MicroRNA-Regulated Protein-Protein Interaction Networks and Their Functions in Breast Cancer

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
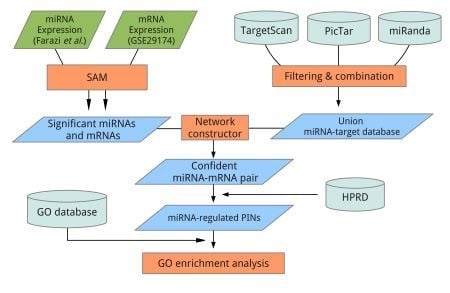
3. Experimental Section
3.1. miRNA Microarray Experiments
3.2. mRNA Expression Profiles
3.3. miRNA Expression Profiles
3.4. Data Analysis
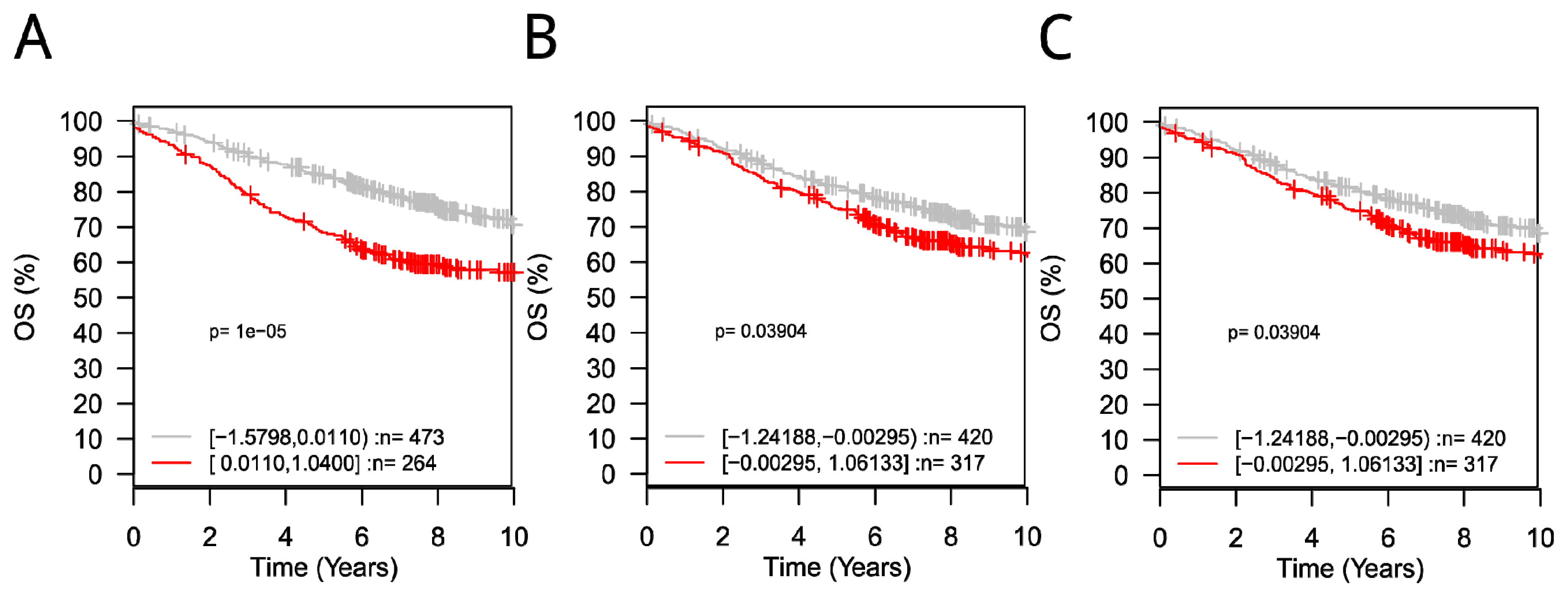
3.5. ROC and GOBO Survival Analysis
4. Conclusions
Supplementary Information

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRBase Accession | miRNA Name | Fold Change |
|---|---|---|
| MIMAT0004761 | hsa-miR-483-5p | 0.01 |
| MIMAT0004552 | hsa-miR-139-3p | 0.01 |
| MIMAT0000738 | hsa-miR-383 | 0.02 |
| MIMAT0002856 | hsa-miR-520d-3p | 0.02 |
| MIMAT0002811 | hsa-miR-202-3p | 0.03 |
| MIMAT0002177 | hsa-miR-486-5p | 0.04 |
| MIMAT0022721 | hsa-miR-1247-3p | 0.05 |
| MIMAT0002175 | hsa-miR-485-5p | 0.06 |
| MIMAT0000265 | hsa-miR-204-5p | 0.07 |
| MIMAT0000752 | hsa-miR-328 | 0.07 |
| MIMAT0000421 | hsa-miR-122-5p | 0.07 |
| MIMAT0000447 | hsa-miR-134 | 0.08 |
| MIMAT0000722 | hsa-miR-370 | 0.09 |
| MIMAT0004513 | hsa-miR-101-5p | 0.09 |
| MIMAT0000446 | hsa-miR-127-3p | 0.10 |
| MIMAT0000097 | hsa-miR-99a-5p | 0.10 |
| MIMAT0004566 | hsa-miR-218-2-3p | 0.10 |
| MIMAT0000729 | hsa-miR-376a-3p | 0.11 |
| MIMAT0009197 | hsa-miR-205-3p | 0.11 |
| MIMAT0004615 | hsa-miR-195-3p | 0.11 |
| MIMAT0005899 | hsa-miR-1247-5p | 0.11 |
| MIMAT0000720 | hsa-miR-376c | 0.12 |
| MIMAT0000762 | hsa-miR-324-3p | 0.12 |
| MIMAT0004679 | hsa-miR-296-3p | 0.12 |
| MIMAT0004614 | hsa-miR-193a-5p | 0.12 |
| MIMAT0003880 | hsa-miR-671-5p | 0.12 |
| MIMAT0004795 | hsa-miR-574-5p | 0.12 |
| MIMAT0004599 | hsa-miR-143-5p | 0.13 |
| MIMAT0000423 | hsa-miR-125b-5p | 0.13 |
| MIMAT0004957 | hsa-miR-760 | 0.13 |
| MIMAT0004911 | hsa-miR-874 | 0.14 |
| MIMAT0004603 | hsa-miR-125b-2-3p | 0.15 |
| MIMAT0004952 | hsa-miR-665 | 0.15 |
| MIMAT0018205 | hsa-miR-3928 | 0.15 |
| MIMAT0004767 | hsa-miR-193b-5p | 0.15 |
| MIMAT0002861 | hsa-miR-518e-3p | 0.15 |
| MIMAT0004604 | hsa-miR-127-5p | 0.16 |
| MIMAT0002807 | hsa-miR-491-5p | 0.16 |
| MIMAT0004689 | hsa-miR-377-5p | 0.16 |
| MIMAT0004762 | hsa-miR-486-3p | 0.16 |
| MIMAT0000732 | hsa-miR-378a-3p | 0.17 |
| MIMAT0017981 | hsa-miR-3605-5p | 0.18 |
| MIMAT0004605 | hsa-miR-129-2-3p | 0.19 |
| MIMAT0006789 | hsa-miR-1468 | 0.20 |
| MIMAT0000737 | hsa-miR-382-5p | 0.21 |
| MIMAT0000077 | hsa-miR-22-3p | 0.21 |
| MIMAT0000089 | hsa-miR-31-5p | 0.21 |
| MIMAT0004612 | hsa-miR-186-3p | 0.21 |
| MIMAT0004592 | hsa-miR-125b-1-3p | 0.22 |
| MIMAT0001639 | hsa-miR-409-3p | 0.22 |
| MIMAT0015032 | hsa-miR-3158-3p | 0.22 |
| MIMAT0004496 | hsa-miR-23a-5p | 0.22 |
| MIMAT0000690 | hsa-miR-296-5p | 0.22 |
| MIMAT0000731 | hsa-miR-378a-5p | 0.23 |
| MIMAT0000448 | hsa-miR-136-5p | 0.23 |
| MIMAT0004796 | hsa-miR-576-3p | 0.23 |
| MIMAT0010133 | hsa-miR-2110 | 0.23 |
| MIMAT0004951 | hsa-miR-887 | 0.23 |
| MIMAT0003239 | hsa-miR-574-3p | 0.25 |
| MIMAT0005901 | hsa-miR-1249 | 0.25 |
| MIMAT0000510 | hsa-miR-320a | 0.26 |
| MIMAT0002172 | hsa-miR-376b | 0.26 |
| MIMAT0000250 | hsa-miR-139-5p | 0.27 |
| MIMAT0005825 | hsa-miR-1180 | 0.27 |
| MIMAT0000437 | hsa-miR-145-5p | 0.28 |
| MIMAT0004601 | hsa-miR-145-3p | 0.28 |
| MIMAT0003322 | hsa-miR-652-3p | 0.28 |
| MIMAT0000756 | hsa-miR-326 | 0.28 |
| MIMAT0000098 | hsa-miR-100-5p | 0.29 |
| MIMAT0003296 | hsa-miR-627 | 0.29 |
| MIMAT0002820 | hsa-miR-497-5p | 0.31 |
| MIMAT0004507 | hsa-miR-92a-1-5p | 0.31 |
| MIMAT0000271 | hsa-miR-214-3p | 0.32 |
| MIMAT0004702 | hsa-miR-339-3p | 0.33 |
| MIMAT0004611 | hsa-miR-185-3p | 0.33 |
| MIMAT0000064 | hsa-let-7c | 0.34 |
| MIMAT0004673 | hsa-miR-29c-5p | 0.35 |
| MIMAT0000733 | hsa-miR-379-5p | 0.35 |
| MIMAT0004594 | hsa-miR-132-5p | 0.35 |
| MIMAT0000765 | hsa-miR-335-5p | 0.35 |
| MIMAT0002819 | hsa-miR-193b-3p | 0.36 |
| MIMAT0000088 | hsa-miR-30a-3p | 0.36 |
| MIMAT0005951 | hsa-miR-1307-3p | 0.36 |
| MIMAT0004597 | hsa-miR-140-3p | 0.37 |
| MIMAT0004556 | hsa-miR-10b-3p | 0.37 |
| MIMAT0000272 | hsa-miR-215 | 0.37 |
| MIMAT0004511 | hsa-miR-99a-3p | 0.37 |
| MIMAT0000443 | hsa-miR-125a-5p | 0.38 |
| MIMAT0004482 | hsa-let-7b-3p | 0.38 |
| MIMAT0000076 | hsa-miR-21-5p | 6.58 |
| NCBI gene ID | Gene Symbol | Fold Change |
|---|---|---|
| 2949 | GSTM5 | 0.06 |
| 10894 | LYVE1 | 0.06 |
| 5950 | RBP4 | 0.07 |
| 762 | CA4 | 0.09 |
| 54997 | TESC | 0.09 |
| 3489 | IGFBP6 | 0.09 |
| 3952 | LEP | 0.09 |
| 213 | ALB | 0.09 |
| 3131 | HLF | 0.10 |
| 4023 | LPL | 0.10 |
| 10633 | RASL10A | 0.11 |
| 364 | AQP7 | 0.11 |
| 1908 | EDN3 | 0.11 |
| 1811 | SLC26A3 | 0.11 |
| 91851 | CHRDL1 | 0.11 |
| 729359 | PLIN4 | 0.13 |
| 1149 | CIDEA | 0.13 |
| 5959 | RDH5 | 0.13 |
| 5348 | FXYD1 | 0.14 |
| 5346 | PLIN1 | 0.14 |
| 10249 | GLYAT | 0.14 |
| 158800 | RHOXF1 | 0.14 |
| 221476 | PI16 | 0.14 |
| 3040 | HBA2 | 0.14 |
| 6939 | TCF15 | 0.14 |
| 79645 | EFCAB1 | 0.14 |
| 80343 | SEL1L2 | 0.14 |
| 9413 | FAM189A2 | 0.15 |
| 26289 | AK5 | 0.15 |
| 25891 | PAMR1 | 0.15 |
| 3679 | ITGA7 | 0.15 |
| 1264 | CNN1 | 0.15 |
| 92304 | SCGB3A1 | 0.15 |
| 2167 | FABP4 | 0.15 |
| 23285 | KIAA1107 | 0.15 |
| 7145 | TNS1 | 0.16 |
| 4881 | NPR1 | 0.16 |
| 1028 | CDKN1C | 0.16 |
| 1036 | CDO1 | 0.16 |
| 130271 | PLEKHH2 | 0.16 |
| 8736 | MYOM1 | 0.16 |
| 8908 | GYG2 | 0.16 |
| 619373 | MBOAT4 | 0.17 |
| 130399 | ACVR1C | 0.17 |
| 1646 | AKR1C2 | 0.17 |
| 80763 | C12orf39 | 0.17 |
| 2159 | F10 | 0.18 |
| 84889 | SLC7A3 | 0.18 |
| 1308 | COL17A1 | 0.18 |
| 83699 | SH3BGRL2 | 0.18 |
| 84417 | C2orf40 | 0.18 |
| 4081 | MAB21L1 | 0.18 |
| 3484 | IGFBP1 | 0.18 |
| 5239 | PGM5 | 0.19 |
| 4969 | OGN | 0.19 |
| 2719 | GPC3 | 0.19 |
| 116362 | RBP7 | 0.19 |
| 948 | CD36 | 0.19 |
| 5764 | PTN | 0.19 |
| 3043 | HBB | 0.19 |
| 56920 | SEMA3G | 0.20 |
| 94274 | PPP1R14A | 0.20 |
| 57447 | NDRG2 | 0.20 |
| 84795 | PYROXD2 | 0.20 |
| 84649 | DGAT2 | 0.20 |
| 2690 | GHR | 0.20 |
| 22802 | CLCA4 | 0.20 |
| 5179 | PENK | 0.20 |
| 6663 | SOX10 | 0.20 |
| 6649 | SOD3 | 0.21 |
| 54922 | RASIP1 | 0.21 |
| 8406 | SRPX | 0.21 |
| 1446 | CSN1S1 | 0.21 |
| 7123 | CLEC3B | 0.22 |
| 9647 | PPM1F | 0.22 |
| 1842 | ECM2 | 0.22 |
| 3909 | LAMA3 | 0.22 |
| 8639 | AOC3 | 0.23 |
| 2934 | GSN | 0.23 |
| 9370 | ADIPOQ | 0.23 |
| 3202 | HOXA5 | 0.23 |
| 9452 | ITM2A | 0.23 |
| 6290 | SAA3P | 0.23 |
| 4604 | MYBPC1 | 0.23 |
| 79785 | RERGL | 0.16 |
| 221091 | LRRN4CL | 0.17 |
| 3991 | LIPE | 0.17 |
| 27175 | TUBG2 | 0.24 |
| 1346 | COX7A1 | 0.24 |
| 6376 | CX3CL1 | 0.24 |
| 50486 | G0S2 | 0.24 |
| 6285 | S100B | 0.24 |
| 443 | ASPA | 0.24 |
| 947 | CD34 | 0.25 |
| 84632 | AFAP1L2 | 0.25 |
| 3866 | KRT15 | 0.25 |
| 147463 | ANKRD29 | 0.25 |
| 2878 | GPX3 | 0.25 |
| 7079 | TIMP4 | 0.25 |
| 54345 | SOX18 | 0.25 |
| 51277 | DNAJC27 | 0.25 |
| 84870 | RSPO3 | 0.25 |
| 55323 | LARP6 | 0.25 |
| 6387 | CXCL12 | 0.25 |
| 137835 | TMEM71 | 0.25 |
| 5212 | VIT | 0.25 |
| 26577 | PCOLCE2 | 0.25 |
| 845 | CASQ2 | 0.25 |
| 6422 | SFRP1 | 0.25 |
| 10351 | ABCA8 | 0.26 |
| 10840 | ALDH1L1 | 0.26 |
| 65983 | GRAMD3 | 0.26 |
| 84327 | ZBED3 | 0.26 |
| 57124 | CD248 | 0.26 |
| 3235 | HOXD9 | 0.26 |
| 2192 | FBLN1 | 0.26 |
| 91653 | BOC | 0.26 |
| 4147 | MATN2 | 0.26 |
| 126669 | SHE | 0.27 |
| 2788 | GNG7 | 0.27 |
| 129804 | FBLN7 | 0.27 |
| 270 | AMPD1 | 0.27 |
| 79656 | BEND5 | 0.27 |
| 58503 | PROL1 | 0.27 |
| 3316 | HSPB2 | 0.27 |
| 729440 | CCDC61 | 0.27 |
| 54438 | GFOD1 | 0.27 |
| 5243 | ABCB1 | 0.27 |
| 1128 | CHRM1 | 0.23 |
| 83878 | USHBP1 | 0.24 |
| 63970 | TP53AIP1 | 0.24 |
| 79192 | IRX1 | 0.28 |
| 3400 | ID4 | 0.28 |
| 57519 | STARD9 | 0.29 |
| 57666 | FBRSL1 | 0.29 |
| 3590 | IL11RA | 0.29 |
| 57664 | PLEKHA4 | 0.29 |
| 197257 | LDHD | 0.29 |
| 66036 | MTMR9 | 0.29 |
| 2321 | FLT1 | 0.29 |
| 126 | ADH1C | 0.29 |
| 1363 | CPE | 0.29 |
| 56131 | PCDHB4 | 0.29 |
| 22915 | MMRN1 | 0.29 |
| 7069 | THRSP | 0.29 |
| 57161 | PELI2 | 0.30 |
| 770 | CA11 | 0.30 |
| 53342 | IL17D | 0.30 |
| 79987 | SVEP1 | 0.30 |
| 857 | CAV1 | 0.30 |
| 222166 | C7orf41 | 0.30 |
| 27190 | IL17B | 0.30 |
| 116159 | CYYR1 | 0.30 |
| 4487 | MSX1 | 0.30 |
| 9068 | ANGPTL1 | 0.30 |
| 10411 | RAPGEF3 | 0.30 |
| 3199 | HOXA2 | 0.30 |
| 2944 | GSTM1 | 0.30 |
| 2920 | CXCL2 | 0.30 |
| 201134 | CEP112 | 0.31 |
| 220001 | VWCE | 0.31 |
| 83888 | FGFBP2 | 0.31 |
| 6366 | CCL21 | 0.31 |
| 6711 | SPTBN1 | 0.31 |
| 85378 | TUBGCP6 | 0.31 |
| 26040 | SETBP1 | 0.31 |
| 4692 | NDN | 0.31 |
| 25890 | ABI3BP | 0.31 |
| 23531 | MMD | 0.31 |
| 30846 | EHD2 | 0.31 |
| 6196 | RPS6KA2 | 0.31 |
| 2009 | EML1 | 0.31 |
| 810 | CALML3 | 0.27 |
| 6898 | TAT | 0.27 |
| 5648 | MASP1 | 0.28 |
| 25999 | CLIP3 | 0.28 |
| 125875 | CLDND2 | 0.28 |
| 7102 | TSPAN7 | 0.28 |
| 1879 | EBF1 | 0.28 |
| 23252 | OTUD3 | 0.28 |
| 5493 | PPL | 0.28 |
| 83987 | CCDC8 | 0.28 |
| 9073 | CLDN8 | 0.28 |
| 221981 | THSD7A | 0.28 |
| 64102 | TNMD | 0.28 |
| 137872 | ADHFE1 | 0.33 |
| 27151 | CPAMD8 | 0.33 |
| 387923 | SERP2 | 0.33 |
| 145581 | LRFN5 | 0.33 |
| 6263 | RYR3 | 0.33 |
| 2354 | FOSB | 0.33 |
| 51302 | CYP39A1 | 0.33 |
| 4128 | MAOA | 0.34 |
| 117248 | GALNTL2 | 0.34 |
| 10268 | RAMP3 | 0.34 |
| 7730 | ZNF177 | 0.34 |
| 10873 | ME3 | 0.34 |
| 7461 | CLIP2 | 0.34 |
| 7049 | TGFBR3 | 0.34 |
| 79901 | CYBRD1 | 0.34 |
| 5152 | PDE9A | 0.34 |
| 50805 | IRX4 | 0.34 |
| 8644 | AKR1C3 | 0.34 |
| 5915 | RARB | 0.34 |
| 2770 | GNAI1 | 0.34 |
| 54996 | 2-Mar | 0.35 |
| 79791 | FBXO31 | 0.35 |
| 54776 | PPP1R12C | 0.35 |
| 9079 | LDB2 | 0.35 |
| 57104 | PNPLA2 | 0.35 |
| 30008 | EFEMP2 | 0.35 |
| 91461 | PKDCC | 0.35 |
| 23368 | PPP1R13B | 0.35 |
| 23461 | ABCA5 | 0.35 |
| 9572 | NR1D1 | 0.35 |
| 23338 | PHF15 | 0.35 |
| 6289 | SAA2 | 0.31 |
| 345275 | HSD17B13 | 0.31 |
| 2701 | GJA4 | 0.32 |
| 112609 | MRAP2 | 0.32 |
| 727 | C5 | 0.32 |
| 477 | ATP1A2 | 0.32 |
| 9627 | SNCAIP | 0.32 |
| 4435 | CITED1 | 0.32 |
| 10974 | C10orf116 | 0.32 |
| 11005 | SPINK5 | 0.32 |
| 80325 | ABTB1 | 0.33 |
| 221395 | GPR116 | 0.33 |
| 10014 | HDAC5 | 0.33 |
| 1489 | CTF1 | 0.37 |
| 35 | ACADS | 0.37 |
| 3749 | KCNC4 | 0.37 |
| 140738 | TMEM37 | 0.37 |
| 2791 | GNG11 | 0.37 |
| 23604 | DAPK2 | 0.37 |
| 10217 | CTDSPL | 0.37 |
| 23550 | PSD4 | 0.37 |
| 4306 | NR3C2 | 0.37 |
| 119587 | CPXM2 | 0.37 |
| 7942 | TFEB | 0.37 |
| 3815 | KIT | 0.37 |
| 1805 | DPT | 0.37 |
| 23242 | COBL | 0.37 |
| 4313 | MMP2 | 0.37 |
| 4139 | MARK1 | 0.37 |
| 9104 | RGN | 0.37 |
| 2329 | FMO4 | 0.37 |
| 25802 | LMOD1 | 0.38 |
| 4239 | MFAP4 | 0.38 |
