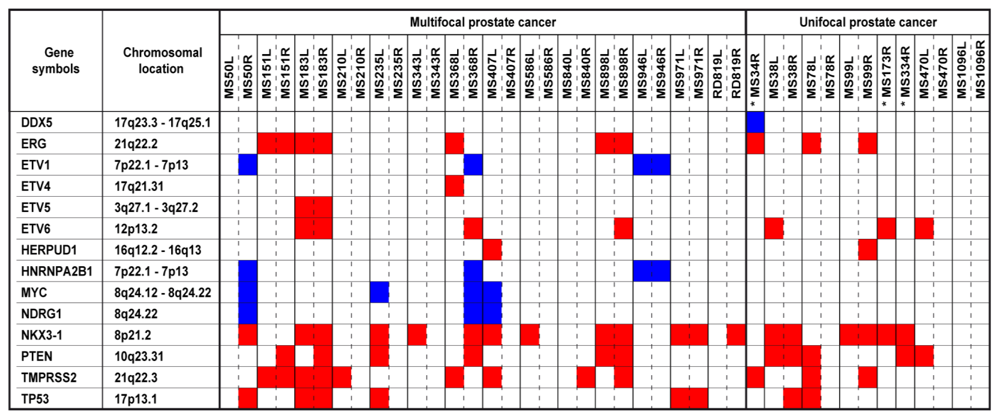
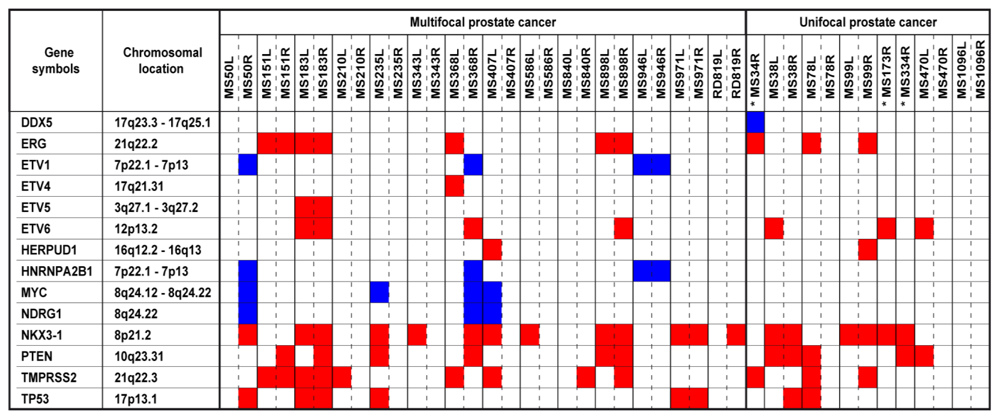
2.4. Copy Number Altered Genes Elucidate Prostate Cancer Heterogeneity
3. Experimental Section
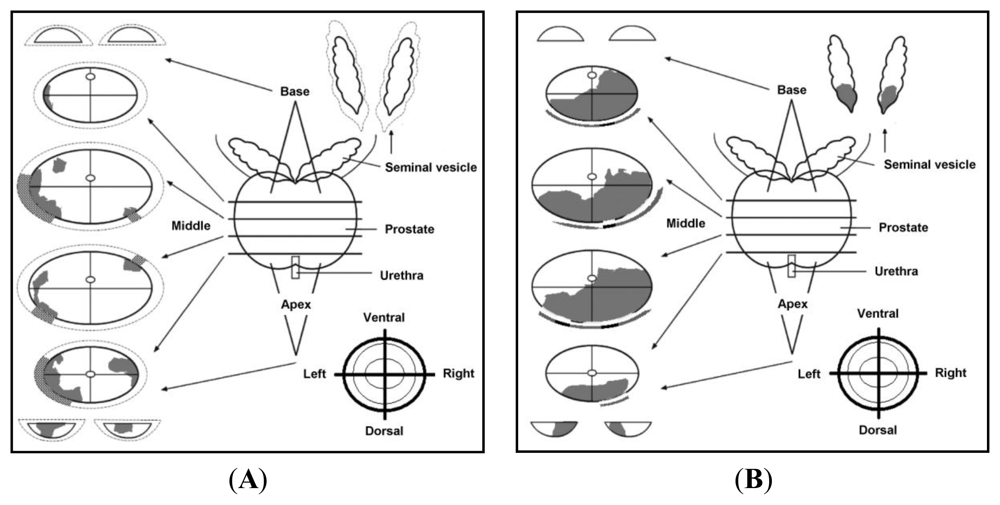
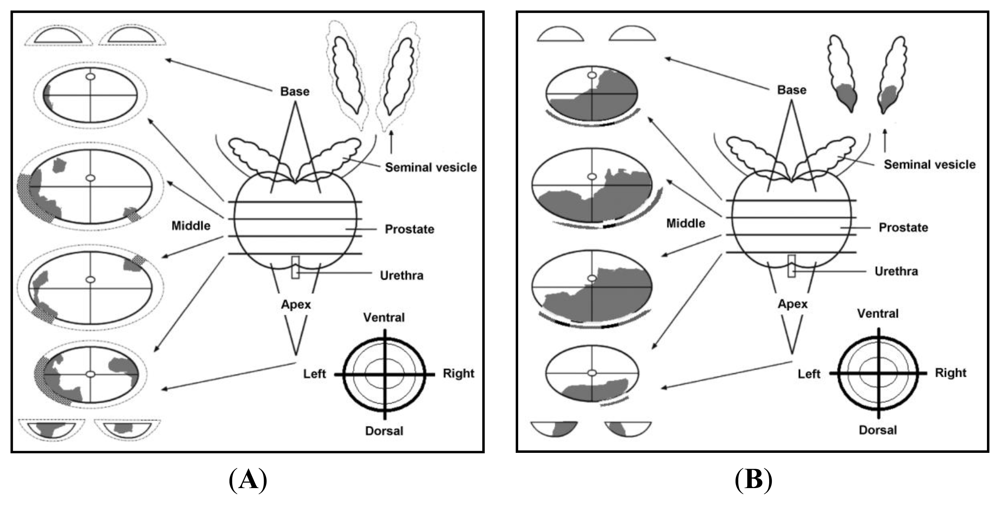
3.1. Sample Acquisition, Handling and Preparation
3.2. DNA Preparation and Quality Control
3.3. Genome-Wide Microarray Analysis
3.4. Data Analysis
4. Conclusions
Supplementary Information
ijms-14-11816-s001.pdfAcknowledgments
Conflict of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global Cancer Statistics. CA: Cancer J. Clin 2011, 61, 69–90. [Google Scholar]
- Schmidt, H.; DeAngelis, G.; Eltze, E.; Gockel, I.; Semjonow, A.; Brandt, B. Asynchronous Growth of Prostate Cancer is Reflected by Circulating Tumor Cells Delivered from Distinct, Even Small Foci, Harboring Loss of Heterozygosity of the PTEN Gene. Cancer Res 2006, 66, 8959–8965. [Google Scholar]
- Arora, R.; Koch, M.O.; Eble, J.N.; Ulbright, T.M.; Li, L.; Cheng, L. Heterogeneity of Gleason Grade in Multifocal Adenocarcinoma of the Prostate. Cancer 2004, 100, 2362–2366. [Google Scholar]
- Meiers, I.; Waters, D.J.; Bostwick, D.G. Preoperative Prediction of Multifocal Prostate Cancer and Application of Focal Therapy: Review 2007. Urology 2007, 70, 3–8. [Google Scholar]
- Djavan, B.; Susani, M.; Bursa, B.; Basharkhah, A.; Simak, R.; Marberger, M. Predictability and Significance of Multifocal Prostate Cancer in the Radical Prostatectomy Specimen. Tech. Urol 1999, 5, 139–142. [Google Scholar]
- Andreoiu, M.; Cheng, L. Multifocal Prostate Cancer: Biologic, Prognostic, and Therapeutic Implications. Hum. Pathol 2010, 41, 781–793. [Google Scholar]
- Ruijter, E.T.; van de Kaa, C.A.; Schalken, J.A.; Debruyne, F.M.; Ruiter, D.J. Histological Grade Heterogeneity in Multifocal Prostate Cancer. Biological and Clinical Implications. J. Pathol 1996, 180, 295–299. [Google Scholar]
