Notch Signaling Pathway Is Activated in Motoneurons of Spinal Muscular Atrophy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
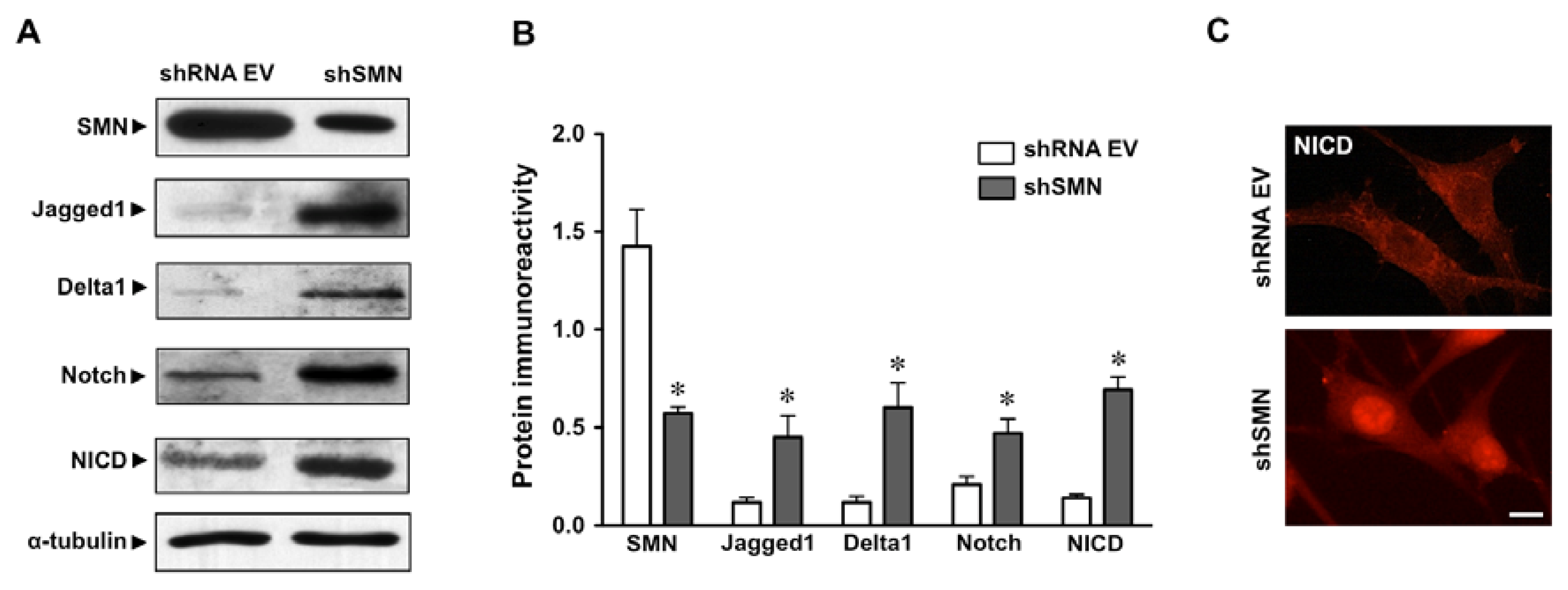
2.1. Increased Notch Signaling in U87MG Astroglioma Cells Depleted of SMN
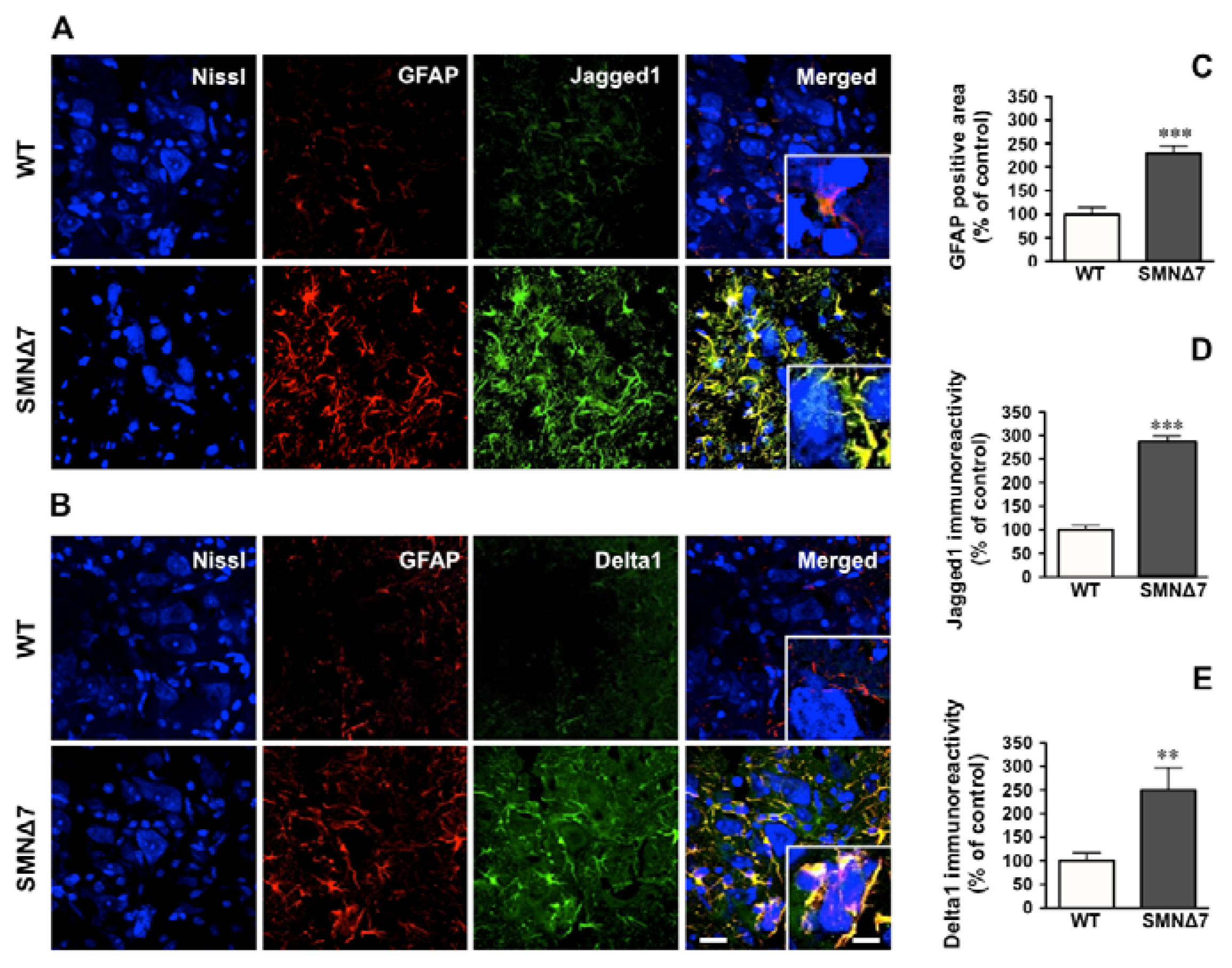
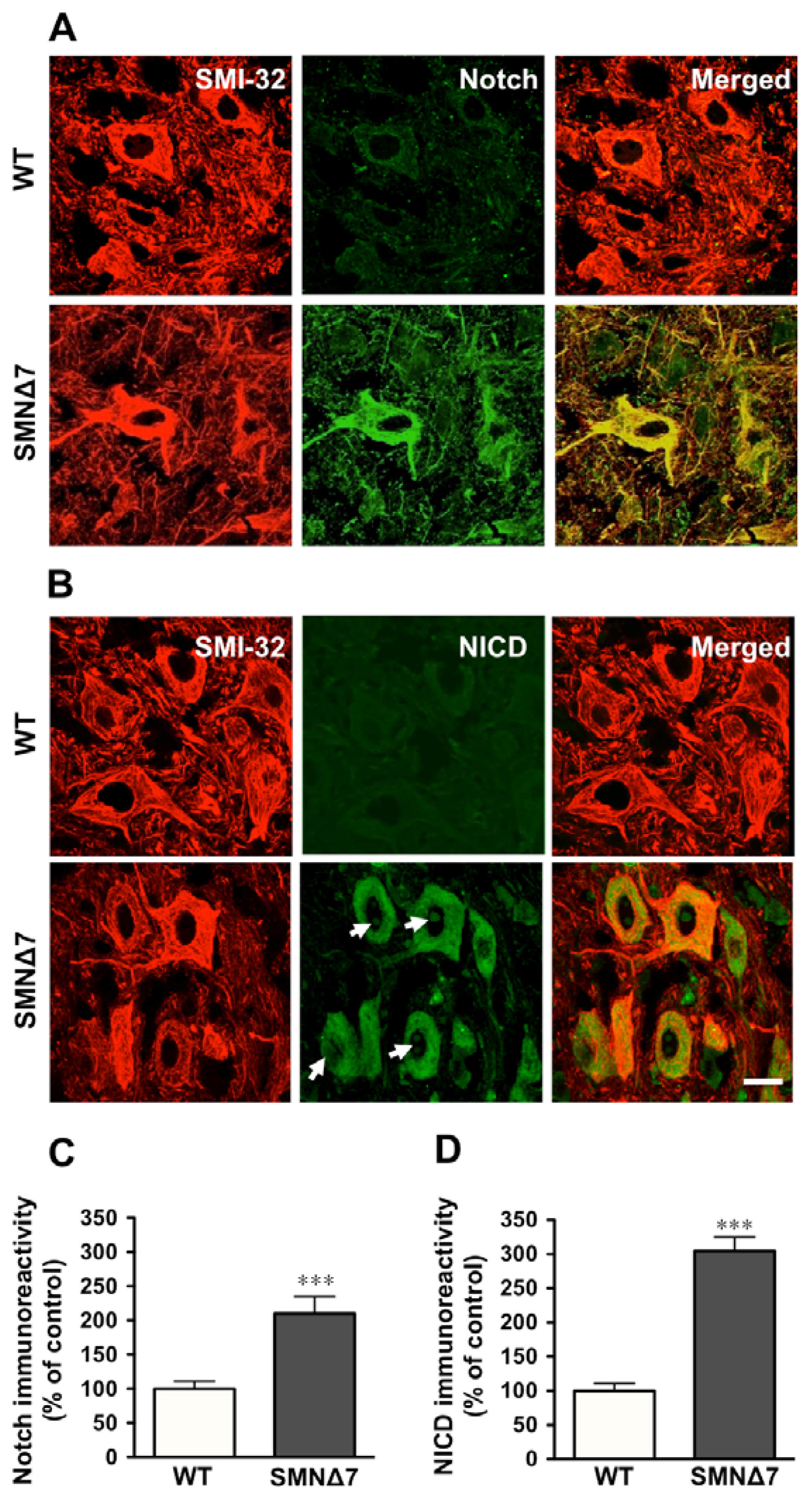
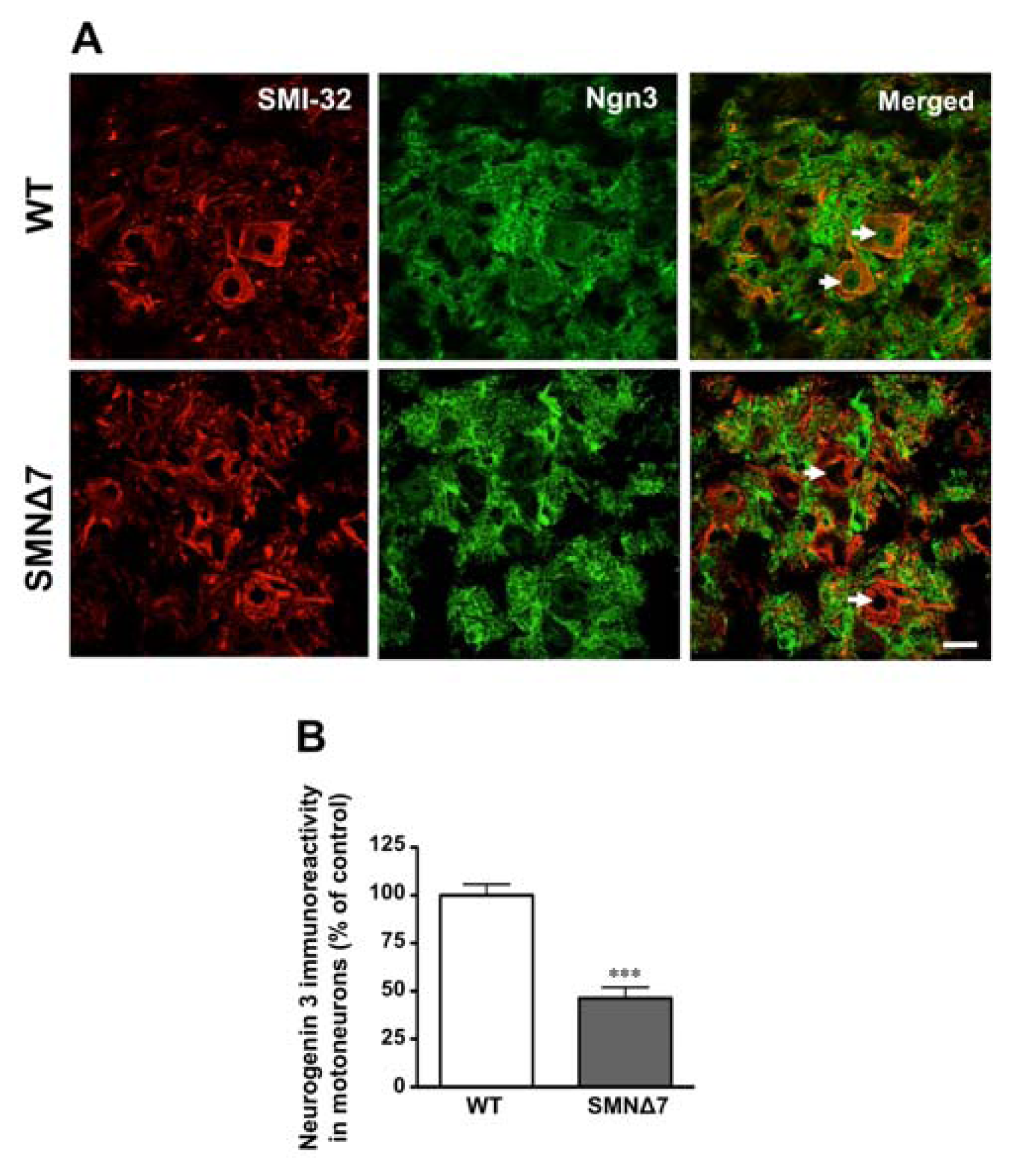
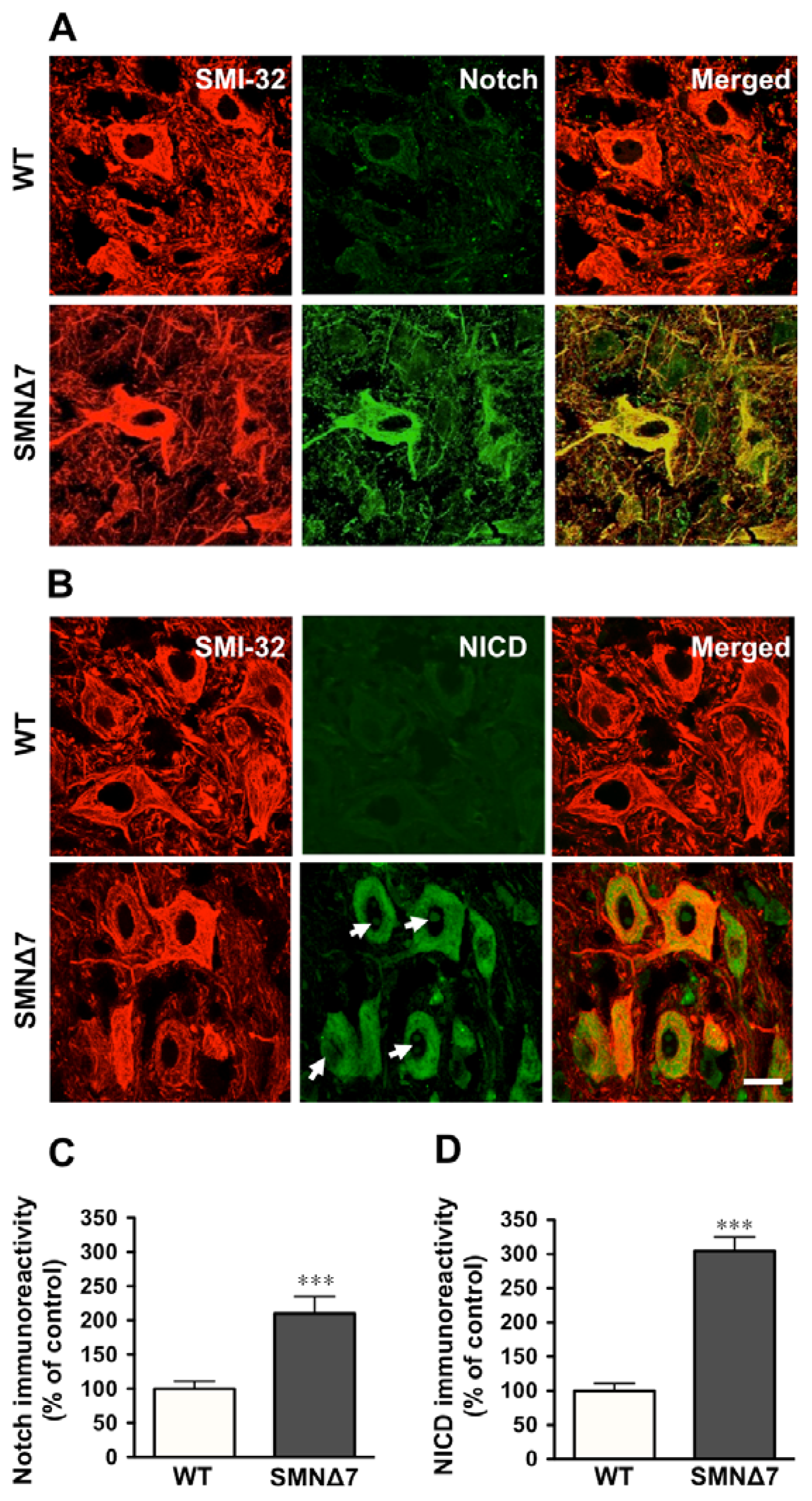
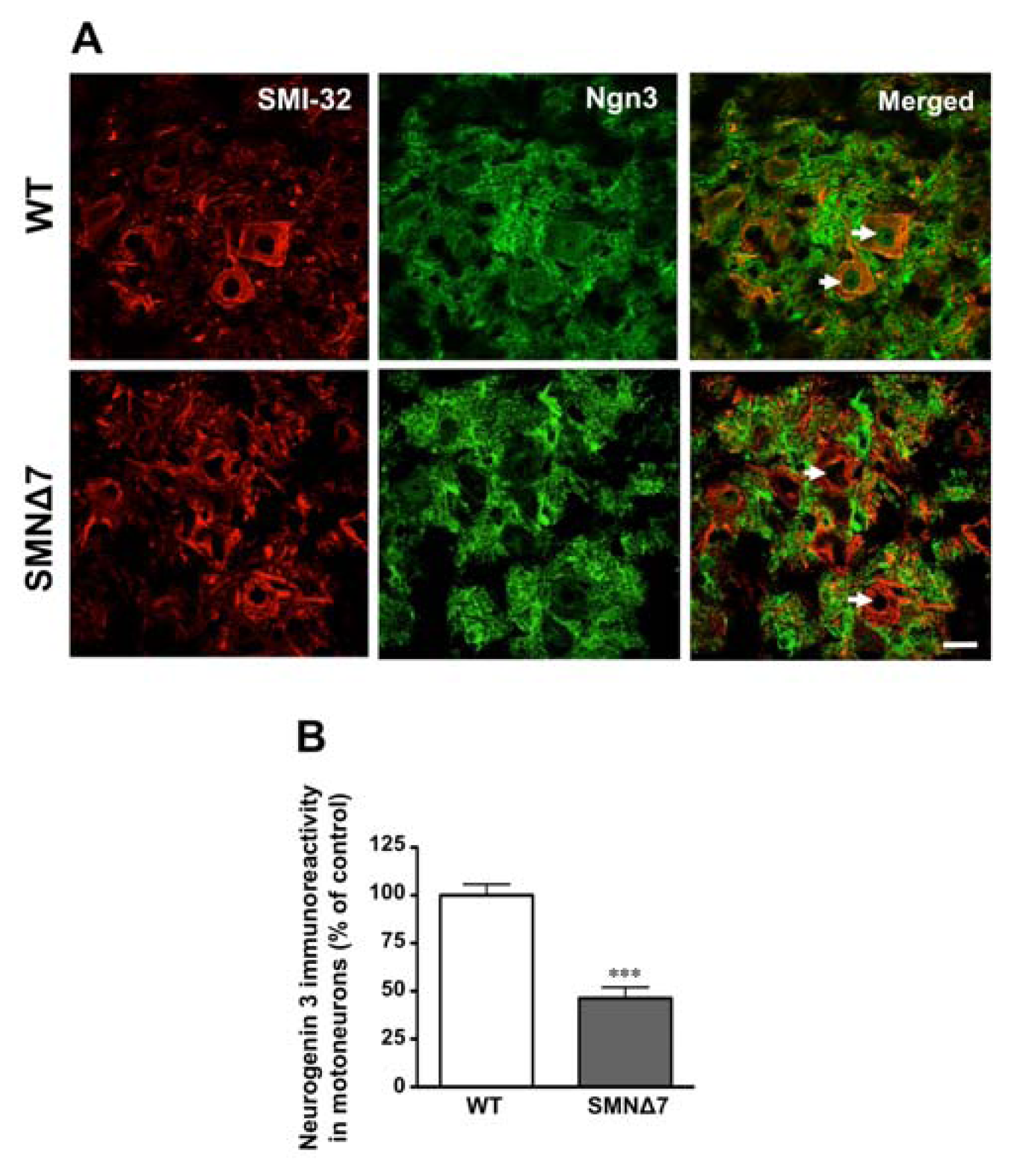
2.2. Increased Notch Signaling in Spinal Cord Motoneurons of the SMNΔ7 Mouse
3. Discussion
4. Experimental Section
4.1. U87MG Cell Culture and Transduction
4.2. Western Blotting
4.3. Mouse Model
4.4. Immunofluorescence and Nissl Staining
4.5. Image Acquisition and Analysis
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis 1996, 3, 97–110. [Google Scholar]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis 2011, 6, 71. [Google Scholar]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar]
- Schrank, B.; Gotz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar]
- Cifuentes-Diaz, C.; Frugier, T.; Tiziano, F.D.; Lacene, E.; Roblot, N.; Joshi, V.; Moreau, M.H.; Melki, J. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol 2001, 152, 1107–1114. [Google Scholar]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet 1997, 16, 265–269. [Google Scholar]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci 2006, 7, 93–102. [Google Scholar]
- Cau, E.; Blader, P. Notch activity in the nervous system: To switch or not switch? Neural Dev 2009, 4, 36. [Google Scholar]