| 10392 | NOD1 | 0.38 |
| 6794 | STK11 | 0.38 |
| 85458 | DIXDC1 | 0.38 |
| 4123 | MAN2C1 | 0.38 |
| 54476 | RNF216 | 0.38 |
| 9920 | KBTBD11 | 0.38 |
| 6329 | SCN4A | 0.38 |
| 10253 | SPRY2 | 0.38 |
| 1910 | EDNRB | 0.38 |
| 9249 | DHRS3 | 0.38 |
| 22869 | ZNF510 | 0.38 |
| 114800 | CCDC85A | 0.35 |
| 2550 | GABBR1 | 0.35 |
| 4638 | MYLK | 0.35 |
| 2327 | FMO2 | 0.35 |
| 139411 | PTCHD1 | 0.35 |
| 10391 | CORO2B | 0.35 |
| 25854 | FAM149A | 0.35 |
| 55701 | ARHGEF40 | 0.36 |
| 1759 | DNM1 | 0.36 |
| 22849 | CPEB3 | 0.36 |
| 57716 | PRX | 0.36 |
| 1628 | DBP | 0.36 |
| 80031 | SEMA6D | 0.36 |
| 259217 | HSPA12A | 0.36 |
| 6909 | TBX2 | 0.36 |
| 1511 | CTSG | 0.36 |
| 79971 | WLS | 0.36 |
| 90865 | IL33 | 0.36 |
| 11343 | MGLL | 0.36 |
| 55800 | SCN3B | 0.36 |
| 1949 | EFNB3 | 0.36 |
| 284217 | LAMA1 | 0.36 |
| 22927 | HABP4 | 0.37 |
| 23645 | PPP1R15A | 0.39 |
| 342574 | KRT27 | 0.39 |
| 83543 | AIF1L | 0.39 |
| 624 | BDKRB2 | 0.39 |
| 347 | APOD | 0.39 |
| 84935 | C13orf33 | 0.39 |
| 858 | CAV2 | 0.39 |
| 5138 | PDE2A | 0.40 |
| 114928 | GPRASP2 | 0.40 |
| 58190 | CTDSP1 | 0.40 |
| 513 | ATP5D | 0.40 |
| 57684 | ZBTB26 | 0.40 |
| 7041 | TGFB1I1 | 0.40 |
| 5787 | PTPRB | 0.40 |
| 7294 | TXK | 0.40 |
| 56301 | SLC7A10 | 0.40 |
| 55937 | APOM | 0.40 |
| 6368 | CCL23 | 0.40 |
| 55020 | TTC38 | 0.40 |
| 134265 | AFAP1L1 | 0.40 |
| 4485 | MST1 | 0.40 |
| 3384 | ICAM2 | 0.38 |
| 8613 | PPAP2B | 0.38 |
| 1950 | EGF | 0.38 |
| 55273 | TMEM100 | 0.38 |
| 6297 | SALL2 | 0.38 |
| 9365 | KL | 0.38 |
| 8863 | PER3 | 0.38 |
| 8404 | SPARCL1 | 0.38 |
| 2202 | EFEMP1 | 0.38 |
| 8369 | HIST1H4G | 0.38 |
| 5187 | PER1 | 0.39 |
| 30815 | ST6GALNAC6 | 0.39 |
| 256364 | EML3 | 0.39 |
| 57381 | RHOJ | 0.39 |
| 761 | CA3 | 0.39 |
| 83989 | FAM172A | 0.39 |
| 1408 | CRY2 | 0.39 |
| 2281 | FKBP1B | 0.39 |
| 51222 | ZNF219 | 0.39 |
| 54540 | FAM193B | 0.39 |
| 4053 | LTBP2 | 0.39 |
| 55184 | DZANK1 | 0.39 |
| 5740 | PTGIS | 0.39 |
| 84814 | PPAPDC3 | 0.42 |
| 79365 | BHLHE41 | 0.42 |
| 316 | AOX1 | 0.42 |
| 23380 | SRGAP2 | 0.42 |
| 84033 | OBSCN | 0.42 |
| 90353 | CTU1 | 0.42 |
| 9013 | TAF1C | 0.42 |
| 474344 | GIMAP6 | 0.42 |
| 84883 | AIFM2 | 0.42 |
| 58480 | RHOU | 0.42 |
| 65982 | ZSCAN18 | 0.42 |
| 666 | BOK | 0.42 |
| 79762 | C1orf115 | 0.42 |
| 525 | ATP6V1B1 | 0.42 |
| 4675 | NAP1L3 | 0.42 |
| 3257 | HPS1 | 0.43 |
| 55781 | RIOK2 | 0.43 |
| 63947 | DMRTC1 | 0.43 |
| 1969 | EPHA2 | 0.43 |
| 25927 | CNRIP1 | 0.43 |
| 57685 | CACHD1 | 0.43 |
| 51559 | NT5DC3 | 0.40 |
| 7169 | TPM2 | 0.40 |
| 51705 | EMCN | 0.40 |
| 8938 | BAIAP3 | 0.40 |
| 10365 | KLF2 | 0.40 |
| 59 | ACTA2 | 0.40 |
| 80309 | SPHKAP | 0.40 |
| 3779 | KCNMB1 | 0.41 |
| 10826 | C5orf4 | 0.41 |
| 219654 | ZCCHC24 | 0.41 |
| 92162 | TMEM88 | 0.41 |
| 7450 | VWF | 0.41 |
| 10266 | RAMP2 | 0.41 |
| 25875 | LETMD1 | 0.41 |
| 1938 | EEF2 | 0.41 |
| 121551 | BTBD11 | 0.41 |
| 2119 | ETV5 | 0.41 |
| 9696 | CROCC | 0.41 |
| 1031 | CDKN2C | 0.41 |
| 9037 | SEMA5A | 0.41 |
| 3397 | ID1 | 0.41 |
| 84707 | BEX2 | 0.41 |
| 57616 | TSHZ3 | 0.41 |
| 1471 | CST3 | 0.41 |
| 55214 | LEPREL1 | 0.41 |
| 3914 | LAMB3 | 0.41 |
| 57478 | USP31 | 0.41 |
| 3783 | KCNN4 | 0.41 |
| 8839 | WISP2 | 0.41 |
| 1583 | CYP11A1 | 0.42 |
| 10124 | ARL4A | 0.42 |
| 738 | C11orf2 | 0.42 |
| 29800 | ZDHHC1 | 0.42 |
| 23135 | KDM6B | 0.44 |
| 171024 | SYNPO2 | 0.44 |
| 10350 | ABCA9 | 0.44 |
| 3691 | ITGB4 | 0.44 |
| 2348 | FOLR1 | 0.44 |
| 11145 | PLA2G16 | 0.44 |
| 554 | AVPR2 | 0.45 |
| 64072 | CDH23 | 0.45 |
| 80177 | MYCT1 | 0.45 |
| 5957 | RCVRN | 0.45 |
| 408 | ARRB1 | 0.45 |
| 29997 | GLTSCR2 | 0.43 |
| 26051 | PPP1R16B | 0.43 |
| 83604 | TMEM47 | 0.43 |
| 2308 | FOXO1 | 0.43 |
| 55225 | RAVER2 | 0.43 |
| 54839 | LRRC49 | 0.43 |
| 122953 | JDP2 | 0.43 |
| 29775 | CARD10 | 0.43 |
| 166 | AES | 0.43 |
| 25924 | MYRIP | 0.43 |
| 2852 | GPER | 0.43 |
| 51421 | AMOTL2 | 0.43 |
| 124936 | CYB5D2 | 0.43 |
| 1294 | COL7A1 | 0.43 |
| 127435 | PODN | 0.43 |
| 84952 | CGNL1 | 0.43 |
| 83483 | PLVAP | 0.43 |
| 1958 | EGR1 | 0.43 |
| 230 | ALDOC | 0.43 |
| 65987 | KCTD14 | 0.43 |
| 4804 | NGFR | 0.44 |
| 64852 | TUT1 | 0.44 |
| 84253 | GARNL3 | 0.44 |
| 5866 | RAB3IL1 | 0.44 |
| 10608 | MXD4 | 0.44 |
| 4211 | MEIS1 | 0.44 |
| 83547 | RILP | 0.44 |
| 9172 | MYOM2 | 0.44 |
| 57192 | MCOLN1 | 0.44 |
| 255877 | BCL6B | 0.44 |
| 56904 | SH3GLB2 | 0.44 |
| 51285 | RASL12 | 0.44 |
| 3425 | IDUA | 0.44 |
| 402117 | VWC2L | 0.46 |
| 81490 | PTDSS2 | 0.46 |
| 283748 | PLA2G4D | 0.46 |
| 23523 | CABIN1 | 0.46 |
| 6146 | RPL22 | 0.46 |
| 85360 | SYDE1 | 0.46 |
| 60468 | BACH2 | 0.46 |
| 57451 | ODZ2 | 0.46 |
| 4013 | VWA5A | 0.46 |
| 339768 | ESPNL | 0.46 |
| 3860 | KRT13 | 0.46 |
| 144699 | FBXL14 | 0.45 |
| 83719 | YPEL3 | 0.45 |
| 22841 | RAB11FIP2 | 0.45 |
| 283927 | NUDT7 | 0.45 |
| 293 | SLC25A6 | 0.45 |
| 90507 | SCRN2 | 0.45 |
| 37 | ACADVL | 0.45 |
| 112744 | IL17F | 0.45 |
| 6709 | SPTAN1 | 0.45 |
| 8086 | AAAS | 0.45 |
| 7423 | VEGFB | 0.45 |
| 64221 | ROBO3 | 0.45 |
| 7273 | TTN | 0.45 |
| 2657 | GDF1 | 0.45 |
| 59271 | C21orf63 | 0.45 |
| 132160 | PPM1M | 0.45 |
| 27244 | SESN1 | 0.45 |
| 51310 | SLC22A17 | 0.45 |
| 4828 | NMB | 0.45 |
| 54360 | CYTL1 | 0.45 |
| 203245 | NAIF1 | 0.45 |
| 23166 | STAB1 | 0.45 |
| 2121 | EVC | 0.45 |
| 116496 | FAM129A | 0.45 |
| 23239 | PHLPP1 | 0.45 |
| 51673 | TPPP3 | 0.45 |
| 64094 | SMOC2 | 0.45 |
| 6383 | SDC2 | 0.45 |
| 2180 | ACSL1 | 0.45 |
| 23770 | FKBP8 | 0.45 |
| 55901 | THSD1 | 0.46 |
| 25895 | METTL21B | 0.46 |
| 23731 | C9orf5 | 0.46 |
| 126393 | HSPB6 | 0.46 |
| 4056 | LTC4S | 0.46 |
| 79825 | CCDC48 | 0.46 |
| 10810 | WASF3 | 0.46 |
| 29911 | HOOK2 | 0.46 |
| 583 | BBS2 | 0.46 |
| 28984 | C13orf15 | 0.46 |
| 1465 | CSRP1 | 0.46 |
| 55258 | THNSL2 | 0.46 |
| 161198 | CLEC14A | 0.46 |
| 3699 | ITIH3 | 0.48 |
| 7094 | TLN1 | 0.46 |
| 4232 | MEST | 0.46 |
| 1410 | CRYAB | 0.46 |
| 57452 | GALNTL1 | 0.47 |
| 63935 | PCIF1 | 0.47 |
| 25873 | RPL36 | 0.47 |
| 9812 | KIAA0141 | 0.47 |
| 51665 | ASB1 | 0.47 |
| 64123 | ELTD1 | 0.47 |
| 6122 | RPL3 | 0.47 |
| 222962 | SLC29A4 | 0.47 |
| 23102 | TBC1D2B | 0.47 |
| 3476 | IGBP1 | 0.47 |
| 93408 | MYL10 | 0.47 |
| 5310 | PKD1 | 0.47 |
| 4628 | MYH10 | 0.47 |
| 221935 | SDK1 | 0.47 |
| 23328 | SASH1 | 0.47 |
| 8522 | GAS7 | 0.47 |
| 10023 | FRAT1 | 0.47 |
| 7301 | TYRO3 | 0.47 |
| 2767 | GNA11 | 0.47 |
| 9457 | FHL5 | 0.47 |
| 4094 | MAF | 0.47 |
| 65268 | WNK2 | 0.47 |
| 54585 | LZTFL1 | 0.47 |
| 375449 | MAST4 | 0.47 |
| 138311 | FAM69B | 0.47 |
| 160622 | GRASP | 0.47 |
| 22837 | COBLL1 | 0.47 |
| 51435 | SCARA3 | 0.47 |
| 217 | ALDH2 | 0.47 |
| 6236 | RRAD | 0.47 |
| 8322 | FZD4 | 0.47 |
| 653275 | CFC1B | 0.47 |
| 10908 | PNPLA6 | 0.47 |
| 57526 | PCDH19 | 0.47 |
| 8424 | BBOX1 | 0.47 |
| 9905 | SGSM2 | 0.48 |
| 10435 | CDC42EP2 | 0.48 |
| 23087 | TRIM35 | 0.48 |
| 60314 | C12orf10 | 0.48 |
| 1073 | CFL2 | 0.48 |
| 5256 | PHKA2 | 0.49 |
| 92922 | CCDC102A | 0.48 |
| 65057 | ACD | 0.48 |
| 9095 | TBX19 | 0.48 |
| 6441 | SFTPD | 0.48 |
| 22846 | VASH1 | 0.48 |
| 51066 | C3orf32 | 0.48 |
| 23179 | RGL1 | 0.48 |
| 4664 | NAB1 | 0.48 |
| 50511 | SYCP3 | 0.48 |
| 6430 | SRSF5 | 0.48 |
| 11078 | TRIOBP | 0.48 |
| 78991 | PCYOX1L | 0.48 |
| 6623 | SNCG | 0.48 |
| 23384 | SPECC1L | 0.48 |
| 53826 | FXYD6 | 0.48 |
| 9397 | NMT2 | 0.48 |
| 6041 | RNASEL | 0.48 |
| 113510 | HELQ | 0.48 |
| 64788 | LMF1 | 0.48 |
| 2217 | FCGRT | 0.48 |
| 79720 | VPS37B | 0.48 |
| 6764 | ST5 | 0.48 |
| 252969 | NEIL2 | 0.48 |
| 8987 | STBD1 | 0.48 |
| 41 | ACCN2 | 0.48 |
| 7905 | REEP5 | 0.48 |
| 5919 | RARRES2 | 0.48 |
| 10544 | PROCR | 0.48 |
| 6876 | TAGLN | 0.48 |
| 8436 | SDPR | 0.49 |
| 23500 | DAAM2 | 0.49 |
| 130132 | RFTN2 | 0.49 |
| 80310 | PDGFD | 0.49 |
| 4215 | MAP3K3 | 0.49 |
| 282775 | OR5J2 | 0.49 |
| 51161 | C3orf18 | 0.49 |
| 29098 | RANGRF | 0.49 |
| 53336 | CPXCR1 | 0.49 |
| 9081 | PRY | 0.49 |
| 9459 | ARHGEF6 | 0.49 |
| 2995 | GYPC | 0.49 |
| 23057 | NMNAT2 | 0.49 |
| 4669 | NAGLU | 0.49 |
| 6452 | SH3BP2 | 0.49 |
| 6237 | RRAS | 0.49 |
| 5288 | PIK3C2G | 0.49 |
| 10252 | SPRY1 | 0.49 |
| 79026 | AHNAK | 0.49 |
| 9693 | RAPGEF2 | 0.49 |
| 51226 | COPZ2 | 0.49 |
| 158326 | FREM1 | 0.49 |
| 1956 | EGFR | 0.49 |
| 5360 | PLTP | 0.49 |
| 290 | ANPEP | 0.49 |
| 1756 | DMD | 0.49 |
| 5118 | PCOLCE | 0.49 |
| 56654 | NPDC1 | 0.49 |
| 9254 | CACNA2D2 | 0.49 |
| 55536 | CDCA7L | 0.49 |
| 124975 | GGT6 | 0.49 |
| 1906 | EDN1 | 0.49 |
| 81029 | WNT5B | 0.49 |
| 2646 | GCKR | 0.49 |
| 9811 | CTIF | 0.50 |
| 145376 | PPP1R36 | 0.50 |
| 222865 | TMEM130 | 0.50 |
| 92999 | ZBTB47 | 0.50 |
| 168002 | DACT2 | 0.50 |
| 6829 | SUPT5H | 0.50 |
| 9992 | KCNE2 | 0.50 |
| 58509 | C19orf29 | 0.50 |
| 79706 | PRKRIP1 | 0.50 |
| 1153 | CIRBP | 0.50 |
| 9639 | ARHGEF10 | 0.50 |
| 4054 | LTBP3 | 0.50 |
| 1120 | CHKB | 0.50 |
| 286046 | XKR6 | 0.50 |
| 9590 | AKAP12 | 0.50 |
| 64115 | C10orf54 | 0.50 |
| 2067 | ERCC1 | 0.50 |
| 7507 | XPA | 0.50 |
| 22897 | CEP164 | 0.50 |
| 652 | BMP4 | 0.50 |
| 55702 | CCDC94 | 0.50 |
| 57613 | KIAA1467 | 0.50 |
| 28514 | DLL1 | 0.50 |
| 169270 | ZNF596 | 0.50 |
| 83982 | IFI27L2 | 0.50 |
| 51458 | RHCG | 0.49 |
| 1112 | FOXN3 | 0.49 |
| 29954 | POMT2 | 0.49 |
| 9612 | NCOR2 | 0.49 |
| 3198 | HOXA1 | 0.49 |
| 5311 | PKD2 | 0.49 |
| 2946 | GSTM2 | 0.49 |
| 2109 | ETFB | 0.49 |
| 56062 | KLHL4 | 0.49 |
| 6915 | TBXA2R | 0.50 |
| 64288 | ZNF323 | 0.50 |
| 5195 | PEX14 | 0.50 |
| 84557 | MAP1LC3A | 0.50 |
| 6164 | RPL34 | 0.50 |
| 8835 | SOCS2 | 0.50 |
| 2735 | GLI1 | 0.50 |
| 26022 | TMEM98 | 0.50 |
| 3908 | LAMA2 | 0.50 |
| 1825 | DSC3 | 0.50 |
| 5730 | PTGDS | 0.50 |
| 162515 | SLC16A11 | 0.51 |
| 274 | BIN1 | 0.51 |
| 79654 | HECTD3 | 0.51 |
| 22863 | ATG14 | 0.51 |
| 25949 | SYF2 | 0.51 |
| 84872 | ZC3H10 | 0.51 |
| 23187 | PHLDB1 | 0.51 |
| 5434 | POLR2E | 0.51 |
| 6181 | RPLP2 | 0.51 |
| 6141 | RPL18 | 0.51 |
| 84747 | UNC119B | 0.51 |
| 23399 | CTDNEP1 | 0.51 |
| 599 | BCL2L2 | 0.51 |
| 197258 | FUK | 0.51 |
| 5207 | PFKFB1 | 0.51 |
| 8131 | NPRL3 | 0.51 |
| 25839 | COG4 | 0.51 |
| 10816 | SPINT3 | 0.51 |
| 60485 | SAV1 | 0.51 |
| 5681 | PSKH1 | 0.51 |
| 80318 | GKAP1 | 0.51 |
| 57088 | PLSCR4 | 0.51 |
| 93129 | ORAI3 | 0.51 |
| 5829 | PXN | 0.51 |
| 2247 | FGF2 | 0.50 |
| 26248 | OR2K2 | 0.50 |
| 84303 | CHCHD6 | 0.50 |
| 3615 | IMPDH2 | 0.50 |
| 1813 | DRD2 | 0.50 |
| 80148 | PQLC1 | 0.50 |
| 390081 | OR52E4 | 0.50 |
| 352954 | GATS | 0.50 |
| 90871 | C9orf123 | 0.50 |
| 50945 | TBX22 | 0.52 |
| 5204 | PFDN5 | 0.52 |