- Eltze, E.; Schmidt, H.; Semjonow, A.; Brandt, B. Translating Genetic Pathways to Protein Networks for Cancer Sub-Typing. Ann. Oncol 2008, 19, 44–47. [Google Scholar]
- Torring, N.; Borre, M.; Sorensen, K.D.; Andersen, C.L.; Wiuf, C.; Orntoft, T.F. Genome-Wide Analysis of Allelic Imbalance in Prostate Cancer using the Affymetrix 50K SNP Mapping Array. Br. J. Cancer 2007, 96, 499–506. [Google Scholar]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural Variation in the Human Genome. Nat. Rev. Genet 2006, 7, 85–97. [Google Scholar]
- Bettendorf, O.; Schmidt, H.; Staebler, A.; Grobholz, R.; Heinecke, A.; Boecker, W.; Hertle, L.; Semjonow, A. Chromosomal Imbalances, Loss of Heterozygosity, and Immunohistochemical Expression of TP53, RB1, and PTEN in Intraductal Cancer, Intraepithelial Neoplasia, and Invasive Adenocarcinoma of the Prostate. Genes Chromosomes Cancer 2008, 47, 565–572. [Google Scholar]
- Ribeiro, F.R.; Diep, C.B.; Jeronimo, C.; Henrique, R.; Lopes, C.; Eknaes, M.; Lingjaerde, O.C.; Lothe, R.A.; Teixeira, M.R. Statistical Dissection of Genetic Pathways Involved in Prostate Carcinogenesis. Genes Chromosomes Cancer 2006, 45, 154–163. [Google Scholar]
- Kallioniemi, O.P.; Visakorpi, T. Genetic Basis and Clonal Evolution of Human Prostate Cancer. Adv. Cancer Res 1996, 68, 225–255. [Google Scholar]
- Boyd, L.K.; Mao, X.; Xue, L.; Lin, D.; Chaplin, T.; Kudahetti, S.C.; Stankiewicz, E.; Yu, Y.; Beltran, L.; Shaw, G.; et al. High-Resolution Genome-Wide Copy-Number Analysis Suggests a Monoclonal Origin of Multifocal Prostate Cancer. Genes Chromosomes Cancer 2012, 51, 579–589. [Google Scholar]
- Robbins, C.M.; Tembe, W.A.; Baker, A.; Sinari, S.; Moses, T.Y.; Beckstrom-Sternberg, S.; Beckstrom-Sternberg, J.; Barrett, M.; Long, J.; Chinnaiyan, A.; et al. Copy Number and Targeted Mutational Analysis Reveals Novel Somatic Events in Metastatic Prostate Tumors. Genome Res 2011, 21, 47–55. [Google Scholar]