- Pierfelice, T.; Alberi, L.; Gaiano, N. Notch in the vertebrate nervous system: An old dog with new tricks. Neuron 2011, 69, 840–855. [Google Scholar]
- D’Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar]
- Kovall, R.A. More complicated than it looks: Assembly of Notch pathway transcription complexes. Oncogene 2008, 27, 5099–5109. [Google Scholar]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar]
- Salama-Cohen, P.; Arevalo, M.-A.; Grantyn, R.; Rodriguez-Tebar, A. Notch and NGF/p75NTR control dendrite morphology and the balance of excitatory/inhibitory synaptic input to hippocampal neurones through neurogenin 3. J. Neurochem 2006, 97, 1269–1278. [Google Scholar]
- Apelqvist, A.; Li, H.; Sommer, L.; Beatus, P.; Anderson, D.J.; Honjo, T.; Hrabe de Angelis, M.; Lendahl, U.; Edlund, H. Notch signalling controls pancreatic cell differentiation. Nature 1999, 400, 877–881. [Google Scholar]
- Serafimidis, I.; Rakatzi, I.; Episkopou, V.; Gouti, M.; Gavalas, A. Novel effectors of directed and Ngn3-mediated differentiation of mouse embryonic stem cells into endocrine pancreas progenitors. Stem Cells 2008, 26, 3–16. [Google Scholar]
- Lewis, J. Notch signalling and the control of cell fate choices in vertebrates. Semin. Cell Dev. Biol 1998, 9, 583–589. [Google Scholar]
- Ables, J.L.; Breunig, J.J.; Eisch, A.J.; Rakic, P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci 2011, 12, 269–283. [Google Scholar]
- Berezovska, O.; Xia, M.Q.; Hyman, B.T. Notch is expressed in adult brain, is coexpressed with presenilin-1, and is altered in Alzheimer disease. J. Neuropathol. Exp. Neurol 1998, 57, 738–745. [Google Scholar]