| 5338 | PLD2 | 0.52 |
| 94 | ACVRL1 | 0.52 |
| 54039 | PCBP3 | 0.52 |
| 7691 | ZNF132 | 0.52 |
| 338 | APOB | 0.52 |
| 84658 | EMR3 | 0.52 |
| 283232 | TMEM80 | 0.52 |
| 5430 | POLR2A | 0.52 |
| 54623 | PAF1 | 0.52 |
| 11070 | TMEM115 | 0.52 |
| 10395 | DLC1 | 0.52 |
| 57140 | RNPEPL1 | 0.52 |
| 79781 | IQCA1 | 0.52 |
| 1838 | DTNB | 0.52 |
| 51386 | EIF3L | 0.52 |
| 56919 | DHX33 | 0.52 |
| 57542 | KLHDC5 | 0.52 |
| 3628 | INPP1 | 0.52 |
| 4520 | MTF1 | 0.52 |
| 8547 | FCN3 | 0.52 |
| 60401 | EDA2R | 0.52 |
| 8082 | SSPN | 0.52 |
| 80755 | AARSD1 | 0.52 |
| 710 | SERPING1 | 0.52 |
| 56246 | MRAP | 0.52 |
| 10555 | AGPAT2 | 0.52 |
| 949 | SCARB1 | 0.52 |
| 23743 | BHMT2 | 0.52 |
| 3910 | LAMA4 | 0.52 |
| 60370 | AVPI1 | 0.52 |
| 5021 | OXTR | 0.52 |
| 55997 | CFC1 | 0.52 |
| 23144 | ZC3H3 | 0.52 |
| 56776 | FMN2 | 0.51 |
| 85456 | TNKS1BP1 | 0.51 |
| 283 | ANG | 0.51 |
| 7035 | TFPI | 0.51 |
| 51232 | CRIM1 | 0.51 |
| 112616 | CMTM7 | 0.51 |
| 22981 | NINL | 0.51 |
| 8727 | CTNNAL1 | 0.51 |
| 9902 | MRC2 | 0.51 |
| 10900 | RUNDC3A | 0.51 |
| 51299 | NRN1 | 0.51 |
| 79632 | FAM184A | 0.52 |
| 80820 | EEPD1 | 0.52 |
| 150709 | ANKAR | 0.52 |
| 6591 | SNAI2 | 0.52 |
| 10129 | FRY | 0.52 |
| 5166 | PDK4 | 0.52 |
| 146433 | IL34 | 0.52 |
| 118812 | MORN4 | 0.53 |
| 10516 | FBLN5 | 0.53 |
| 9463 | PICK1 | 0.53 |
| 127495 | LRRC39 | 0.53 |
| 7753 | ZNF202 | 0.53 |
| 79827 | CLMP | 0.53 |
| 203260 | CCDC107 | 0.53 |
| 83657 | DYNLRB2 | 0.53 |
| NCBI gene ID | Gene Symbol | Fold Change |
|---|---|---|
| 1300 | COL10A1 | 42.74 |
| 3007 | HIST1H1D | 29.72 |
| 8366 | HIST1H4B | 25.58 |
| 6286 | S100P | 25.19 |
| 1301 | COL11A1 | 24.72 |
| 3627 | CXCL10 | 17.83 |
| 4283 | CXCL9 | 15.88 |
| 1387 | CREBBP | 12.83 |
| 27299 | ADAMDEC1 | 12.78 |
| 54986 | ULK4 | 12.46 |
| 55771 | PRR11 | 12.02 |
| 54790 | TET2 | 11.25 |
| 6241 | RRM2 | 10.60 |
| 3433 | IFIT2 | 10.49 |
| 6999 | TDO2 | 9.73 |
| 1656 | DDX6 | 9.72 |
| 55088 | C10orf118 | 9.37 |
| 9648 | GCC2 | 9.24 |
| 6696 | SPP1 | 8.92 |
| 2803 | GOLGA4 | 8.57 |
| 83540 | NUF2 | 7.73 |
| 10112 | KIF20A | 7.66 |
| 9833 | MELK | 7.59 |
| 55165 | CEP55 | 7.50 |
| 10142 | AKAP9 | 7.44 |
| 9447 | AIM2 | 7.42 |
| 54443 | ANLN | 5.79 |
| 6710 | SPTB | 5.71 |
| 7272 | TTK | 5.64 |
| 10635 | RAD51AP1 | 5.49 |
| 4069 | LYZ | 5.37 |
| 55183 | RIF1 | 5.34 |
| 891 | CCNB1 | 5.34 |
| 91543 | RSAD2 | 5.31 |
| 81610 | FAM83D | 5.24 |
| 64581 | CLEC7A | 5.10 |
| 10051 | SMC4 | 5.02 |
| 4085 | MAD2L1 | 4.96 |
| 55872 | PBK | 4.83 |
| 991 | CDC20 | 4.82 |
| 9221 | NOLC1 | 4.74 |
| 2124 | EVI2B | 4.66 |
| 375248 | ANKRD36 | 4.66 |
| 1164 | CKS2 | 4.64 |
| 1230 | CCR1 | 4.62 |
| 890 | CCNA2 | 4.56 |
| 127933 | UHMK1 | 4.49 |
| 10274 | STAG1 | 4.45 |
| 597 | BCL2A1 | 4.43 |
| 55355 | HJURP | 4.41 |
| 54210 | TREM1 | 4.36 |
| 253558 | LCLAT1 | 4.26 |
| 2706 | GJB2 | 7.33 |
| 6498 | SKIL | 7.13 |
| 219285 | SAMD9L | 7.06 |
| 10261 | IGSF6 | 7.01 |
| 2335 | FN1 | 6.95 |
| 699 | BUB1 | 6.75 |
| 1058 | CENPA | 6.75 |
| 332 | BIRC5 | 6.73 |
| 51203 | NUSAP1 | 6.59 |
| 259266 | ASPM | 6.54 |
| 1063 | CENPF | 6.49 |
| 165918 | RNF168 | 6.44 |
| 9232 | PTTG1 | 6.34 |
| 5996 | RGS1 | 6.07 |
| 29089 | UBE2T | 5.96 |
| 22974 | TPX2 | 5.94 |
| 4321 | MMP12 | 5.91 |
| 983 | CDK1 | 5.89 |
| 85444 | LRRCC1 | 5.87 |
| 29121 | CLEC2D | 3.83 |
| 4090 | SMAD5 | 3.80 |
| 2123 | EVI2A | 3.80 |
| 57695 | USP37 | 3.79 |
| 133418 | EMB | 3.76 |
| 4131 | MAP1B | 3.76 |
| 9787 | DLGAP5 | 3.75 |
| 9768 | KIAA0101 | 3.74 |
| 54625 | PARP14 | 3.73 |
| 2215 | FCGR3B | 3.71 |
| 9134 | CCNE2 | 3.70 |
| 3117 | HLA-DQA1 | 3.68 |
| 10380 | BPNT1 | 3.67 |
| 79056 | PRRG4 | 3.63 |
| 10673 | TNFSF13B | 3.63 |
| 8467 | SMARCA5 | 3.61 |
| 115908 | CTHRC1 | 3.61 |
| 3428 | IFI16 | 3.61 |
| 1520 | CTSS | 3.61 |
| 10797 | MTHFD2 | 3.57 |
| 55681 | SCYL2 | 3.57 |
| 9749 | PHACTR2 | 3.57 |
| 94240 | EPSTI1 | 3.56 |
| 64151 | NCAPG | 3.51 |
| 25879 | DCAF13 | 3.51 |
| 1033 | CDKN3 | 4.24 |
| 79801 | SHCBP1 | 4.23 |
| 126731 | C1orf96 | 4.21 |
| 6772 | STAT1 | 4.20 |
| 55729 | ATF7IP | 4.14 |
| 6713 | SQLE | 4.14 |
| 157570 | ESCO2 | 4.10 |
| 79871 | RPAP2 | 4.09 |
| 9493 | KIF23 | 4.09 |
| 4751 | NEK2 | 4.05 |
| 10631 | POSTN | 4.03 |
| 23515 | MORC3 | 4.02 |
| 7153 | TOP2A | 4.02 |
| 10403 | NDC80 | 4.00 |
| 10915 | TCERG1 | 3.99 |
| 57650 | KIAA1524 | 3.99 |
| 23049 | SMG1 | 3.93 |
| 80231 | CXorf21 | 3.87 |
| 5111 | PCNA | 3.86 |
| 79682 | MLF1IP | 3.11 |
| 29123 | ANKRD11 | 3.09 |
| 5429 | POLH | 3.09 |
| 701 | BUB1B | 3.07 |
| 200030 | NBPF11 | 3.06 |
| 55677 | IWS1 | 3.06 |
| 160418 | TMTC3 | 3.04 |
| 9147 | NEMF | 3.04 |
| 11320 | MGAT4A | 3.04 |
| 5238 | PGM3 | 3.03 |
| 2820 | GPD2 | 3.02 |
| 388886 | FAM211B | 3.01 |
| 7852 | CXCR4 | 3.00 |
| 57082 | CASC5 | 2.99 |
| 22926 | ATF6 | 2.98 |
| 7594 | ZNF43 | 2.98 |
| 968 | CD68 | 2.97 |
| 7171 | TPM4 | 2.96 |
| 11004 | KIF2C | 2.96 |
| 10808 | HSPH1 | 2.95 |
| 84909 | C9orf3 | 2.94 |
| 1894 | ECT2 | 2.93 |
| 1629 | DBT | 2.92 |
| 116969 | ART5 | 2.90 |
| 3227 | HOXC11 | 2.88 |
| 116064 | LRRC58 | 3.47 |
| 29899 | GPSM2 | 3.47 |
| 135114 | HINT3 | 3.45 |
| 27333 | GOLIM4 | 3.43 |
| 55839 | CENPN | 3.43 |
| 23213 | SULF1 | 3.41 |
| 81671 | VMP1 | 3.39 |
| 9889 | ZBED4 | 3.36 |
| 3092 | HIP1 | 3.34 |
| 51512 | GTSE1 | 3.34 |
| 92797 | HELB | 3.34 |
| 51426 | POLK | 3.30 |
| 5611 | DNAJC3 | 3.30 |
| 6596 | HLTF | 3.28 |
| 9910 | RABGAP1L | 3.25 |
| 528 | ATP6V1C1 | 3.23 |
| 3833 | KIFC1 | 3.23 |
| 197131 | UBR1 | 3.20 |
| 29923 | HILPDA | 3.20 |
| 28998 | MRPL13 | 3.19 |
| 58527 | C6orf115 | 3.19 |
| 79000 | C1orf135 | 3.19 |
| 9857 | CEP350 | 3.18 |
| 84296 | GINS4 | 3.18 |
| 81034 | SLC25A32 | 3.15 |
| 55723 | ASF1B | 3.14 |
| 7110 | TMF1 | 3.14 |
| 84081 | NSRP1 | 3.14 |
| 23075 | SWAP70 | 3.12 |
| 6726 | SRP9 | 2.69 |
| 55215 | FANCI | 2.68 |
| 57590 | WDFY1 | 2.67 |
| 55142 | HAUS2 | 2.66 |
| 23047 | PDS5B | 2.66 |
| 5373 | PMM2 | 2.66 |
| 11065 | UBE2C | 2.66 |
| 23085 | ERC1 | 2.66 |
| 389197 | C4orf50 | 2.65 |
| 11260 | XPOT | 2.65 |
| 29980 | DONSON | 2.65 |
| 64399 | HHIP | 2.64 |
| 6453 | ITSN1 | 2.63 |
| 29108 | PYCARD | 2.63 |
| 9877 | ZC3H11A | 2.62 |
| 3149 | HMGB3 | 2.87 |
| 10437 | IFI30 | 2.87 |
| 57489 | ODF2L | 2.87 |
| 2151 | F2RL2 | 2.86 |
| 23215 | PRRC2C | 2.85 |
| 128710 | C20orf94 | 2.85 |
| 23594 | ORC6 | 2.84 |
| 5205 | ATP8B1 | 2.83 |
| 51430 | C1orf9 | 2.80 |
| 57405 | SPC25 | 2.80 |
| 112401 | BIRC8 | 2.80 |
| 3606 | IL18 | 2.80 |
| 115362 | GBP5 | 2.80 |
| 50515 | CHST11 | 2.79 |
| 83461 | CDCA3 | 2.79 |
| 10744 | PTTG2 | 2.78 |
| 51765 | MST4 | 2.77 |
| 10926 | DBF4 | 2.76 |
| 27125 | AFF4 | 2.75 |
| 10615 | SPAG5 | 2.75 |
| 55143 | CDCA8 | 2.74 |
| 51602 | NOP58 | 2.74 |
| 51478 | HSD17B7 | 2.73 |
| 2209 | FCGR1A | 2.73 |
| 9958 | USP15 | 2.72 |
| 5469 | MED1 | 2.72 |
| 8813 | DPM1 | 2.70 |
| 6731 | SRP72 | 2.70 |
| 9991 | PTBP3 | 2.70 |
| 79866 | BORA | 2.41 |
| 7072 | TIA1 | 2.40 |
| 55632 | G2E3 | 2.40 |
| 2213 | FCGR2B | 2.40 |
| 3987 | LIMS1 | 2.39 |
| 829 | CAPZA1 | 2.39 |
| 26973 | CHORDC1 | 2.38 |
| 435 | ASL | 2.38 |
| 29979 | UBQLN1 | 2.38 |
| 8548 | BLZF1 | 2.37 |
| 9694 | TTC35 | 2.37 |
| 55055 | ZWILCH | 2.36 |
| 4481 | MSR1 | 2.36 |
| 10213 | PSMD14 | 2.35 |
| 9966 | TNFSF15 | 2.35 |
| 81624 | DIAPH3 | 2.62 |
| 79723 | SUV39H2 | 2.61 |
| 55789 | DEPDC1B | 2.61 |
| 10097 | ACTR2 | 2.59 |
| 23036 | ZNF292 | 2.58 |
| 22936 | ELL2 | 2.57 |
| 8477 | GPR65 | 2.57 |
| 23397 | NCAPH | 2.57 |
| 3015 | H2AFZ | 2.54 |
| 55749 | CCAR1 | 2.53 |
| 25937 | WWTR1 | 2.52 |
| 360023 | ZBTB41 | 2.51 |
| 5080 | PAX6 | 2.51 |
| 4193 | MDM2 | 2.51 |
| 24137 | KIF4A | 2.51 |
| 9212 | AURKB | 2.51 |
| 168850 | ZNF800 | 2.50 |
| 55109 | AGGF1 | 2.49 |
| 23185 | LARP4B | 2.49 |
| 51571 | FAM49B | 2.49 |
| 51077 | FCF1 | 2.49 |
| 23167 | EFR3A | 2.49 |
| 23468 | CBX5 | 2.48 |
| 5396 | PRRX1 | 2.48 |
| 10096 | ACTR3 | 2.47 |
| 10308 | ZNF267 | 2.47 |
| 6782 | HSPA13 | 2.47 |
| 3832 | KIF11 | 2.47 |
| 917 | CD3G | 2.47 |
| 80821 | DDHD1 | 2.46 |
| 52 | ACP1 | 2.46 |
| 4179 | CD46 | 2.46 |
| 10499 | NCOA2 | 2.44 |
| 60558 | GUF1 | 2.44 |
| 55676 | SLC30A6 | 2.43 |
| 6646 | SOAT1 | 2.43 |
| 5440 | POLR2K | 2.43 |
| 84955 | NUDCD1 | 2.42 |
| 54739 | XAF1 | 2.42 |
| 84295 | PHF6 | 2.23 |
| 7295 | TXN | 2.23 |
| 2710 | GK | 2.23 |
| 10905 | MAN1A2 | 2.22 |
| 6780 | STAU1 | 2.22 |
| 51582 | AZIN1 | 2.35 |
| 54843 | SYTL2 | 2.34 |
| 9039 | UBA3 | 2.33 |
| 933 | CD22 | 2.33 |
| 5685 | PSMA4 | 2.33 |
| 9885 | OSBPL2 | 2.33 |
| 9262 | STK17B | 2.33 |
| 56942 | C16orf61 | 2.32 |
| 10767 | HBS1L | 2.32 |
| 87178 | PNPT1 | 2.32 |
| 6303 | SAT1 | 2.32 |
| 7316 | UBC | 2.32 |
| 4205 | MEF2A | 2.32 |
| 85465 | EPT1 | 2.31 |
| 84640 | USP38 | 2.31 |
| 5810 | RAD1 | 2.30 |
| 64397 | ZFP106 | 2.29 |
| 5706 | PSMC6 | 2.29 |
| 22948 | CCT5 | 2.29 |
| 10672 | GNA13 | 2.29 |
| 339344 | MYPOP | 2.28 |
| 7292 | TNFSF4 | 2.28 |
| 57103 | C12orf5 | 2.28 |
| 388403 | YPEL2 | 2.28 |
| 54876 | DCAF16 | 2.27 |
| 113235 | SLC46A1 | 2.27 |
| 11177 | BAZ1A | 2.27 |
| 339175 | METTL2A | 2.26 |
| 26586 | CKAP2 | 2.26 |
| 55785 | FGD6 | 2.26 |
| 24145 | PANX1 | 2.25 |
| 253461 | ZBTB38 | 2.25 |
| 23232 | TBC1D12 | 2.25 |
| 995 | CDC25C | 2.25 |
| 55974 | SLC50A1 | 2.25 |
| 472 | ATM | 2.25 |
| 23008 | KLHDC10 | 2.24 |
| 10024 | TROAP | 2.24 |
| 9521 | EEF1E1 | 2.24 |
| 7402 | UTRN | 2.09 |
| 55589 | BMP2K | 2.08 |
| 158747 | MOSPD2 | 2.08 |
| 56886 | UGGT1 | 2.07 |
| 203100 | HTRA4 | 2.07 |
| 10282 | BET1 | 2.22 |
| 134430 | WDR36 | 2.21 |
| 4299 | AFF1 | 2.21 |
| 6747 | SSR3 | 2.21 |
| 7334 | UBE2N | 2.21 |
| 5965 | RECQL | 2.21 |
| 4605 | MYBL2 | 2.2 |
| 6093 | ROCK1 | 2.19 |
| 161725 | OTUD7A | 2.19 |
| 23518 | R3HDM1 | 2.18 |
| 2239 | GPC4 | 2.18 |
| 28977 | MRPL42 | 2.18 |
| 64859 | OBFC2A | 2.18 |
| 3845 | KRAS | 2.18 |
| 51388 | NIP7 | 2.18 |
| 7586 | ZKSCAN1 | 2.18 |
| 10762 | NUP50 | 2.17 |
| 7328 | UBE2H | 2.17 |
| 10730 | YME1L1 | 2.17 |
| 23093 | TTLL5 | 2.17 |
| 6790 | AURKA | 2.17 |
| 22889 | KIAA0907 | 2.17 |
| 10875 | FGL2 | 2.17 |
| 23161 | SNX13 | 2.17 |
| 9169 | SCAF11 | 2.16 |
| 1788 | DNMT3A | 2.15 |
| 9088 | PKMYT1 | 2.15 |
| 23033 | DOPEY1 | 2.13 |
| 89882 | TPD52L3 | 2.13 |
| 6556 | SLC11A1 | 2.13 |
| 64216 | TFB2M | 2.13 |
| 3071 | NCKAP1L | 2.13 |
| 51068 | NMD3 | 2.13 |
| 509 | ATP5C1 | 2.13 |
| 953 | ENTPD1 | 2.13 |
| 51105 | PHF20L1 | 2.13 |
| 5062 | PAK2 | 2.13 |
| 9205 | ZMYM5 | 2.12 |
| 55157 | DARS2 | 2.12 |
| 8520 | HAT1 | 2.11 |
| 79739 | TTLL7 | 2.11 |
| 9495 | AKAP5 | 2.10 |