- Holcomb, I.N.; Grove, D.I.; Kinnunen, M.; Friedman, C.L.; Gallaher, I.S.; Morgan, T.M.; Sather, C.L.; Delrow, J.J.; Nelson, P.S.; Lange, P.H.; et al. Genomic Alterations Indicate Tumor Origin and Varied Metastatic Potential of Disseminated Cells from Prostate Cancer Patients. Cancer Res 2008, 68, 5599–5608. [Google Scholar]
- El Gammal, A.T.; Bruchmann, M.; Zustin, J.; Isbarn, H.; Hellwinkel, O.J.; Kollermann, J.; Sauter, G.; Simon, R.; Wilczak, W.; Schwarz, J.; et al. Chromosome 8p Deletions and 8q Gains are Associated with Tumor Progression and Poor Prognosis in Prostate Cancer. Clin. Cancer Res 2010, 16, 56–64. [Google Scholar]
- Takahashi, S.; Qian, J.; Brown, J.A.; Alcaraz, A.; Bostwick, D.G.; Lieber, M.M.; Jenkins, R.B. Potential Markers of Prostate Cancer Aggressiveness Detected by Fluorescence in Situ Hybridization in Needle Biopsies. Cancer Res 1994, 54, 3574–3579. [Google Scholar]
- Jenkins, R.B.; Qian, J.; Lieber, M.M.; Bostwick, D.G. Detection of c-Myc Oncogene Amplification and Chromosomal Anomalies in Metastatic Prostatic Carcinoma by Fluorescence in Situ Hybridization. Cancer Res 1997, 57, 524–531. [Google Scholar]
- Ruijter, E.T.; Miller, G.J.; van de Kaa, C.A.; van Bokhoven, A.; Bussemakers, M.J.; Debruyne, F.M.; Ruiter, D.J.; Schalken, J.A. Molecular Analysis of Multifocal Prostate Cancer Lesions. J. Pathol 1999, 188, 271–277. [Google Scholar]
- Rice, K.R.; Furusato, B.; Chen, Y.; McLeod, D.G.; Sesterhenn, I.A.; Brassell, S.A. Clinicopathological Behavior of Single Focus Prostate Adenocarcinoma. J. Urol 2009, 182, 2689–2694. [Google Scholar]
- Wellcome Trust Sanger Institute Cancer Genome Project. Cancer Gene Census. Available online: http://www.sanger.ac.uk/genetics/CGP/Census (accessed on 3 February 2013).