- Lathia, J.D.; Mattson, M.P.; Cheng, A. Notch: From neural development to neurological disorders. J. Neurochem 2008, 107, 1471–1481. [Google Scholar]
- Nagarsheth, M.H.; Viehman, A.; Lippa, S.M.; Lippa, C.F. Notch-1 immunoexpression is increased in Alzheimer’s and Pick’s disease. J. Neurol. Sci 2006, 244, 111–116. [Google Scholar]
- Fischer, D.F.; van Dijk, R.; Sluijs, J.A.; Nair, S.M.; Racchi, M.; Levelt, C.N.; van Leeuwen, F.W.; Hol, E.M. Activation of the Notch pathway in down syndrome: Cross-talk of Notch and APP. FASEB J 2005, 19, 1451–1458. [Google Scholar]
- Berezovska, O.; McLean, P.; Knowles, R.; Frosh, M.; Lu, F.M.; Lux, S.E.; Hyman, B.T. Notch1 inhibits neurite outgrowth in postmitotic primary neurons. Neuroscience 1999, 93, 433–439. [Google Scholar]
- Bowerman, M.; Shafey, D.; Kothary, R. Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J. Mol. Neurosci 2007, 32, 120–131. [Google Scholar]
- Redmond, L.; Oh, S.R.; Hicks, C.; Weinmaster, G.; Ghosh, A. Nuclear Notch1 signaling and the regulation of dendritic development. Nat. Neurosci 2000, 3, 30–40. [Google Scholar]
- Liu, H.; Beauvais, A.; Baker, A.N.; Tsilfidis, C.; Kothary, R. Smn deficiency causes neuritogenesis and neurogenesis defects in the retinal neurons of a mouse model of spinal muscular atrophy. Dev. Neurobiol 2011, 71, 153–169. [Google Scholar]