| 3181 | HNRNPA2B 1 | 2.10 |
| 55279 | ZNF654 | 2.07 |
| 54499 | TMCO1 | 2.07 |
| 81930 | KIF18A | 2.07 |
| 142686 | ASB14 | 2.06 |
| 55209 | SETD5 | 2.06 |
| 9736 | USP34 | 2.04 |
| 116285 | ACSM1 | 2.04 |
| 2201 | FBN2 | 2.04 |
| 963 | CD53 | 2.04 |
| 55159 | RFWD3 | 2.03 |
| 9871 | SEC24D | 2.03 |
| 9887 | SMG7 | 2.02 |
| 23376 | UFL1 | 2.02 |
| 79646 | PANK3 | 2.01 |
| 50613 | UBQLN3 | 2.00 |
| 201595 | STT3B | 2.00 |
| 59345 | GNB4 | 1.99 |
| 5876 | RABGGTB | 1.99 |
| 79820 | CATSPERB | 1.99 |
| 6637 | SNRPG | 1.99 |
| 51330 | TNFRSF12A | 1.99 |
| 9928 | KIF14 | 1.99 |
| 286097 | EFHA2 | 1.98 |
| 9131 | AIFM1 | 1.98 |
| 488 | ATP2A2 | 1.98 |
| 23042 | PDXDC1 | 1.98 |
| 7114 | TMSB4X | 1.98 |
| 9123 | SLC16A3 | 1.98 |
| 54454 | ATAD2B | 1.97 |
| 23143 | LRCH1 | 1.97 |
| 4212 | MEIS2 | 1.97 |
| 1457 | CSNK2A1 | 1.97 |
| 80012 | PHC3 | 1.97 |
| 128497 | SPATA25 | 1.96 |
| 186 | AGTR2 | 1.96 |
| 53981 | CPSF2 | 1.96 |
| 56996 | SLC12A9 | 1.96 |
| 1584 | CYP11B1 | 1.96 |
| 133619 | PRRC1 | 1.96 |
| 4288 | MKI67 | 1.96 |
| 9014 | TAF1B | 1.96 |
| 55858 | TMEM165 | 1.96 |
| 2212 | FCGR2A | 1.96 |
| 389898 | UBE2NL | 2.10 |
| 29850 | TRPM5 | 2.10 |
| 3070 | HELLS | 2.10 |
| 331 | XIAP | 2.09 |
| 55751 | TMEM184C | 2.09 |
| 2146 | EZH2 | 2.09 |
| 26057 | ANKRD17 | 1.95 |
| 128061 | C1orf131 | 1.95 |
| 64090 | GAL3ST2 | 1.94 |
| 130507 | UBR3 | 1.93 |
| 2298 | FOXD4 | 1.93 |
| 123169 | LEO1 | 1.93 |
| 57187 | THOC2 | 1.93 |
| 148789 | B3GALNT2 | 1.93 |
| 58508 | MLL3 | 1.92 |
| 5701 | PSMC2 | 1.92 |
| 148066 | ZNRF4 | 1.92 |
| 6670 | SP3 | 1.92 |
| 10075 | HUWE1 | 1.96 |
| 220988 | HNRNPA3 | 1.96 |
| 80146 | UXS1 | 1.95 |
| 122011 | CSNK1A1L | 1.95 |
| 150468 | CKAP2L | 1.95 |
| 84624 | FNDC1 | 1.95 |
| 7332 | UBE2L3 | 1.92 |
| 3336 | HSPE1 | 1.92 |
| 54800 | KLHL24 | 1.92 |
| 2290 | FOXG1 | 1.91 |
| 50848 | F11R | 1.91 |
| 10627 | MYL12A | 1.91 |
| 5074 | PAWR | 1.91 |
| 6476 | SI | 1.91 |
| 1009 | CDH11 | 1.90 |
| 29066 | ZC3H7A | 1.90 |
| 51319 | RSRC1 | 1.90 |
| miRBase Accession | miRNA name | AUC | p-value |
|---|---|---|---|
| MIMAT0002856 | hsa-miR-520d-3p | 0.49 | 0.549112 |
| MIMAT0000265 | hsa-miR-204-5p | 0.98 | 6.47 × 10−10 *** |
| MIMAT0000272 | hsa-miR-215 | 0.21 | 0.999782 |
| MIMAT0000271 | hsa-miR-214-3p | 0.68 | 0.010387 * |
| MIMAT0002820 | hsa-miR-497-5p | 0.99 | 2.75 × 10−10 *** |
| MIMAT0000076 | hsa-miR-21-5p | 0.78 | 0.000184 *** |
| MIMAT0000738 | hsa-miR-383 | 0.60 | 0.106284 |
| MIMAT0000423 | hsa-miR-125b-5p | 0.99 | 2.48 × 10−10 *** |
| MIMAT0000064 | hsa-let-7c | 0.93 | 3.79 × 10−8 *** |
| MIMAT0000089 | hsa-miR-31-5p | 0.80 | 8.63 × 10−5 *** |
| MIMAT0000077 | hsa-miR-22-3p | 0.27 | 0.99749 |
| MIMAT0000098 | hsa-miR-100-5p | 0.98 | 5.55 × 10−10 *** |
| MIMAT0000097 | hsa-miR-99a-5p | 0.99 | 2.55 × 10−10 *** |
| MIMAT0000443 | hsa-miR-125a-5p | 0.31 | 0.990694 |
| MIMAT0002819 | hsa-miR-193b-3p | 0.41 | 0.86128 |
| MIMAT0000250 | hsa-miR-139-5p | 0.99 | 2.42 × 10−10 *** |
| MIMAT0000437 | hsa-miR-145-5p | 0.96 | 3.14 × 10−9 *** |
| MIMAT0000421 | hsa-miR-122-5p | 0.48 | 0.597483 |
| miRBase Accession | miR name | Total gene count | L0 count | L1 count |
|---|---|---|---|---|
| MIMAT0002819 | hsa-miR-193b-3p | 16 | 1 | 15 |
| MIMAT0000250 | hsa-miR-139-5p | 28 | 10 | 18 |
| MIMAT0000437 | hsa-miR-145-5p | 86 | 22 | 64 |
| MIMAT0000423 | hsa-miR-125b-5p | 211 | 16 | 195 |
| MIMAT0000443 | hsa-miR-125a-5p | 206 | 14 | 192 |
| MIMAT0000097 | hsa-miR-99a-5p | 14 | 1 | 13 |
| MIMAT0000265 | hsa-miR-204-5p | 64 | 18 | 46 |
| MIMAT0000076 | hsa-miR-21-5p | 91 | 16 | 75 |
| MIMAT0000064 | hsa-let-7c | 96 | 20 | 76 |
| MIMAT0000421 | hsa-miR-122-5p | 5 | 3 | 2 |
| MIMAT0000098 | hsa-miR-100-5p | 14 | 1 | 13 |
| MIMAT0000272 | hsa-miR-215 | 3 | 3 | 0 |
| MIMAT0000271 | hsa-miR-214-3p | 14 | 8 | 6 |
| MIMAT0000738 | hsa-miR-383 | 34 | 3 | 31 |
| MIMAT0002856 | hsa-miR-520d-3p | 146 | 23 | 123 |
| MIMAT0000077 | hsa-miR-22-3p | 46 | 11 | 35 |
| MIMAT0002820 | hsa-miR-497-5p | 267 | 32 | 235 |
| MIMAT0000089 | hsa-miR-31-5p | 34 | 3 | 31 |
| MIMAT0002856(hsa-miR-520d-3p) | ||
|---|---|---|
| GO term | Genes | Adj. p-value |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | SH3KBP1, HDAC2, RET, ABI1, LYN, GRB2, SORBS1, CLTC, CLTA, CDC42, CASP9, RAF1, SRC, AP2A1, AP2B1, MAPK3, ARHGEF7, PRKCA, RPS6, PRKAR2B, MAPK1, ARHGEF6, CDK1, SH3GL2, EIF4G1, HDAC1, ECT2, MKNK1, CASP3, PRKACA, ADRB2, PRKAR2A, EIF4B, SHC1, RAC1 | 2.77 × 10−29 |
| GO:0048011, Nerve growth factor receptor signaling pathway | HDAC2, GRB2, CLTC, CLTA, CASP9, RAF1, SRC, AP2A1, AP2B1, MAPK3, ARHGEF7, PRKCA, PRKAR2B, MAPK1, ARHGEF6, CDK1, SH3GL2, HDAC1, ECT2, CASP3, PRKACA, PRKAR2A, SHC1, RAC1 | 5.09 × 10−24 |
| GO:0007173, Epidermal growth factor receptor signaling pathway | SH3KBP1, GRB2, CLTC, CLTA, CDC42, CASP9, RAF1, SRC, AP2A1, AP2B1, MAPK3, ARHGEF7, PRKCA, PRKAR2B, MAPK1, CDK1, SH3GL2, PRKACA, PRKAR2A, SHC1 | 2.96 × 10−22 |
| GO:0043067, Regulation of programmed cell death | HDAC2, STK17B, ESR1, ABL1, LYN, TP53, GABRB3, PAK2, LCK, CASP9, RAF1, PLK1, ARHGEF7, PRKCA, RPS6, SH3RF1, MAPK1, IFT57, ARHGAP10, ARHGEF6, CDK1, APAF1, HDAC1, ECT2, CASP3, SOX10, EP300, ARAF, TFAP2A, ADRB2, HCK, KLHL20, CASP8, HIP1, RAC1 | 4.76 × 10−19 |
| GO:0042058, Regulation of epidermal growth factor receptor signaling pathway | SH3KBP1, ESR1, GRB2, CLTC, CLTA, CDC42, AP2A1, AP2B1, ARHGEF7, SH3GL2, SHC1 | 2.36 × 10−12 |
| GO:0008543, Fibroblast growth factor receptor signaling pathway | GRB2, CASP9, RAF1, SRC, MAPK3, PRKCA, PRKAR2B, MAPK1, CDK1, MKNK1, PRKACA, PRKAR2A, SHC1 | 2.39 × 10−12 |
| GO:0043068, Positive regulation of programmed cell death | STK17B, ABL1, LYN, TP53, LCK, CASP9, ARHGEF7, PRKCA, RPS6, SH3RF1, MAPK1, ARHGEF6, APAF1, ECT2, CASP3, EP300, TFAP2A, ADRB2, CASP8, HIP1, RAC1 | 3.09 × 10−12 |
| GO:0010942, Positive regulation of cell death | STK17B, ABL1, LYN, TP53, LCK, CASP9, ARHGEF7, PRKCA, RPS6, SH3RF1, MAPK1, ARHGEF6, APAF1, ECT2, CASP3, EP300, TFAP2A, ADRB2, CASP8, HIP1, RAC1 | 4.49 × 10−12 |
| GO:0006917, Induction of apoptosis | STK17B, ABL1, TP53, LCK, CASP9, ARHGEF7, PRKCA, SH3RF1, MAPK1, ARHGEF6, APAF1, ECT2, CASP3, EP300, CASP8, HIP1, RAC1 | 6.91 × 10−11 |
| GO:0012502, Induction of programmed cell death | STK17B, ABL1, TP53, LCK, CASP9, ARHGEF7, PRKCA, SH3RF1, MAPK1, ARHGEF6, APAF1, ECT2, CASP3, EP300, CASP8, HIP1, RAC1 | 7.42 × 10−11 |
| GO:0042059, Negative regulation of epidermal growth factor receptor signaling pathway | SH3KBP1, GRB2, CLTC, CLTA, CDC42, AP2A1, AP2B1, ARHGEF7, SH3GL2 | 8.63 × 10−11 |
| GO:0015630, Microtubule cytoskeleton | STMN1, SORBS1, SMAD4, CLTC, CDC42, LCK, RACGAP1, PLK1, PRKAR2B, YES1, MAPK1, IFT57, CDK1, ECT2, PRKACA, RB1, EP300, CCNB1, CHAF1B, TFAP2A, CASP8, PRKAR2A | 4.75 × 10−10 |
| GO:0060548, Negative regulation of cell death | HDAC2, ESR1, TP53, SMAD4, RAF1, PLK1, PRKCA, RPS6, SH3RF1, CDK1, HDAC1, CASP3, SOX10, ARAF, TFAP2A, HCK, KLHL20 | 6.27 × 10−8 |
| GO:0008286, Insulin receptor signaling pathway | GRB2, SORBS1, RAF1, MAPK3, RPS6, MAPK1, CDK1, EIF4G1, EIF4B, SHC1 | 2.15 × 10−7 |
| GO:0043069, Negative regulation of programmed cell death | HDAC2, ESR1, TP53, RAF1, PLK1, PRKCA, RPS6, SH3RF1, CDK1, HDAC1, CASP3, SOX10, ARAF, TFAP2A, HCK, KLHL20 | 3.13 × 10−7 |
| GO:0008284, Positive regulation of cell proliferation | HDAC2, ESR1, LYN, CDC42, E2F1, PRKCA, MAPK1, CDK1, RHOG, HDAC1, NCK1, SOX10, CCNB1, ADRB2, HCK, SHC1 | 8.34 × 10−7 |
| GO:0051988, Regulation of attachment of spindle microtubules to kinetochore | CDC42, RACGAP1, ECT2, CCNB1 | 3.27 × 10−5 |
| GO:0008629, Induction of apoptosis by intracellular signals | ABL1, TP53, CASP9, APAF1, CASP3, EP300, CASP8 | 5.83 × 10−5 |
| MIMAT0002820(hsa-miR-497-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0043067, Regulation of programmed cell death | ESR1, MEN1, ABL1, HIPK3, PPARGC1A, SIAH1, SH3RF1, PAK2, LCK, MED1, PPARG, CBX4, ARHGEF7, YWHAB, RXRA, ACVR1, MAPK1, CASP3, CASP6, AR, PTPRF, MDM2, BRCA1, MLH1, RAB27A, PIAS4, FAF1, RAC1, VHL, SKI, NR4A1, LYN, TP53, PSMC2, GATA1, GATA6, GATA3, RAF1, CDKN1B, PLK1, PSMD11, HOXA13, RPS6, ESR2, ARHGAP10, ARHGEF6, SMAD3, SKIL, RYR2, PSEN1, HCK, TRAF2 | 2.67 × 10−25 |
| GO:0043068, Positive regulation of programmed cell death | MEN1, ABL1, SIAH1, SH3RF1, LCK, PPARG, ARHGEF7, YWHAB, RXRA, MAPK1, CASP3, CASP6, PTPRF, BRCA1, MLH1, RAB27A, PIAS4, FAF1, RAC1, NR4A1, LYN, TP53, GATA6, CDKN1B, HOXA13, RPS6, ESR2, ARHGEF6, SMAD3, RYR2, PSEN1, TRAF2 | 1.74 × 10−17 |
| GO:0010942, Positive regulation of cell death | MEN1, ABL1, SIAH1, SH3RF1, LCK, PPARG, ARHGEF7, YWHAB, RXRA, MAPK1, CASP3, CASP6, PTPRF, BRCA1, MLH1, RAB27A, PIAS4, FAF1, RAC1, NR4A1, LYN, TP53, GATA6, CDKN1B, HOXA13, RPS6, ESR2, ARHGEF6, SMAD3, RYR2, PSEN1, TRAF2 | 3.08 × 10−17 |
| GO:0008285, Negative regulation of cell proliferation | MEN1, MED1, PPARG, RXRA, CASP3, AR, PTPRF, VDR, VHL, SKI, LYN, TP53, TOB1, GATA1, GATA3, RAF1, HNF4A, CDKN1B, BRD7, MED25, ESR2, ABI1, SMAD1, SMAD2, SMAD3, SMAD4, SOX7 | 3.85 × 10−14 |
| GO:0015629, Actin cytoskeleton | ABL1, SORBS1, FLNA, SEPT7, ANLN, MACF1, HAP1, SH3PXD2A, IQGAP2, BRCA1, ACTC1, ACTA1, MYL2, MYLK, SORBS2, ARPC4, ARPC5, ACTR2, ACTR3, ARPC1B, WASF1, WASF2, HCK | 2.52 × 10−13 |
| GO:0006917, Induction of apoptosis | ABL1, SH3RF1, LCK, PPARG, ARHGEF7, YWHAB, MAPK1, CASP3, CASP6, BRCA1, MLH1, RAB27A, RAC1, NR4A1, TP53, CDKN1B, ARHGEF6, SMAD3, RYR2, PSEN1, TRAF2 | 9.69 × 10−11 |
| GO:0012502, Induction of programmed cell death | ABL1, SH3RF1, LCK, PPARG, ARHGEF7, YWHAB, MAPK1, CASP3, CASP6, BRCA1, MLH1, RAB27A, RAC1, NR4A1, TP53, CDKN1B, ARHGEF6, SMAD3, RYR2, PSEN1, TRAF2 | 1.06 × 10−10 |
| GO:0007178, Transmembrane receptor protein serine/threonine kinase signaling pathway | ACVR1, SMURF2, SKI, GDF6, BMP6, ZNF8, GATA4, HNF4A, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, RYR2 | 1.22 × 10−10 |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | SORBS1, CDC42, SRC, MAPK3, ARHGEF7, YWHAB, MAPK1, SH3GL2, CASP3, MDM2, EIF4G1, RAC1, SH3KBP1, NR4A1, LYN, GRB2, RAF1, CDKN1B, RPS6, ABI1, ARHGEF6, MKNK1, PSEN1, EIF4B | 1.67 × 10−10 |
| GO:0090092, Regulation of transmembrane receptor protein serine/threonine kinase signaling pathway | MEN1, ACVR1, SMURF2, SKI, GDF6, TP53, BMP6, GATA4, GATA6, HOXA13, SMAD2, SMAD3, SMAD4, SKIL | 7.51 × 10−10 |
| GO:0030509, BMP signaling pathway | ACVR1, SMURF2, SKI, GDF6, BMP6, ZNF8, SMAD1, SMAD4, SMAD5, RYR2 | 3.25 × 10−9 |