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A Census of Human Cancer Genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar]
- Eminaga, O.; Hinkelammert, R.; Semjonow, A.; Neumann, J.; Abbas, M.; Koepke, T.; Bettendorf, O.; Eltze, E.; Dugas, M. Clinical Map Document Based on XML (cMDX): Document Architecture with Mapping Feature for Reporting and Analysing Prostate Cancer in Radical Prostatectomy Specimens. BMC Med. Inform. Decis. Mak 2010, 10, 71. [Google Scholar]
- Maxwell Promega reagents and systems. Available online: http://www.promega.com (accessed on 10 June 2010).
- Affymetrix Genotyping Console, version 4.1.2. In Software for creating custom SQLite format annotations; Affymetrix; Santa Clara, CA, USA, 2011.
- Partek Genomic Suite Software, version 6.6. In Software for analyzing statistics; Partek Inc.: St. Louis, MO, USA, 2012.
- Optimizing Copy Number Segmentation in Partek; Partek Inc.: St. Louis, MO, USA, 2009.
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequences (RefSeq): A Curated Non-Redundant Sequence Database of Genomes, Transcripts and Proteins. Nucleic Acids Res 2007, 35, 61–65. [Google Scholar]
- User Guide: Associating a Spreadsheet with an Annotation File in Partek Genomics Suite; Partek Inc.: St. Louis, MO, USA, 2010.
- User Guide: Hierarchical Clustering Analysis of Microarray Data; Partek Inc.: St. Louis, MO, USA, 2009.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Patient age at time of surgery (years) | Total Gleason score | Gleason score of individual focus | Pathological stage | Clinical stage | Total tumor volume (cm3) | Tumor volume of individual focus (cm3) | Focality | Number of tumor foci in prostate |
|---|---|---|---|---|---|---|---|---|---|
| MS50L | 66 | 4 + 3 | 3 + 4 | pT3a | cT2c | 2.1 | 0.7 | multifocal | 2 |
| MS50R | 4 + 3 | 1.4 | |||||||
| MS151L | 61 | 4 + 3 | 4 + 3/3 + 3 | pT3b | cT2c | 2.85 | 0.76 | multifocal | 2 |
| MS151R | 4 + 3 | 2.09 | |||||||
| MS183L | 59 | 4 + 3 | 3 + 4 | pT3a | cT2b | 7.35 | 0.35 | multifocal | 2 |
| MS183R | 3 + 4 | 7.0 | |||||||
| MS210L | 62 | 3 + 3 | 3 + 3 | pT3b | cT2c | 8.64 | 4.16 | multifocal | 2 |
| MS210R | 3 + 3 | 4.48 | |||||||
| MS235L | 65 | 4 + 5 | 4 + 4/4 + 5 | pT3a | cT2b | 12.5 | 12.0 | multifocal | 2 |
| MS235R | 3 + 3/3 + 4 | 0.5 | |||||||
| MS343L | 57 | 2 + 3 | 3 + 3 | pT2b | cT2b | 1.33 | 0.95 | multifocal | 3 |
| MS343R | 3 + 4/3 + 3 | 0.19 | |||||||
| MS368L | 65 | 4 + 5 | 3 + 4 | pT3c | cT2b | 13.76 | 2.56 | multifocal | 2 |
| MS368R | 4 + 5 | 11.2 | |||||||
| MS407L | 51 | 3 + 4 | 3 + 4/4 + 3 | pT2b | cT2c | 1.35 | 0.81 | multifocal | 2 |
| MS407R | 3 + 3/3 + 4 | 0.54 | |||||||
| MS586L | 53 | 3 + 2 | 3 + 3 | pT2c | cT1c | 4.55 | 0.7 | multifocal | 5 |
| MS586R | 3 + 3 | 1.4 | |||||||