- Hashimoto-Torii, K.; Torii, M.; Sarkisian, M.R.; Bartley, C.M.; Shen, J.; Radtke, F.; Gridley, T.; Sestan, N.; Rakic, P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron 2008, 60, 273–284. [Google Scholar]
- Caraballo-Miralles, V.; Cardona-Rossinyol, A.; Garcera, A.; Villalonga, P.; Soler, R.M.; Olmos, G.; Llado, J. SMN deficiency attenuates migration of U87MG astroglioma cells through the activation of RhoA. Mol. Cell. Neurosci 2012, 49, 282–289. [Google Scholar]
- Giniger, E.; Jan, L.Y.; Jan, Y.N. Specifying the path of the intersegmental nerve of the Drosophila embryo: A role for Delta and Notch. Development 1993, 117, 431–440. [Google Scholar]
- McWhorter, M.L.; Monani, U.R.; Burghes, A.H.; Beattie, C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol 2003, 162, 919–931. [Google Scholar]
- De Bivort, B.L.; Guo, H.F.; Zhong, Y. Notch signaling is required for activity-dependent synaptic plasticity at the Drosophila neuromuscular junction. J. Neurogenet 2009, 23, 395–404. [Google Scholar]
- Torres-Benito, L.; Neher, M.F.; Cano, R.; Ruiz, R.; Tabares, L. SMN requirement for synaptic vesicle, active zone and microtubule postnatal organization in motor nerve terminals. PLoS One 2011, 6, e26164. [Google Scholar]
- Morga, E.; Mouad-Amazzal, L.; Felten, P.; Heurtaux, T.; Moro, M.; Michelucci, A.; Gabel, S.; Grandbarbe, L.; Heuschling, P. Jagged1 regulates the activation of astrocytes via modulation of NFκB and JAK/STAT/SOCS pathways. Glia 2009, 57, 1741–1753. [Google Scholar]