| GO:0060548, Negative regulation of cell death | ESR1, HIPK3, PPARGC1A, SH3RF1, MED1, CBX4, ACVR1, CASP3, AR, MDM2, VHL, TP53, GATA1, GATA6, GATA3, RAF1, CDKN1B, PLK1, RPS6, SMAD3, SMAD4, PSEN1, HCK | 7.88 × 10−9 |
| GO:0007173, Epidermal growth factor receptor signaling pathway | CDC42, SRC, MAPK3, ARHGEF7, YWHAB, MAPK1, SH3GL2, MDM2, SH3KBP1, NR4A1, GRB2, RAF1, CDKN1B | 9.22 × 10−9 |
| GO:0030521, Androgen receptor signaling pathway | PPARGC1A, MED14, MED1, AR, BRCA1, MED12, PIAS1, RAN, NR1I3 | 1.42 × 10−8 |
| GO:0043069, Negative regulation of programmed cell death | ESR1, HIPK3, PPARGC1A, SH3RF1, MED1, CBX4, ACVR1, CASP3, AR, MDM2, VHL, TP53, GATA1, GATA6, GATA3, RAF1, CDKN1B, PLK1, RPS6, SMAD3, PSEN1, HCK | 2.44 × 10−8 |
| GO:0048011, Nerve growth factor receptor signaling pathway | SRC, MAPK3, ARHGEF7, YWHAB, MAPK1, SH3GL2, CASP3, MDM2, RAC1, NR4A1, GRB2, RAF1, CDKN1B, ARHGEF6, PSEN1 | 2.64 × 10−8 |
| GO:0032956, Regulation of actin cytoskeleton organization | ABL1, LRP1, ARPC4, ARPC5, ACTR3, ARPC1B, SMAD3, NCK1, SORBS3, HCK, LIMK1 | 6.42 × 10−6 |
| GO:0008543, Fibroblast growth factor receptor signaling pathway | SRC, MAPK3, YWHAB, MAPK1, MDM2, NR4A1, GRB2, RAF1, CDKN1B, MKNK1 | 9.06 × 10−6 |
| GO:0042059, Negative regulation of epidermal growth factor receptor signaling pathway | CDC42, ARHGEF7, SH3GL2, PTPRF, SH3KBP1, GRB2, PSEN1 | 1.11 × 10−5 |
| GO:0042058, Regulation of epidermal growth factor receptor signaling pathway | ESR1, CDC42, ARHGEF7, SH3GL2, PTPRF, SH3KBP1, GRB2, PSEN1 | 1.17 × 10−5 |
| GO:0007179, Transforming growth factor beta receptor signaling pathway | ACVR1, SMURF2, SKI, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5 | 1.67 × 10−5 |
| GO:0015630, Microtubule cytoskeleton | STMN1, RIF1, SORBS1, CDC42, LCK, RACGAP1, YES1, YWHAB, MAPK1, SEPT7, KIF23, CDC16, MACF1, BRCA1, FEZ1, NCOR1, PLK1, CHD3, SMAD4, CEP350, CDC27, PSEN1 | 2.14 × 10−5 |
| GO:0017015, Regulation of transforming growth factor beta receptor signaling pathway | MEN1, SMURF2, SKI, TP53, SMAD2, SMAD3, SMAD4, SKIL | 2.30 × 10−5 |
| GO:0070302, Regulation of stress-activated protein kinase signaling cascade | MEN1, ZEB2, HIPK3, SH3RF1, CDC42, MAPK3, MAPK1, LYN, NCOR1, TRAF2 | 2.74 × 10−5 |
| GO:0001959, Regulation of cytokine-mediated signaling pathway | HSP90AB1, MED1, PPARG, PTPRF, NR1H2, PIAS1, IL36RN, HIPK1 | 6.35 × 10−5 |
| GO:0008284, Positive regulation of cell proliferation | ESR1, CDC42, MED1, RARA, MAPK1, AR, MDM2, NR4A1, LYN, FZR1, BMP6, GATA1, GATA4, GATA6, CDKN1B, NCK1, HCLS1, HCK | 7.29 × 10−5 |
| MIMAT0000423(hsa-miR-125b-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0043067, Regulation of programmed cell death | HMGA2, PML, PRNP, FGF2, XRCC4, BRCA1, IGFBP3, HDAC3, CTNNB1, CD5, CDK1, NKX2-5, MEF2C, PRKCI, CASP2, PSMA4, PSMA3, CFDP1, CAV1, FAF1, YWHAB, HIF1A, RELA, TCF7L2, TNFSF12, PSEN2, TP53, TOP2A, TNFRSF4, BID, MYC, JUN, OGT, CDKN1A, HOXA13, RNF7, PPP2R4, HDAC2, HDAC1, SNCA, PTEN, NFKBIA, IFI16, NOL3, TRAF2, HSP90B1 | 4.56 × 10−24 |
| GO:0060548, Negative regulation of cell death | HMGA2, PRNP, FGF2, XRCC4, HDAC3, CTNNB1, CDK1, NKX2-5, MEF2C, PRKCI, CFDP1, HIF1A, RELA, TCF7L2, PSEN2, TP53, TNFRSF4, MYC, JUN, CDKN1A, RNF7, HDAC2, HDAC1, SNCA, PTEN, MGMT, NFKBIA, NOL3, HSP90B1 | 8.04 × 10−17 |
| GO:0008284, Positive regulation of cell proliferation | HMGA2, FGF2, XRCC4, CDC25B, CTNNB1, EGR1, AGGF1, CDK1, NKX2-5, MEF2C, PRKCI, IRS1, HIF1A, RELA, HCLS1, TNFSF12, ARNT, PTPRC, TNFSF4, TNFRSF4, MYC, JUN, FGF1, CDKN1A, HDAC2, HDAC1, NOLC1, PTEN | 2.20 × 10−15 |
| GO:0043069, Negative regulation of programmed cell death | HMGA2, PRNP, XRCC4, HDAC3, CTNNB1, CDK1, NKX2-5, MEF2C, PRKCI, CFDP1, HIF1A, RELA, TCF7L2, PSEN2, TP53, TNFRSF4, MYC, JUN, CDKN1A, RNF7, HDAC2, HDAC1, SNCA, PTEN, NFKBIA, NOL3, HSP90B1 | 3.62 × 10−15 |
| GO:0043068, Positive regulation of programmed cell death | HMGA2, PML, BRCA1, IGFBP3, CTNNB1, CD5, MEF2C, PRKCI, CASP2, CAV1, FAF1, YWHAB, TNFSF12, PSEN2, TP53, TOP2A, BID, JUN, OGT, CDKN1A, HOXA13, RNF7, PPP2R4, PTEN, IFI16, TRAF2 | 4.51 × 10−14 |
| GO:0010942, Positive regulation of cell death | HMGA2, PML, BRCA1, IGFBP3, CTNNB1, CD5, MEF2C, PRKCI, CASP2, CAV1, FAF1, YWHAB, TNFSF12, PSEN2, TP53, TOP2A, BID, JUN, OGT, CDKN1A, HOXA13, RNF7, PPP2R4, PTEN, IFI16, TRAF2 | 7.15 × 10−14 |
| GO:0006916, Anti-apoptosis | PRNP, HDAC3, CDK1, NKX2-5, MEF2C, PRKCI, CFDP1, RELA, TCF7L2, PSEN2, RNF7, HDAC1, SNCA, NFKBIA, NOL3, HSP90B1 | 1.25 × 10−10 |
| GO:0008285, Negative regulation of cell proliferation | SERPINF1, SRF, PML, PRNP, FGF2, CSNK2B, IGFBP3, CTNNB1, CAV1, HMGA1, VDR, CDH5, HSF1, COL18A1, TP53, MYC, JUN, CDKN1A, PAK1, PTEN | 2.08 × 10−9 |
| GO:0015630, Microtubule cytoskeleton | STMN1, KIF1C, RANGAP1, CDC25B, BRCA1, HDAC3, CTNNB1, PIN4, HSPH1, RANBP9, CDK1, SPTAN1, YWHAQ, DVL1, FKBP4, YWHAB, CCDC85B, MAPT, PSEN2, TOP2A, SPIB, MYC, OGT, APEX1, PAFAH1B1 | 2.24 × 10−9 |
| GO:0048011, Nerve growth factor receptor signaling pathway | HDAC3, CDK1, MEF2C, PRKCI, CASP2, IRS1, YWHAB, RELA, PSEN2, HDAC2, HDAC1, PTEN, ATF1, NFKBIA | 1.99 × 10−8 |
| GO:0050678, Regulation of epithelial cell proliferation | SERPINF1, PGR, FGF2, CTNNB1, AGGF1, CAV1, HIF1A, TNFSF12, ARNT, MYC, JUN, FGF1, PTEN | 3.41 × 10−8 |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | FGF2, HDAC3, CDK1, MEF2C, PRKCI, CASP2, IRS1, FIBP, PTPN1, YWHAB, RELA, PSEN2, FGF1, HDAC2, HDAC1, PTEN, ATF1, NFKBIA, EIF4EBP1 | 6.39 × 10−8 |
| GO:0006917, Induction of apoptosis | PML, BRCA1, CD5, CASP2, CAV1, YWHAB, TNFSF12, PSEN2, TP53, BID, OGT, CDKN1A, RNF7, PTEN, IFI16, TRAF2 | 1.68 × 10−7 |
| GO:0012502, Induction of programmed cell death | PML, BRCA1, CD5, CASP2, CAV1, YWHAB, TNFSF12, PSEN2, TP53, BID, OGT, CDKN1A, RNF7, PTEN, IFI16, TRAF2 | 1.81 × 10−7 |
| GO:0035666, TRIF-dependent toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 2.40 × 10−6 |
| GO:0034138, Toll-like receptor 3 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 2.71 × 10−6 |
| GO:0051693, Actin filament capping | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 3.43 × 10−6 |
| GO:0002756, MyD88- independent toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 3.82 × 10−6 |
| GO:0034134, Toll-like receptor 2 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 5.33 × 10−6 |
| GO:0034130, Toll-like receptor 1 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 5.33 × 10−6 |
| GO:0030835, Negative regulation of actin filament depolymerization | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 5.58 × 10−6 |
| GO:0015629, Actin cytoskeleton | WAS, CDH1, BRCA1, SPTB, SPTBN1, SPTAN1, CTDP1, STX1A, SPTA1, PAK1, SNCA, ADD1, EPB41, EPB49 | 5.58 × 10−6 |
| GO:0002755, MyD88-dependent toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 7.66 × 10−6 |
| GO:0034142, Toll-like receptor 4 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 1.14 × 10−5 |
| GO:0050679, Positive regulation of epithelial cell proliferation | FGF2, CTNNB1, AGGF1, HIF1A, TNFSF12, ARNT, MYC, JUN, FGF1 | 1.14 × 10−5 |
| MIMAT0000076(hsa-miR-21-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0030834, Regulation of actin filament depolymerization | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 1.27 × 10−5 |
| GO:0030837, Negative regulation of actin filament polymerization | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 2.71 × 10−5 |
| GO:0002224, Toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 2.96 × 10−5 |
| GO:0008629, Induction of apoptosis by intracellular signals | PML, BRCA1, YWHAB, TP53, BID, CDKN1A, RNF7, IFI16 | 3.52 × 10−5 |
| GO:0043067, Regulation of programmed cell death | SPRY2, TP53, ADAMTSL4, ETS1, TDGF1, RAF1, HOXA5, HOXA13, MSX1, MSX2, NKX2-5, CBL, INHBB, COL4A3, ACVR1C, TRAF2 | 1.92 × 10−5 |
| GO:0007173, Epidermal growth factor receptor signaling pathway | SPRY2, SPRY1, GRB2, PTPN11, TDGF1, RAF1, CBL | 9.44 × 10−5 |
| MIMAT0000250(hsa-miR-139-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0035583, Negative regulation of transforming growth factor beta receptor signaling pathway by extracellular sequestering of TGFbeta | LTBP1, FBN1, FBN2 | 2.61 × 10−5 |
| MIMAT0000089(hsa-miR-31-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0007187, G-protein signaling, coupled to cyclic nucleotide second messenger | GNA12, GNA13, DRD5, MTNR1A, S1PR3, TSHR, S1PR4 | 8.74 × 10−7 |
| GO:0019935, Cyclic-nucleotidemediated signaling | GNA12, GNA13, DRD5, MTNR1A, S1PR3, TSHR, S1PR4 | 1.60 × 10−6 |
| GO:0007188, G-protein signaling, coupled to cAMP nucleotide second messenger | GNA12, GNA13, DRD5, S1PR3, TSHR, S1PR4 | 6.82 × 10−6 |
| GO:0048011, Nerve growth factor receptor signaling pathway | ARHGEF1, PRKCD, ARHGEF12, PRKACA, PRKCE, MCF2, ARHGEF11 | 6.93 × 10−6 |
| GO:0019933, CAMP-mediated signaling | GNA12, GNA13, DRD5, S1PR3, TSHR, S1PR4 | 1.05 × 10−5 |
| GO:0043067, Regulation of programmed cell death | PTK2B, PRKCD, TGFBR1, ARHGEF12, CTNNB1, PRKCE, TIA1, MCF2, FASTK, ARHGEF11, F2R | 1.25 × 10−5 |
| GO:0003376, Sphingosine-1- phosphate signaling pathway | S1PR3, S1PR2, S1PR4 | 3.22 × 10−5 |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | PTK2B, ARHGEF1, PRKCD, ARHGEF12, PRKACA, PRKCE, MCF2, ARHGEF11 | 5.20 × 10−5 |
| GO:0043068, Positive regulation of programmed cell death | TGFBR1, ARHGEF12, CTNNB1, PRKCE, TIA1, MCF2, FASTK, ARHGEF11 | 7.67 × 10−5 |
| GO:0010942, Positive regulation of cell death | TGFBR1, ARHGEF12, CTNNB1, PRKCE, TIA1, MCF2, FASTK, ARHGEF11 | 8.78 × 10−5 |
| GO:0006917, Induction of apoptosis | TGFBR1, ARHGEF12, PRKCE, TIA1, MCF2, FASTK, ARHGEF11 | 9.32 × 10−5 |
| GO:0012502, Induction of programmed cell death | TGFBR1, ARHGEF12, PRKCE, TIA1, MCF2, FASTK, ARHGEF11 | 9.51 × 10−5 |
| MIMAT0000437(hsa-miR-145-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0030509, BMP signaling pathway | BMP6, ZNF8, ACVR1, SMAD1, SMAD4, RYR2, SMAD5, SMURF2, GDF6 | 1.37 × 10−11 |
| GO:0007178, Transmembrane receptor protein serine/threonine kinase signaling pathway | BMP6, ZNF8, ACVR1, SMAD1, SMAD4, RYR2, SMAD5, SMURF2, GDF6 | 2.56 × 10−8 |
| GO:0090092, Regulation of transmembrane receptor protein serine/threonine kinase signaling pathway | MEN1, TP53, BMP6, HOXA13, ACVR1, SMAD4, SULF1, SMURF2, GDF6 | 8.22 × 10−8 |
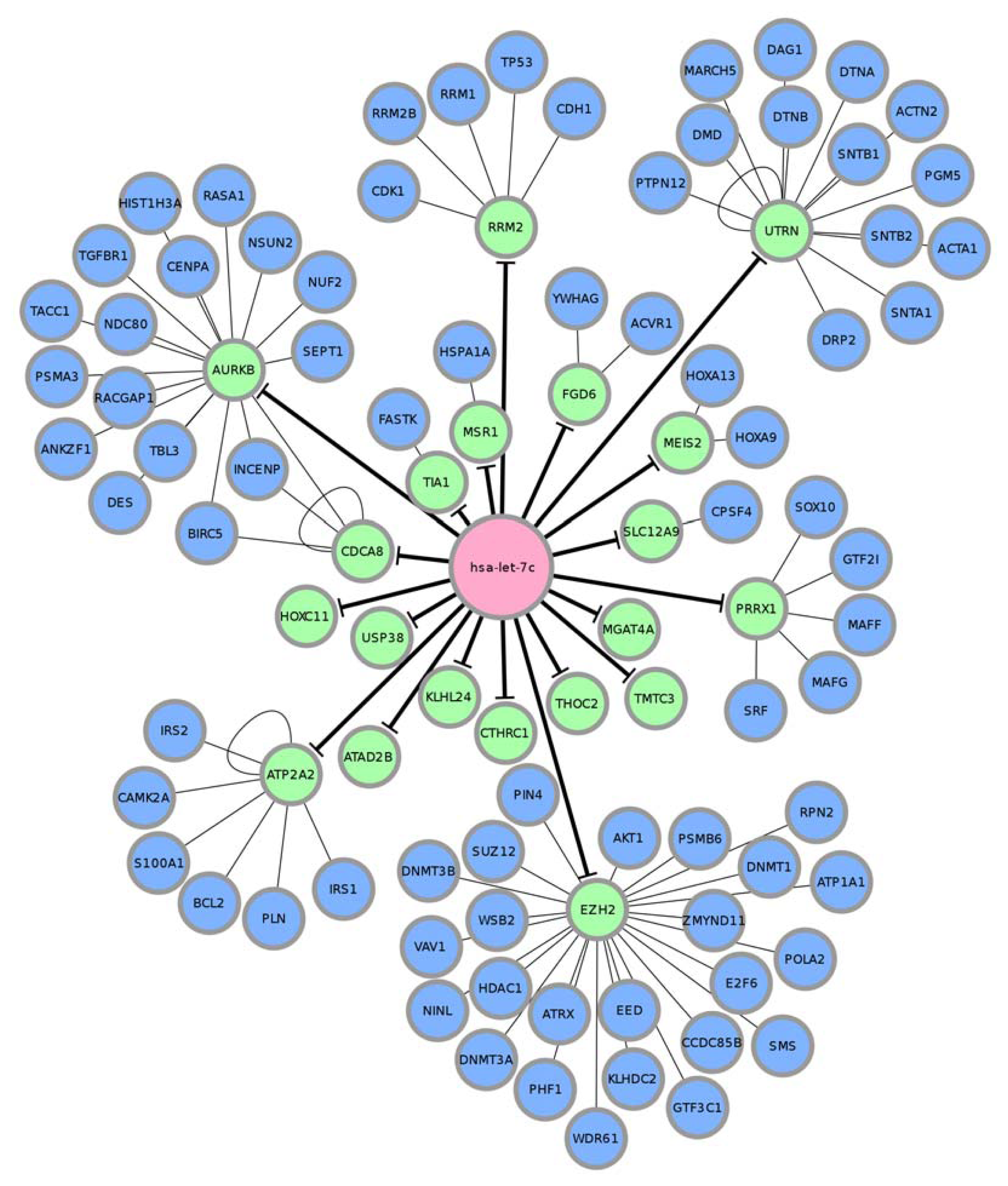
| MIMAT0000064(hsa-let-7c) | ||
| GO term | Genes | Adj.p-value |
| GO:0043067, Regulation of programmed cell death | RRM2B, ACVR1, TP53, RASA1, TGFBR1, PSMA3, BIRC5, ACTN2, HOXA13, IRS2, FASTK, VAV1, PSMB6, BCL2, CDK1, HDAC1, SOX10, TIA1, AKT1, AURKB | 3.43 × 10−8 |