| MS840L | 66 | 4 + 3 | 3 + 4 | pT3a | cT2c | 2.64 | 1.44 | multifocal | 4 |
| MS840R | 3 + 4 | 0.48 | |||||||
| MS898L | 54 | 4 + 3 | 3 + 4/3 + 3 | pT2c | cT1c | 3.5 | 0.35 | multifocal | 3 |
| MS898R | 3 + 4 | 2.8 | |||||||
| MS946L | 61 | 3 + 2 | 3 + 3 | pT3a | cT2b | 6.75 | 6.3 | multifocal | 2 |
| MS946R | 3 + 3 | 0.45 | |||||||
| MS971L | 50 | 4 + 5 | 3 + 4 | pT3a | cT2a | 1.26 | 1.08 | multifocal | 2 |
| MS971R | 3 + 4 | 0.18 | |||||||
| RD819L | 52 | NA | 3 + 3 | pT3a | cT1c | NA | NA | multifocal | 2 |
| RD819R | 3 + 3 | NA | |||||||
| *MS34L | 71 | 4 + 3 | NA | pT2a | cT1c | 4.56 | unifocal | 1 | |
| MS34R | 4 + 3 | ||||||||
| MS38L | 62 | 3 + 4 | 3 + 3 | pT3c | cT2c | 22.4 | unifocal | 1 | |
| MS38R | 3 + 3 | ||||||||
| MS78L | 66 | 3 + 3 | 3 + 3 | pT3a | cT2b | 3.08 | unifocal | 1 | |
| MS78R | 3 + 3 | ||||||||
| MS99L | 57 | 3 + 4 | 3 + 3 | pT3a | cT2c | 4.42 | unifocal | 1 | |
| MS99R | 3 + 3 | ||||||||
| *MS173L | 59 | 3 + 3 | NA | pT3b | cT2b | 1.2 | unifocal | 1 | |
| MS173R | 3 + 3 | ||||||||
| *MS334L | 67 | 4 + 4 | NA | pT3c | cT2c | 15.96 | unifocal | 1 | |
| MS334R | 3 + 3 | ||||||||
| MS470L | 64 | 3 + 4 | 3 + 3 | pT3a | cT2b | 11.27 | unifocal | 1 | |
| MS470R | 3 + 3 | ||||||||
| MS1096L | 63 | 5+4 | 3+4/4+4 | pT4 | NA | 45.75 | unifocal | 1 | |
| MS1096R | 3+3 | ||||||||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ibeawuchi, C.; Schmidt, H.; Voss, R.; Titze, U.; Abbas, M.; Neumann, J.; Eltze, E.; Hoogland, A.M.; Jenster, G.; Brandt, B.; et al. Genome-Wide Investigation of Multifocal and Unifocal Prostate Cancer — Are They Genetically Different? Int. J. Mol. Sci. 2013, 14, 11816-11829. https://doi.org/10.3390/ijms140611816
Ibeawuchi C, Schmidt H, Voss R, Titze U, Abbas M, Neumann J, Eltze E, Hoogland AM, Jenster G, Brandt B, et al. Genome-Wide Investigation of Multifocal and Unifocal Prostate Cancer — Are They Genetically Different? International Journal of Molecular Sciences. 2013; 14(6):11816-11829. https://doi.org/10.3390/ijms140611816
Chicago/Turabian StyleIbeawuchi, Chinyere, Hartmut Schmidt, Reinhard Voss, Ulf Titze, Mahmoud Abbas, Joerg Neumann, Elke Eltze, Agnes Marije Hoogland, Guido Jenster, Burkhard Brandt, and et al. 2013. "Genome-Wide Investigation of Multifocal and Unifocal Prostate Cancer — Are They Genetically Different?" International Journal of Molecular Sciences 14, no. 6: 11816-11829. https://doi.org/10.3390/ijms140611816
APA StyleIbeawuchi, C., Schmidt, H., Voss, R., Titze, U., Abbas, M., Neumann, J., Eltze, E., Hoogland, A. M., Jenster, G., Brandt, B., & Semjonow, A. (2013). Genome-Wide Investigation of Multifocal and Unifocal Prostate Cancer — Are They Genetically Different? International Journal of Molecular Sciences, 14(6), 11816-11829. https://doi.org/10.3390/ijms140611816