- Givogri, M.I.; de Planell, M.; Galbiati, F.; Superchi, D.; Gritti, A.; Vescovi, A.; de Vellis, J.; Bongarzone, E.R. Notch signaling in astrocytes and neuroblasts of the adult subventricular zone in health and after cortical injury. Dev. Neurosci 2006, 28, 81–91. [Google Scholar]
- Araki, S.; Hayashi, M.; Tamagawa, K.; Saito, M.; Kato, S.; Komori, T.; Sakakihara, Y.; Mizutani, T.; Oda, M. Neuropathological analysis in spinal muscular atrophy type II. Acta Neuropathol 2003, 106, 441–448. [Google Scholar]
- Garcia-Cabezas, M.A.; Garcia-Alix, A.; Martin, Y.; Gutierrez, M.; Hernandez, C.; Rodriguez, J.I.; Morales, C. Neonatal spinal muscular atrophy with multiple contractures, bone fractures, respiratory insufficiency and 5q13 deletion. Acta Neuropathol 2004, 107, 475–478. [Google Scholar]
- Kuru, S.; Sakai, M.; Konagaya, M.; Yoshida, M.; Hashizume, Y.; Saito, K. An autopsy case of spinal muscular atrophy type III (Kugelberg-Welander disease). Neuropathology 2009, 29, 63–67. [Google Scholar]
- Dachs, E.; Hereu, M.; Piedrafita, L.; Casanovas, A.; Caldero, J.; Esquerda, J.E. Defective neuromuscular junction organization and postnatal myogenesis in mice with severe spinal muscular atrophy. J. Neuropathol. Exp. Neurol 2011, 70, 444–461. [Google Scholar]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet 2005, 14, 845–857. [Google Scholar]
- Barbeito, L. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Rev 2004, 47, 263–274. [Google Scholar]
- Papadimitriou, D.; Le Verche, V.; Jacquier, A.; Ikiz, B.; Przedborski, S.; Re, D.B. Inflammation in ALS and SMA: Sorting out the good from the evil. Neurobiol. Dis 2010, 37, 493–502. [Google Scholar]