| GO:0043069, Negative regulation of programmed cell death | RRM2B, ACVR1, TP53, RASA1, TGFBR1, BIRC5, IRS2, BCL2, CDK1, HDAC1, SOX10, AKT1, AURKB | 6.58 × 10−6 |
| GO:0060548, Negative regulation of cell death | RRM2B, ACVR1, TP53, RASA1, TGFBR1, BIRC5, IRS2, BCL2, CDK1, HDAC1, SOX10, AKT1, AURKB | 8.92 × 10−6 |
| GO:0015630, Microtubule cytoskeleton | INCENP, SNTB2, SEPT1, TACC1, BIRC5, RACGAP1, PIN4, CDCA8, CDK1, PHF1, AKT1, AURKB, NINL, CCDC85B | 5.15 × 10−5 |
| GO:0043067, Regulation of programmed cell death | HMGA2, PML, PRNP, FGF2, XRCC4, BRCA1, IGFBP3, HDAC3, CTNNB1, CD5, CDK1, NKX2-5, MEF2C, PRKCI, CASP2, PSMA4, PSMA3, CFDP1, CAV1, FAF1, YWHAB, HIF1A, RELA, TCF7L2, TNFSF12, PSEN2, TP53, TOP2A, TNFRSF4, BID, MYC, JUN, OGT, CDKN1A, RNF7, PPP2R4, HDAC2, HDAC1, SNCA, PTEN, NFKBIA, IFI16, NOL3, TRAF2, HSP90B1 | 1.62 × 10−23 |
| GO:0060548, Negative regulation of cell death | HMGA2, PRNP, FGF2, XRCC4, HDAC3, CTNNB1, CDK1, NKX2-5, MEF2C, PRKCI, CFDP1, HIF1A, RELA, TCF7L2, PSEN2, TP53, TNFRSF4, MYC, JUN, CDKN1A, RNF7, HDAC2, HDAC1, SNCA, PTEN, MGMT, NFKBIA, NOL3, HSP90B1 | 4.53 × 10−17 |
| GO:0008284, Positive regulation of cell proliferation | HMGA2, FGF2, XRCC4, CDC25B, CTNNB1, EGR1, AGGF1, CDK1, NKX2-5, MEF2C, PRKCI, IRS1, HIF1A, RELA, HCLS1, TNFSF12, ARNT, PTPRC, TNFSF4, TNFRSF4, MYC, JUN, FGF1, CDKN1A, HDAC2, HDAC1, NOLC1, PTEN | 1.23 × 10−15 |
| MIMAT0000443(hsa-miR-125a-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0043069, Negative regulation of programmed cell death | HMGA2, PRNP, XRCC4, HDAC3, CTNNB1, CDK1, NKX2-5, MEF2C, PRKCI, CFDP1, HIF1A, RELA, TCF7L2, PSEN2, TP53, TNFRSF4, MYC, JUN, CDKN1A, RNF7, HDAC2, HDAC1, SNCA, PTEN, NFKBIA, NOL3, HSP90B1 | 2.02 × 10−15 |
| GO:0043068, Positive regulation of programmed cell death | HMGA2, PML, BRCA1, IGFBP3, CTNNB1, CD5, MEF2C, PRKCI, CASP2, CAV1, FAF1, YWHAB, TNFSF12, PSEN2, TP53, TOP2A, BID, JUN, OGT, CDKN1A, RNF7, PPP2R4, PTEN, IFI16, TRAF2 | 2.80 × 10−13 |
| GO:0010942, Positive regulation of cell death | HMGA2, PML, BRCA1, IGFBP3, CTNNB1, CD5, MEF2C, PRKCI, CASP2, CAV1, FAF1, YWHAB, TNFSF12, PSEN2, TP53, TOP2A, BID, JUN, OGT, CDKN1A, RNF7, PPP2R4, PTEN, IFI16, TRAF2 | 4.35 × 10−13 |
| GO:0006916, Anti-apoptosis | PRNP, HDAC3, CDK1, NKX2-5, MEF2C, PRKCI, CFDP1, RELA, TCF7L2, PSEN2, RNF7, HDAC1, SNCA, NFKBIA, NOL3, HSP90B1 | 9.04 × 10−11 |
| GO:0008285, Negative regulation of cell proliferation | SERPINF1, SRF, PML, PRNP, FGF2, CSNK2B, IGFBP3, CTNNB1, CAV1, HMGA1, VDR, CDH5, HSF1, COL18A1, TP53, MYC, JUN, CDKN1A, PAK1, PTEN | 1.32 × 10−9 |
| GO:0015630, Microtubule cytoskeleton | STMN1, KIF1C, RANGAP1, CDC25B, BRCA1, HDAC3, CTNNB1, PIN4, HSPH1, RANBP9, CDK1, SPTAN1, YWHAQ, DVL1, FKBP4, YWHAB, CCDC85B, MAPT, PSEN2, TOP2A, SPIB, MYC, OGT, APEX1, PAFAH1B1 | 1.32 × 10−9 |
| GO:0048011, Nerve growth factor receptor signaling pathway | HDAC3, CDK1, MEF2C, PRKCI, CASP2, IRS1, YWHAB, RELA, PSEN2, HDAC2, HDAC1, PTEN, ATF1, NFKBIA | 1.50 × 10−8 |
| GO:0050678, Regulation of epithelial cell proliferation | SERPINF1, PGR, FGF2, CTNNB1, AGGF1, CAV1, HIF1A, TNFSF12, ARNT, MYC, JUN, FGF1, PTEN | 2.66 × 10−8 |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | FGF2, HDAC3, CDK1, MEF2C, PRKCI, CASP2, IRS1, FIBP, PTPN1, YWHAB, RELA, PSEN2, FGF1, HDAC2, HDAC1, PTEN, ATF1, NFKBIA, EIF4EBP1 | 4.37 × 10−8 |
| GO:0006917, Induction of apoptosis | PML, BRCA1, CD5, CASP2, CAV1, YWHAB, TNFSF12, PSEN2, TP53, BID, OGT, CDKN1A, RNF7, PTEN, IFI16, TRAF2 | 1.22 × 10−7 |
| GO:0012502, Induction of programmed cell death | PML, BRCA1, CD5, CASP2, CAV1, YWHAB, TNFSF12, PSEN2, TP53, BID, OGT, CDKN1A, RNF7, PTEN, IFI16, TRAF2 | 1.31 × 10−7 |
| GO:0035666, TRIF-dependent toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 2.01 × 10−6 |
| GO:0034138, Toll-like receptor 3 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 2.27 × 10−6 |
| GO:0051693, Actin filament capping | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 2.97 × 10−6 |
| GO:0002756, MyD88- independent toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 3.20 × 10−6 |
| GO:0015629, Actin cytoskeleton | WAS, CDH1, BRCA1, SPTB, SPTBN1, SPTAN1, CTDP1, STX1A, SPTA1, PAK1, SNCA, ADD1, EPB41, EPB49 | 4.25 × 10−6 |
| GO:0034134, Toll-like receptor 2 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 4.43 × 10−6 |
| GO:0034130, Toll-like receptor 1 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 4.43 × 10−6 |
| GO:0030835, Negative regulation of actin filament depolymerization | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 4.72 × 10−6 |
| GO:0002755, MyD88-dependent toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 6.41 × 10−6 |
| GO:0050679, Positive regulation of epithelial cell proliferation | FGF2, CTNNB1, AGGF1, HIF1A, TNFSF12, ARNT, MYC, JUN, FGF1 | 9.31 × 10−6 |
| GO:0034142, Toll-like receptor 4 signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 9.31 × 10−6 |
| GO:0030834, Regulation of actin filament depolymerization | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 1.09 × 10−5 |
| GO:0030837, Negative regulation of actin filament polymerization | SPTB, SPTBN1, SPTAN1, SPTA1, ADD1, EPB49 | 2.37 × 10−5 |
| GO:0002224, Toll-like receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 2.48 × 10−5 |
| GO:0008629, Induction of apoptosis by intracellular signals | PML, BRCA1, YWHAB, TP53, BID, CDKN1A, RNF7, IFI16 | 2.92 × 10−5 |
| GO:0050851, Antigen receptor-mediated signaling pathway | WAS, MEF2C, RELA, PSEN2, PTPRC, PAK1, PTEN, NFKBIA | 8.55 × 10−5 |
| GO:0002221, Pattern recognition receptor signaling pathway | ATF2, CDK1, MEF2C, FOS, RELA, JUN, ATF1, NFKBIA | 9.15 × 10−5 |
| GO:0001936, Regulation of endothelial cell proliferation | FGF2, AGGF1, CAV1, HIF1A, TNFSF12, ARNT, JUN | 9.47 × 10−5 |
| MIMAT0000077(hsa-miR-22-3p) | ||
| GO term | Genes | Adj.p-value |
| GO:0035583, Negative regulation of transforming growth factor beta receptor signaling pathway by extracellular sequestering of TGFbeta | FBN2, FBN1, LTBP1 | 1.45 × 10−5 |
| MIMAT0000265(hsa-miR-204-5p) | ||
| GO term | Genes | Adj.p-value |
| GO:0035583, Negative regulation of transforming growth factor beta receptor signaling pathway by extracellular sequestering of TGFbeta | FBN1, FBN2, LTBP1 | 6.31 × 10−5 |
| miRNA | GO term | p-value |
|---|---|---|
| MIMAT0000064(hsa-let-7c) | GO:0015630, Microtubule cytoskeleton | 9.97 × 10−6*** |
| GO:0043067, Regulation of programmed cell death | 0.268067 | |
| GO:0043069, Negative regulation of programmed cell death | 0.0390439* | |
| GO:0060548, Negative regulation of cell death | 0.0390439* | |
| MIMAT0000076(hsa-miR-21-5p) | GO:0007173, Epidermal growth factor receptor signaling pathway | 0.721139 |
| GO:0043067, Regulation of programmed cell death | 0.266312 | |
| MIMAT0000077(hsa-miR-22-3p) | GO:0035583, Negative regulation of transforming growth factor beta receptor signaling pathway by extracellular sequestering of TGFbeta | 0.940727 |
| MIMAT0000089(hsa-miR-31-5p) | GO:0003376, Sphingosine-1-phosphate signaling pathway | 0.062202 |
| GO:0006917, Induction of apoptosis | 0.048584* | |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | 0.050408 | |
| GO:0007187, G-protein signaling, coupled to cyclic nucleotide second messenger | 0.289466 | |
| GO:0007188, G-protein signaling, coupled to cAMP nucleotide second messenger | 0.687572 | |
| GO:0010942, Positive regulation of cell death | 0.356228 | |
| GO:0012502, Induction of programmed cell death | 0.048584* | |
| GO:0019933, CAMP-mediated signaling | 0.687572 | |
| GO:0019935, Cyclic-nucleotide-mediated signaling | 0.289466 | |
| GO:0043067, Regulation of programmed cell death | 0.694486 | |
| GO:0043068, Positive regulation of programmed cell death | 0.356228 | |
| GO:0048011, Nerve growth factor receptor signaling pathway | 0.154543 | |
| MIMAT0000250(hsa-miR-139-5p) | GO:0035583, Negative regulation of transforming growth factor beta receptor signaling pathway by extracellular sequestering of TGFbeta | 0.940727 |
| MIMAT0000265(hsa-miR-204-5p) | GO:0035583, Negative regulation of transforming growth factor beta receptor signaling pathway by extracellular sequestering of TGFbeta | 0.940727 |
| MIMAT0000423(hsa-miR-125b-5p) | GO:0002224, Toll-like receptor signaling pathway | 0.380928 |
| GO:0002755, MyD88-dependent toll-like receptor signaling pathway | 0.380928 | |
| GO:0002756, MyD88-independent toll-like receptor signaling pathway | 0.380928 | |
| GO:0006916, Anti-apoptosis | 0.0593 | |
| GO:0006917, Induction of apoptosis | 0.618064 | |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | 0.776269 | |
| GO:0008284, Positive regulation of cell proliferation | 0.882324 | |
| GO:0008285, Negative regulation of cell proliferation | 0.883393 | |
| GO:0008629, Induction of apoptosis by intracellular signals | 0.073118 | |
| GO:0010942, Positive regulation of cell death | 0.972892 | |
| GO:0012502, Induction of programmed cell death | 0.618064 | |
| GO:0015629, Actin cytoskeleton | 0.596528 | |
| GO:0015630, Microtubule cytoskeleton | 0.028245* | |
| GO:0030834, Regulation of actin filament depolymerization | 0.654383 | |
| GO:0030835, Negative regulation of actin filament depolymerization | 0.654383 | |
| GO:0030837, Negative regulation of actin filament polymerization | 0.654383 | |
| GO:0034130, Toll-like receptor 1 signaling pathway | 0.380928 | |
| GO:0034134, Toll-like receptor 2 signaling pathway | 0.380928 | |
| GO:0034138, Toll-like receptor 3 signaling pathway | 0.380928 | |
| GO:0034142, Toll-like receptor 4 signaling pathway | 0.380928 | |
| GO:0035666, TRIF-dependent toll-like receptor signaling pathway | 0.380928 | |
| GO:0043067, Regulation of programmed cell death | 0.643418 | |
| GO:0043068, Positive regulation of programmed cell death | 0.972892 | |
| GO:0043069, Negative regulation of programmed cell death | 0.492576 | |
| GO:0048011, Nerve growth factor receptor signaling pathway | 0.171634 | |
| GO:0050678, Regulation of epithelial cell proliferation | 0.002205** | |
| GO:0050679, Positive regulation of epithelial cell proliferation | 0.205483 | |
| GO:0051693, Actin filament capping | 0.654383 | |
| GO:0060548, Negative regulation of cell death | 0.413665 | |
| MIMAT0000437(hsa-miR-145-5p) | GO:0007178, Transmembrane receptor protein serine/threonine kinase signaling pathway | 0.196953 |
| GO:0030509, BMP signaling pathway | 0.196953 | |
| GO:0090092, Regulation of transmembrane receptor protein serine/threonine kinase signaling pathway | 0.843529 | |
| MIMAT0000443(hsa-miR-125a-5p) | GO:0001936, Regulation of endothelial cell proliferation | 0.115146 |
| GO:0002221, Pattern recognition receptor signaling pathway | 0.380928 | |
| GO:0002224, Toll-like receptor signaling pathway | 0.380928 | |
| GO:0002755, MyD88-dependent toll-like receptor signaling pathway | 0.380928 | |
| GO:0002756, MyD88-independent toll-like receptor signaling pathway | 0.380928 | |
| GO:0006916, Anti-apoptosis | 0.0593 | |
| GO:0006917, Induction of apoptosis | 0.618064 | |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | 0.776269 | |
| GO:0008284, Positive regulation of cell proliferation | 0.882324 | |
| GO:0008285, Negative regulation of cell proliferation | 0.883393 | |
| GO:0008629, Induction of apoptosis by intracellular signals | 0.073118 | |
| GO:0010942, Positive regulation of cell death | 0.972892 | |
| GO:0012502, Induction of programmed cell death | 0.618064 | |
| GO:0015629, Actin cytoskeleton | 0.596528 | |
| GO:0015630, Microtubule cytoskeleton | 0.028245* | |
| GO:0030834, Regulation of actin filament depolymerization | 0.654383 | |
| GO:0030835, Negative regulation of actin filament depolymerization | 0.654383 | |
| GO:0030837, Negative regulation of actin filament polymerization | 0.654383 | |
| GO:0034130, Toll-like receptor 1 signaling pathway | 0.380928 | |
| GO:0034134, Toll-like receptor 2 signaling pathway | 0.380928 | |
| GO:0034138, Toll-like receptor 3 signaling pathway | 0.380928 | |
| GO:0034142, Toll-like receptor 4 signaling pathway | 0.380928 | |
| GO:0035666, TRIF-dependent toll-like receptor signaling pathway | 0.380928 | |
| GO:0043067, Regulation of programmed cell death | 0.643418 | |
| GO:0043068, Positive regulation of programmed cell death | 0.972892 | |
| GO:0043069, Negative regulation of programmed cell death | 0.492576 | |
| GO:0048011, Nerve growth factor receptor signaling pathway | 0.171634 | |
| GO:0050678, Regulation of epithelial cell proliferation | 0.002205** | |
| GO:0050679, Positive regulation of epithelial cell proliferation | 0.205483 | |
| GO:0050851, Antigen receptor-mediated signaling pathway | 0.103325 | |
| GO:0051693, Actin filament capping | 0.654383 | |
| GO:0060548, Negative regulation of cell death | 0.413665 | |
| MIMAT0002820(hsa-miR-497-5p) | GO:0001959, Regulation of cytokine-mediated signaling pathway | 0.06699 |
| GO:0006917, Induction of apoptosis | 0.142401 | |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | 0.837635 | |
| GO:0007173, Epidermal growth factor receptor signaling pathway | 0.387447 | |
| GO:0007178, Transmembrane receptor protein serine/threonine kinase signaling pathway | 0.240167 | |
| GO:0007179, Transforming growth factor beta receptor signaling pathway | 0.017876* | |
| GO:0008284, Positive regulation of cell proliferation | 0.430255 | |
| GO:0008285, Negative regulation of cell proliferation | 0.149994 | |
| GO:0008543, Fibroblast growth factor receptor signaling pathway | 0.521978 | |
| GO:0010942, Positive regulation of cell death | 0.237692 | |
| GO:0012502, Induction of programmed cell death | 0.142401 | |
| GO:0015629, Actin cytoskeleton | 0.228804 | |
| GO:0015630, Microtubule cytoskeleton | 0.18331 | |
| GO:0017015, Regulation of transforming growth factor beta receptor signaling pathway | 0.128773 | |
| GO:0030509, BMP signaling pathway | 0.39837 | |
| GO:0030521, Androgen receptor signaling pathway | 0.383811 | |
| GO:0032956, Regulation of actin cytoskeleton organization | 0.91762 | |
| GO:0042058, Regulation of epidermal growth factor receptor signaling pathway | 0.934045 | |
| GO:0042059, Negative regulation of epidermal growth factor receptor signaling pathway | 0.789492 | |
| GO:0043067, Regulation of programmed cell death | 0.111544 | |
| GO:0043068, Positive regulation of programmed cell death | 0.237692 | |
| GO:0043069, Negative regulation of programmed cell death | 0.856892 | |
| GO:0048011, Nerve growth factor receptor signaling pathway | 0.471986 | |
| GO:0060548, Negative regulation of cell death | 0.667437 | |
| GO:0070302, Regulation of stress-activated protein kinase signaling cascade | 0.561032 | |
| GO:0090092, Regulation of transmembrane receptor protein serine/threonine kinase signaling pathway | 0.182314 | |
| MIMAT0002856(hsa-miR-520d-3p) | GO:0006917, Induction of apoptosis | 0.489781 |
| GO:0007169, Transmembrane receptor protein tyrosine kinase signaling pathway | 0.171689 | |
| GO:0007173, Epidermal growth factor receptor signaling pathway | 0.449696 | |
| GO:0008284, Positive regulation of cell proliferation | 0.05916 | |
| GO:0008286, Insulin receptor signaling pathway | 0.237933 | |
| GO:0008543, Fibroblast growth factor receptor signaling pathway | 0.159318 | |
| GO:0008629, Induction of apoptosis by intracellular signals | 0.502822 | |
| GO:0010942, Positive regulation of cell death | 0.076906 | |
| GO:0012502, Induction of programmed cell death | 0.489781 | |
| GO:0015630, Microtubule cytoskeleton | 0.000292*** | |
| GO:0042058, Regulation of epidermal growth factor receptor signaling pathway | 0.762633 | |
| GO:0042059, Negative regulation of epidermal growth factor receptor signaling pathway | 0.826854 | |
| GO:0043067, Regulation of programmed cell death | 0.740092 | |
| GO:0043068, Positive regulation of programmed cell death | 0.076906 | |
| GO:0043069, Negative regulation of programmed cell death | 0.067499 | |
| GO:0048011, Nerve growth factor receptor signaling pathway | 0.014926* | |
| GO:0051988, Regulation of attachment of spindle microtubules to kinetochore | 7.48 × 10−6*** | |
| GO:0060548, Negative regulation of cell death | 0.213035 | |
| Sample Name | ER | PR | HER | TNM | Stage | Grade |
|---|---|---|---|---|---|---|
| S621T1 | 0 | 0 | 0 | pT1N0M0 | I | 2 |
| S434T1 | 1 | 0 | 1 | T2N1M0 | IIB | 3 |
| S403T1 | 0 | 1 | 0 | T2N1M0 | IIB | 2 |
| S459T1 | 1 | 0 | 0 | T4N0M1 | IV | 1 |
| S455N1 | 1 | 0 | 0 | T3N3M1 | IV | 3 |
| S545T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 3 |
| S173N1 | 0 | 0 | 1 | T2N3M0 | IIIC | 3 |
| S363T1 | 1 | 1 | 0 | T2N1M0 | IIB | 1 |
| S909T1 | 1 | 1 | 1 | pT3N3aM0 | IIIC | (Unknown) |
| S645T1 | 0 | 1 | 1 | pT1bN0M0 | I | 3 |
| S898T1 | 1 | 0 | 0 | pT2N0(i-)M0 | IIA | (Unknown) |
| S201T1 | 1 | 0 | 0 | T1N1M0 | IIA | 2 |
| S631T1 | 1 | 1 | 1 | T2N3aM1 | IV | 2 |
| S303T1 | 0 | 0 | 1 | T2N0M0 | IIA | 2 |
| S502T1 | 0 | 0 | 0 | pT3N0M0 | IIB | 3 |
| S498N1 | 1 | 1 | 1 | pT1cN1aM0 | IIA | 2 |
| S536T1 | 1 | 0 | 1 | T1cN1miM0 | IIA | 2 |
| S660T1 | 0 | 0 | 1 | T1N0M0 | I | 3 |
| S358N1 | 0 | 0 | 1 | T2pN2M0 | IIIA | 2 |
| S665T1 | 0 | 0 | 0 | T2N3M0 | IIIC | 3 |
| S475T1 | 0 | 0 | 1 | pT1cN0M0 | I | 3 |
| S423T1 | 1 | 1 | 0 | T2N3M0 | IIIC | 1 |
| S507T1 | 0 | 0 | 0 | pT1cN1aM0 | IIA | 3 |
| S891T1 | 1 | 0 | 0 | pT2NxM0 | IIA | 2 |
| S422T1 | 1 | 1 | 0 | T2N0M0 | IIA | 1 |
| S961T1 | 0 | 0 | 1 | T2N2aM0 | IIIA | 2 |
| S622T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 2 |
| S454T1 | 0 | 0 | 0 | T1cN0M0 | I | 2 |
| S433T1 | 1 | 0 | 0 | T2N0M0 | IIA | 1 |
| S673T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 2 |
| S450T1 | 0 | 0 | 1 | T2N1M0 | IIB | 3 |
| S430T1 | 1 | 1 | 0 | T2N3M0 | IIIC | 2 |
| S437T1 | 1 | 0 | 0 | T3N1M0 | IIIA | 3 |
| S574T1 | 0 | 0 | 0 | T2N0M0 | IIA | 2 |
| S401T1 | 1 | 1 | 0 | T1N0M0 | I | 1 |
| S427N1 | 0 | 0 | 0 | T3N1M0 | IIIA | 2 |
| S894T1 | 0 | 0 | 0 | pT1cN0M0 | I | 2 |
| S929T1 | 1 | 0 | 0 | pT2N0M0 | IIA | 2 |
| S173T1 | 0 | 0 | 1 | T2N3M0 | IIIC | 3 |
| S622N1 | 0 | 0 | 0 | pT2N0M0 | IIA | 2 |
| S562T1 | 1 | 1 | 1 | pT1aN0M0 | I | 3 |
| S602T1 | 0 | 0 | 0 | pT1cN0M0 | I | 3 |
| S490T1 | 1 | 1 | 1 | pT2N0M0 | IIA | 2 |
| S677T1 | 0 | 0 | 0 | pT2N1M0 | IIB | 3 |
| S881T1 | 0 | 0 | 0 | pT2N1aM0 | IIB | 3 |
| S619T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 3 |
| S446T1 | 0 | 0 | 0 | T2N2M0 | IIIA | 3 |
| S446T2 | 0 | 0 | 0 | T2N2M0 | IIIA | 3 |
| S453T1 | 1 | 1 | 1 | T2N1M0 | IIB | 2 |
| S562N1 | 1 | 1 | 1 | pT1aN0M0 | I | 3 |
| S557T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 3 |
| S594T1 | 1 | 1 | 1 | pT2N3aM0 | IIIC | 3 |
| S582T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 3 |
| S358T1 | 0 | 0 | 1 | T2pN2M0 | IIIA | 2 |
| S368T1 | 0 | 0 | 1 | T3N3M0 | IIIC | 2 |
| S175T1 | 1 | 1 | 0 | T3N3Mx | IIIC | 3 |
| S357T1 | 0 | 0 | 0 | T2pN1M0 | IIIC | 3 |
| S653T1 | 0 | 0 | 0 | pT3N3aM0 | IIIC | 2 |
| S722T1 | 1 | 1 | 1 | pT1N0M0 | I | 3 |
| S593T1 | 0 | 0 | 0 | pT1N0M0 | I | 3 |
| S543T1 | 1 | 1 | 1 | pT2N1M0 | IIB | 2 |
| S498T1 | 1 | 1 | 1 | pT1cN1aM0 | IIA | 2 |
| S389T1 | 1 | 0 | 0 | T2N1M0 | IIB | 3 |
| S614T1 | 0 | 1 | 1 | TxN0M1 | IIIA | (Unknown) |
| S536N1 | 1 | 0 | 1 | T1cN1miM0 | IIA | 2 |
| S462T1 | 1 | 1 | 0 | T3N3M0 | IIIC | 2 |
| S477T1 | 0 | 0 | 0 | pT1cN0M0 | I | 3 |
| S917T1 | 0 | 0 | 0 | T2N0M0 | IIA | (Unknown) |
| S213T1 | 0 | 0 | 1 | T1cN1M0 | IIA | 3 |
| S628T1 | 0 | 0 | 0 | T1N0M0 | I | 2 |
| S291T1 | 0 | 0 | 0 | T3pN2M0 | IIIA | 3 |
| S593N1 | 0 | 0 | 0 | pT1N0M0 | I | 3 |
| S418T1 | 1 | 1 | 0 | T2N3M0 | IIIC | 2 |
| S420T1 | 1 | 1 | 0 | T1N0M0 | I | 2 |
| S363N1 | 1 | 1 | 0 | T2N1M0 | IIB | 1 |
| S629T1 | 1 | 0 | 1 | pT1N0M0 | I | 2 |
| S586T1 | 1 | 1 | 1 | pT2N3aM0 | IIIC | 2 |
| S415T1 | 0 | 1 | 0 | T2N2M0 | IIIA | 2 |
| S439T1 | 1 | 1 | 0 | T2N2M0 | IIIA | 2 |
| S941N1 | 0 | 0 | 0 | pT1cN1aM0 | IIA | 3 |
| S893T1 | 0 | 0 | 0 | pT2N1micM0 | IIB | 3 |
| S380T1 | 1 | 0 | 0 | T2N1M0 | IIB | 2 |
| S906N1 | 1 | 1 | 1 | pT3N3aM0 | IIIC | 2 |
| S918T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 3 |
| S400T1 | 0 | 0 | 1 | T2N0M0 | IIA | 2 |
| S328T1 | 0 | 0 | 0 | T1cpN0M0 | I | 3 |
| S367T1 | 1 | 0 | 0 | T1N1M0 | IIIA | 1 |
| S420N1 | 1 | 1 | 0 | T1N0M0 | I | 2 |
| S922T1 | 0 | 0 | 0 | pT1cN0(i-)M0 | I | 3 |
| S896T1 | 0 | 0 | 1 | pT2N1aM0 | IIB | 2 |
| S410T1 | 0 | 1 | 1 | T1N0M0 | I | 3 |
| S572T1 | 0 | 0 | 1 | T4N1aM0 | IIIC | 3 |
| S448T1 | 0 | 0 | 0 | T1N0M0 | I | 3 |
| S207T1 | 0 | 0 | 1 | T2N0M0 | IIA | 3 |
| S604T1 | 0 | 0 | 1 | pT2N1M0 | IIB | 3 |
| S379T1 | 0 | 0 | 1 | T2N2M0 | IIIA | 2 |
| S906T1 | 1 | 1 | 1 | pT3N3aM0 | IIIC | 2 |
| S941T2 | 0 | 0 | 0 | pT1cN1aM0 | IIA | 3 |
| S941T1 | 0 | 0 | 0 | pT1cN1aM0 | IIA | 3 |
| S375T1 | 1 | 1 | 0 | T1N0M0 | I | 2 |
| S427T1 | 0 | 0 | 0 | T3N1M0 | IIIA | 2 |
| S417T1 | 0 | 0 | 0 | pT2N0M0 | IIA | 3 |