- Zhang, Y.; Zhang, J.; Navrazhina, K.; Argaw, A.T.; Zameer, A.; Gurfein, B.T.; Brosnan, C.F.; John, G.R. TGFbeta1 induces Jagged1 expression in astrocytes via ALK5 and Smad3 and regulates the balance between oligodendrocyte progenitor proliferation and differentiation. Glia 2010, 58, 964–974. [Google Scholar]
- Katsuno, M.; Adachi, H.; Banno, H.; Suzuki, K.; Tanaka, F.; Sobue, G. Transforming growth factor-beta signaling in motor neuron diseases. Curr. Mol. Med 2011, 11, 48–56. [Google Scholar]
- Kamei, N.; Kwon, S.M.; Ishikawa, M.; Ii, M.; Nakanishi, K.; Yamada, K.; Hozumi, K.; Kawamoto, A.; Ochi, M.; Asahara, T. Endothelial progenitor cells promote astrogliosis following spinal cord injury through Jagged1-dependent Notch signaling. J. Neurotrauma 2012, 29, 1758–1769. [Google Scholar]
- Ross, D.A.; Kadesch, T. Consequences of Notch-mediated induction of Jagged1. Exp. Cell Res 2004, 296, 173–182. [Google Scholar]
- Alberi, L.; Liu, S.; Wang, Y.; Badie, R.; Smith-Hicks, C.; Wu, J.; Pierfelice, T.J.; Abazyan, B.; Mattson, M.P.; Kuhl, D.; et al. Activity-induced Notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron 2011, 69, 437–444. [Google Scholar]
- Wu, G.; Lyapina, S.; Das, I.; Li, J.; Gurney, M.; Pauley, A.; Chui, I.; Deshaies, R.J.; Kitajewski, J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol. Cell. Biol 2001, 21, 7403–7415. [Google Scholar]
- de Celis, J.F.; Bray, S. Feed-back mechanisms affecting notch activation at the dorsoventral boundary in the Drosophila wing. Development 1997, 124, 3241–3251. [Google Scholar]
- Simon-Areces, J.; Membrive, G.; Garcia-Fernandez, C.; Garcia-Segura, L.M.; Arevalo, M.A. Neurogenin 3 cellular and subcellular localization in the developing and adult hippocampus. J. Comp. Neurol 2010, 518, 1814–1824. [Google Scholar]
- Simic, G.; Mladinov, M.; Seso Simic, D.; Jovanov Milosevic, N.; Islam, A.; Pajtak, A.; Barisic, N.; Sertic, J.; Lucassen, P.J.; Hof, P.R.; et al. Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol 2008, 115, 313–326. [Google Scholar]
- McGovern, V.L.; Gavrilina, T.O.; Beattie, C.E.; Burghes, A.H. Embryonic motor axon development in the severe SMA mouse. Hum. Mol. Genet 2008, 17, 2900–2909. [Google Scholar]
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol 2011, 31, 876–882. [Google Scholar]
- Ferrón, S.R.; Marqués-Torrejón, M.A.; Mira, H.; Flores, I.; Taylor, K.; Blasco, M.A.; Fariñas, I. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci 2009, 29, 14394–14407. [Google Scholar]
- Arumugam, T.V.; Chan, S.L.; Jo, D.G.; Yilmaz, G.; Tang, S.C.; Cheng, A.; Gleichmann, M.; Okun, E.; Dixit, V.D.; Chigurupati, S.; et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat. Med 2006, 12, 621–623. [Google Scholar]
- Grandbarbe, L.; Michelucci, A.; Heurtaux, T.; Hemmer, K.; Morga, E.; Heuschling, P. Notch signaling modulates the activation of microglial cells. Glia 2007, 55, 1519–1530. [Google Scholar]
- Jurynczyk, M.; Selmaj, K. Notch: A new player in MS mechanisms. J. Neuroimmunol 2010, 218, 3–11. [Google Scholar]