| S180T1 | 1 | 0 | 0 | T4NxM0 | IIIC | |
| S455T1 | 1 | 0 | 0 | T3N3M1 | IV | |
| S887T1 | 0 | 0 | 0 | pT2N0M0 | IIA | |
| S445T1 | 1 | 0 | 1 | T2N1M0 | IIB | |
| S469T1 | 1 | 1 | 0 | T2N0M0 | IIA | |
| S469T2 | 1 | 1 | 0 | T2N0M0 | IIA | |
| S483T1 | 1 | 1 | 0 | T2N3M0 | IIIC | |
| S444T1 | 1 | 1 | 0 | T1NxM0 | (Unknown) | |
| S909N1 | 1 | 1 | 1 | pT3N3aM0 | IIIC | |
| S464T1 | 1 | 1 | 0 | T1N0M0 | I | |
| S894N1 | 0 | 0 | 0 | pT1cN0M0 | I | |
| S698T1 | 1 | 1 | 1 | pT1cN0M0 | I | |
| S452T1 | 0 | 0 | 0 | T1N0M0 | I | |
| S474T1 | (Unknown) | (Unknown) | (Unknown) | (Unknown) | (Unknown) |
Supplementary Information
ijms-14-11560-s001.pdfAcknowledgments
Conflict of Interest
References
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomark. Prev 2010, 19, 1893–1907. [Google Scholar]
- Veronesi, U.; Boyle, P.; Goldhirsch, A.; Orecchia, R.; Viale, G. Breast cancer. Lancet 2005, 365, 1727–1741. [Google Scholar]
- Cho, E.; Spiegelman, D.; Hunter, D.J.; Chen, W.Y.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C. Premenopausal fat intake and risk of breast cancer. J. Natl. Cancer Inst 2003, 95, 1079–1085. [Google Scholar]
- Huttenhofer, A.; Schattner, P.; Polacek, N. Non-coding RNAs: Hope or hype? Trends Genet 2005, 21, 289–297. [Google Scholar]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol 2009, 10, 126–139. [Google Scholar]
- Kwak, P.B.; Iwasaki, S.; Tomari, Y. The microRNA pathway and cancer. Cancer Sci 2010, 101, 2309–2315. [Google Scholar]
- Hutvagner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar]
- Mourelatos, Z.; Dostie, J.; Paushkin, S.; Sharma, A.; Charroux, B.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 2002, 16, 720–728. [Google Scholar]
- Nelson, P.T.; Hatzigeorgiou, A.G.; Mourelatos, Z. miRNP:mRNA association in polyribosomes in a human neuronal cell line. RNA 2004, 10, 387–394. [Google Scholar]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol 2007, 302, 1–12. [Google Scholar]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar]
- Rask, L.; Balslev, E.; Jorgensen, S.; Eriksen, J.; Flyger, H.; Moller, S.; Hogdall, E.; Litman, T.; Nielsen, B.S. High expression of miR-21 in tumor stroma correlates with increased cancer cell proliferation in human breast cancer. APMIS 2011, 119, 663–673. [Google Scholar]
- Zaman, M.S.; Shahryari, V.; Deng, G.; Thamminana, S.; Saini, S.; Majid, S.; Chang, I.; Hirata, H.; Ueno, K.; Yamamura, S.; et al. Up-regulation of microRNA-21 correlates with lower kidney cancer survival. PLoS One 2012, 7, e31060. [Google Scholar]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Shi, X.B.; Zou, J.X.; Chen, H.; Zhang, J.; Chen, X.; Luo, J.; et al. MicroRNA let-7c is downregulated in prostate cancer and suppresses prostate cancer growth. PLoS One 2012, 7, e32832. [Google Scholar]
- Wang, X.F.; Shi, Z.M.; Wang, X.R.; Cao, L.; Wang, Y.Y.; Zhang, J.X.; Yin, Y.; Luo, H.; Kang, C.S.; Liu, N.; et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J. Cancer Res. Clin. Oncol 2012, 138, 573–584. [Google Scholar]
- Zhang, Y.; Yan, L.X.; Wu, Q.N.; Du, Z.M.; Chen, J.; Liao, D.Z.; Huang, M.Y.; Hou, J.H.; Wu, Q.L.; Zeng, M.S.; et al. miR-125b is methylated and functions as a tumor suppressor by regulating the ETS1 proto-oncogene in human invasive breast cancer. Cancer Res 2011, 71, 3552–3562. [Google Scholar]
- Sachdeva, M.; Mo, Y.Y. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res 2010, 70, 378–387. [Google Scholar]
- Guo, X.; Wu, Y.; Hartley, R.S. MicroRNA-125a represses cell growth by targeting HuR in breast cancer. RNA Biol 2009, 6, 575–583. [Google Scholar]
- Jin, H.; Tuo, W.; Lian, H.; Liu, Q.; Zhu, X.Q.; Gao, H. Strategies to identify microRNA targets: New advances. N. Biotechnol 2010, 27, 734–738. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009, 19, 92–105. [Google Scholar]
- Huang, J.C.; Babak, T.; Corson, T.W.; Chua, G.; Khan, S.; Gallie, B.L.; Hughes, T.R.; Blencowe, B.J.; Frey, B.J.; Morris, Q.D. Using expression profiling data to identify human microRNA targets. Nat. Methods 2007, 4, 1045–1049. [Google Scholar]
- Liu, B.; Li, J.; Tsykin, A.; Liu, L.; Gaur, A.B.; Goodall, G.J. Exploring complex miRNA-mRNA interactions with Bayesian networks by splitting-averaging strategy. BMC Bioinforma 2009, 10, 408. [Google Scholar]
- Tseng, C.W.; Lin, C.C.; Chen, C.N.; Huang, H.C.; Juan, H.F. Integrative network analysis reveals active microRNAs and their functions in gastric cancer. BMC Syst. Biol 2011, 5, 99. [Google Scholar]
- Farazi, T.A.; Horlings, H.M.; Ten Hoeve, J.J.; Mihailovic, A.; Halfwerk, H.; Morozov, P.; Brown, M.; Hafner, M.; Reyal, F.; van Kouwenhove, M.; et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res 2011, 71, 4443–4453. [Google Scholar]
- Derfoul, A.; Juan, A.H.; Difilippantonio, M.J.; Palanisamy, N.; Ried, T.; Sartorelli, V. Decreased microRNA-214 levels in breast cancer cells coincides with increased cell proliferation, invasion and accumulation of the Polycomb Ezh2 methyltransferase. Carcinogenesis 2011, 32, 1607–1614. [Google Scholar]
- Heyn, H.; Engelmann, M.; Schreek, S.; Ahrens, P.; Lehmann, U.; Kreipe, H.; Schlegelberger, B.; Beger, C. MicroRNA miR-335 is crucial for the BRCA1 regulatory cascade in breast cancer development. Int. J. Cancer 2011, 129, 2797–2806. [Google Scholar]
- Sempere, L.F.; Christensen, M.; Silahtaroglu, A.; Bak, M.; Heath, C.V.; Schwartz, G.; Wells, W.; Kauppinen, S.; Cole, C.N. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 2007, 67, 11612–11620. [Google Scholar]
- Zhu, X.M.; Wu, L.J.; Xu, J.; Yang, R.; Wu, F.S. Let-7c microRNA expression and clinical significance in hepatocellular carcinoma. J. Int. Med. Res 2011, 39, 2323–2329. [Google Scholar]
- Iyevleva, A.G.; Kuligina, E.; Mitiushkina, N.V.; Togo, A.V.; Miki, Y.; Imyanitov, E.N. High level of miR-21, miR-10b, and miR-31 expression in bilateral vs. unilateral breast carcinomas. Breast Cancer Res. Treat 2012, 131, 1049–1059. [Google Scholar]
- Rutnam, Z.J.; Yang, B.B. The involvement of microRNAs in malignant transformation. Histol. Histopathol 2012, 27, 1263–1270. [Google Scholar]
- Liu, H. MicroRNAs in breast cancer initiation and progression. Cell Mol. Life Sci 2012, 69, 3587–3599. [Google Scholar]
- Zhang, Z.J.; Ma, S.L. miRNAs in breast cancer tumorigenesis (Review). Oncol. Rep 2012, 27, 903–910. [Google Scholar]
- Mathivanan, S.; Periaswamy, B.; Gandhi, T.K.; Kandasamy, K.; Suresh, S.; Mohmood, R.; Ramachandra, Y.L.; Pandey, A. An evaluation of human protein-protein interaction data in the public domain. BMC Bioinforma 2006, 7, S19. [Google Scholar]
- Halbert, C.H.; Kumanyika, S.; Bowman, M.; Bellamy, S.L.; Briggs, V.; Brown, S.; Bryant, B.; Delmoor, E.; Johnson, J.C.; Purnell, J.; et al. Participation rates and representativeness of African Americans recruited to a health promotion program. Health Educ. Res 2010, 25, 6–13. [Google Scholar]
- Nicolas Diaz-Chico, B.; German Rodriguez, F.; Gonzalez, A.; Ramirez, R.; Bilbao, C.; Cabrera de Leon, A.; Aguirre Jaime, A.; Chirino, R.; Navarro, D.; Diaz-Chico, J.C. Androgens and androgen receptors in breast cancer. J. Steroid Biochem. Mol. Biol 2007, 105, 1–15. [Google Scholar]
- Sethi, N.; Kang, Y. Dysregulation of developmental pathways in bone metastasis. Bone 2011, 48, 16–22. [Google Scholar]
- Liao, S.J.; Zhou, Y.H.; Yuan, Y.; Li, D.; Wu, F.H.; Wang, Q.; Zhu, J.H.; Yan, B.; Wei, J.J.; Zhang, G.M.; et al. Triggering of Toll-like receptor 4 on metastatic breast cancer cells promotes alphavbeta3-mediated adhesion and invasive migration. Breast Cancer Res. Treat 2011, 133, 853–863. [Google Scholar]
- Jiang, P.; Enomoto, A.; Takahashi, M. Cell biology of the movement of breast cancer cells: Intracellular signalling and the actin cytoskeleton. Cancer Lett 2009, 284, 122–130. [Google Scholar]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol 2011, 18, 1139–1146. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Lall, S.; Grun, D.; Krek, A.; Chen, K.; Wang, Y.L.; Dewey, C.N.; Sood, P.; Colombo, T.; Bray, N.; Macmenamin, P.; et al. A genome-wide map of conserved microRNA targets in C. elegans. Curr. Biol 2006, 16, 460–471. [Google Scholar]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res 2008, 36, D149–D153. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res 2009, 37, D767–D772. [Google Scholar]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat 2001, 29, 1165–1188. [Google Scholar]
- Ringner, M.; Fredlund, E.; Hakkinen, J.; Borg, A.; Staaf, J. GOBO: Gene expression-based outcome for breast cancer online. PLoS One 2011, 6, e17911. [Google Scholar]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar]
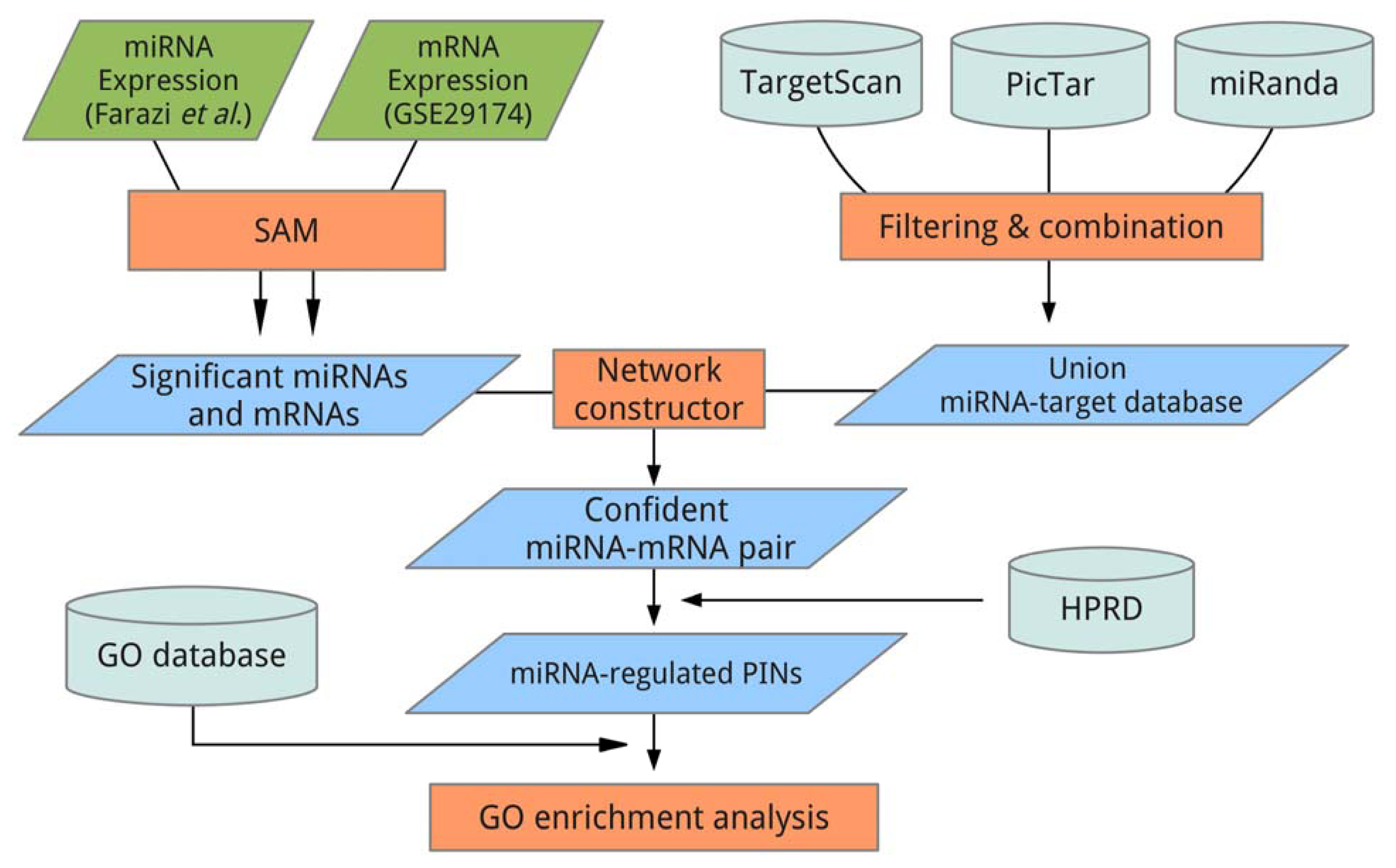
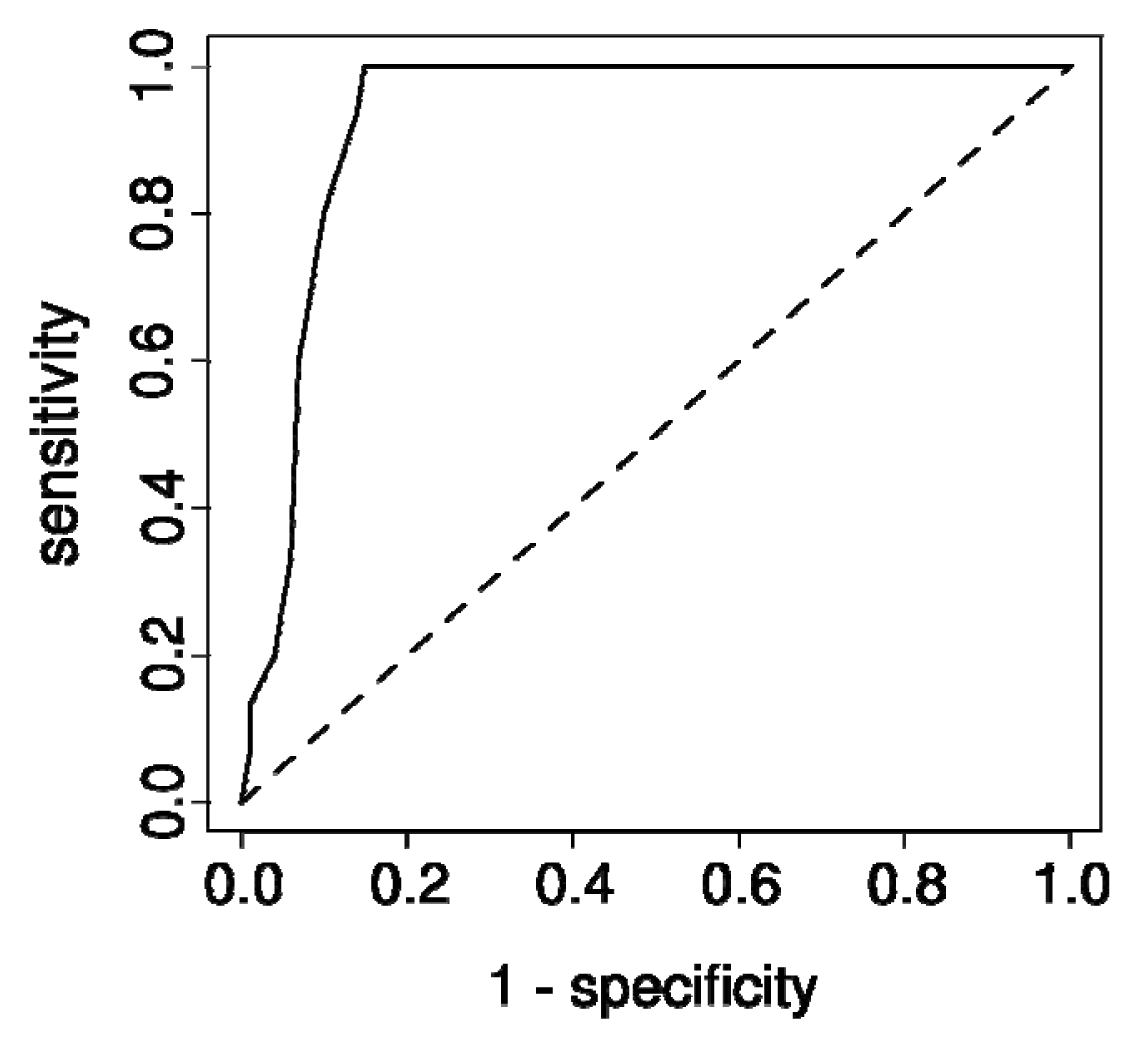
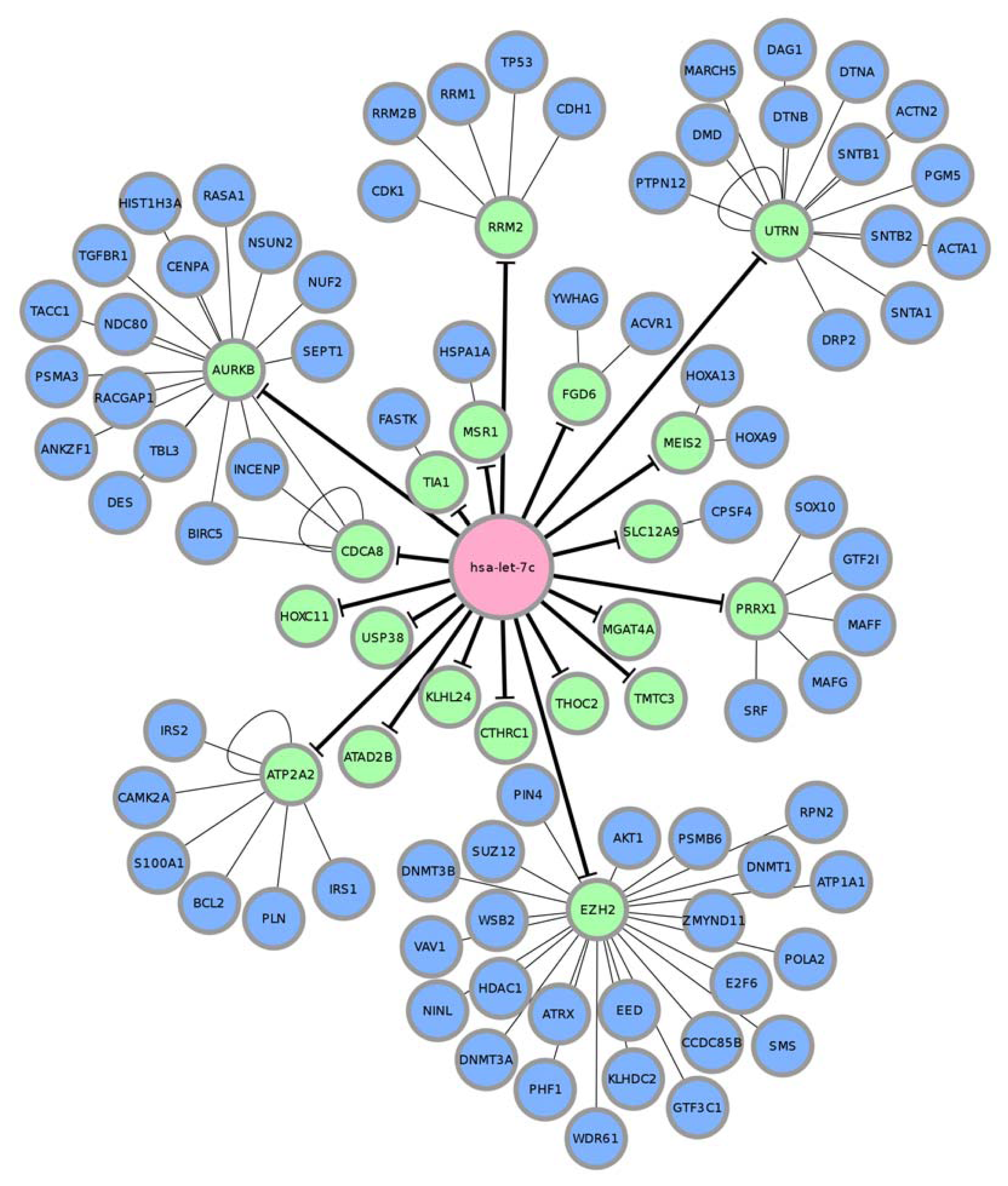
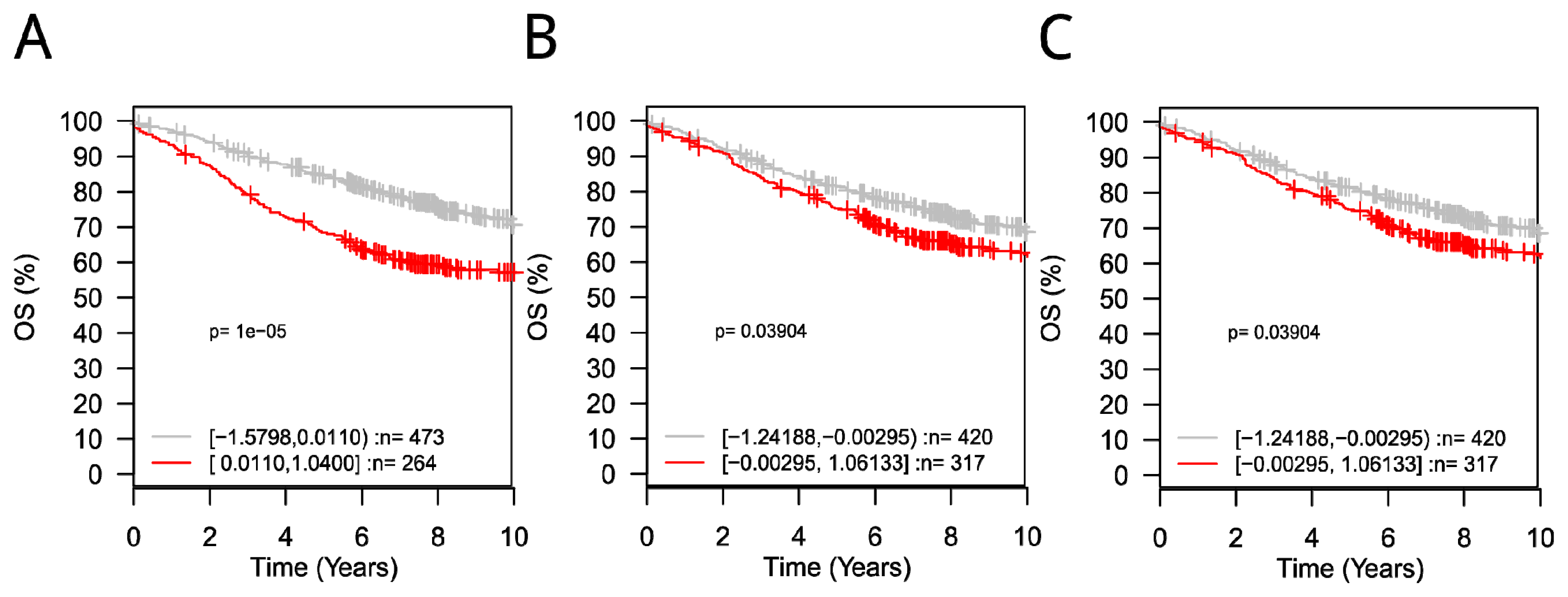
| MIMAT0000064(hsa-let-7c) | ||
|---|---|---|
| GO term | Genes | Adj. p-value |
| GO:0043067, Regulation of programmed cell death | RRM2B, ACVR1, TP53, RASA1, TGFBR1, PSMA3, BIRC5, ACTN2, HOXA13, IRS2, FASTK, VAV1, PSMB6, BCL2, CDK1, HDAC1, SOX10, TIA1, AKT1, AURKB | 3.43 × 10−8 |
| GO:0043069, Negative regulation of programmed cell death | RRM2B, ACVR1, TP53, RASA1, TGFBR1, BIRC5, IRS2, BCL2, CDK1, HDAC1, SOX10, AKT1, AURKB | 6.58 × 10−6 |
| GO:0060548, Negative regulation of cell death | RRM2B, ACVR1, TP53, RASA1, TGFBR1, BIRC5, IRS2, BCL2, CDK1, HDAC1, SOX10, AKT1, AURKB | 8.92 × 10−6 |
| GO:0015630, Microtubule cytoskeleton | INCENP, SNTB2, SEPT1, TACC1, BIRC5, RACGAP1, PIN4, CDCA8, CDK1, PHF1, AKT1, AURKB, NINL, CCDC85B | 5.15 × 10−5 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, C.-H.; Kuo, W.-H.; Lin, C.-C.; Oyang, Y.-J.; Huang, H.-C.; Juan, H.-F. MicroRNA-Regulated Protein-Protein Interaction Networks and Their Functions in Breast Cancer. Int. J. Mol. Sci. 2013, 14, 11560-11606. https://doi.org/10.3390/ijms140611560
Lee C-H, Kuo W-H, Lin C-C, Oyang Y-J, Huang H-C, Juan H-F. MicroRNA-Regulated Protein-Protein Interaction Networks and Their Functions in Breast Cancer. International Journal of Molecular Sciences. 2013; 14(6):11560-11606. https://doi.org/10.3390/ijms140611560
Chicago/Turabian StyleLee, Chia-Hsien, Wen-Hong Kuo, Chen-Ching Lin, Yen-Jen Oyang, Hsuan-Cheng Huang, and Hsueh-Fen Juan. 2013. "MicroRNA-Regulated Protein-Protein Interaction Networks and Their Functions in Breast Cancer" International Journal of Molecular Sciences 14, no. 6: 11560-11606. https://doi.org/10.3390/ijms140611560