- Kume, T. Ligand-dependent Notch signaling in vascular formation. Adv. Exp. Med. Biol 2012, 727, 210–222. [Google Scholar]
- Garcera, A.; Mincheva, S.; Gou-Fabregas, M.; Caraballo-Miralles, V.; Llado, J.; Comella, J.X.; Soler, R.M. A new model to study spinal muscular atrophy: Neurite degeneration and cell death is counteracted by BCL-X(L) overexpression in motoneurons. Neurobiol. Dis 2011, 42, 415–426. [Google Scholar]
- Mir, M.; Asensio, V.J.; Tolosa, L.; Gou-Fabregas, M.; Soler, R.M.; Lladó, J.; Olmos, G. Tumor necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and motoneuron death in rat spinal cord embryonic explants. Neuroscience 2009, 162, 959–971. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Caraballo-Miralles, V.; Cardona-Rossinyol, A.; Garcera, A.; Torres-Benito, L.; Soler, R.M.; Tabares, L.; Lladó, J.; Olmos, G. Notch Signaling Pathway Is Activated in Motoneurons of Spinal Muscular Atrophy. Int. J. Mol. Sci. 2013, 14, 11424-11437. https://doi.org/10.3390/ijms140611424
Caraballo-Miralles V, Cardona-Rossinyol A, Garcera A, Torres-Benito L, Soler RM, Tabares L, Lladó J, Olmos G. Notch Signaling Pathway Is Activated in Motoneurons of Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2013; 14(6):11424-11437. https://doi.org/10.3390/ijms140611424
Chicago/Turabian StyleCaraballo-Miralles, Víctor, Andrea Cardona-Rossinyol, Ana Garcera, Laura Torres-Benito, Rosa M. Soler, Lucía Tabares, Jerònia Lladó, and Gabriel Olmos. 2013. "Notch Signaling Pathway Is Activated in Motoneurons of Spinal Muscular Atrophy" International Journal of Molecular Sciences 14, no. 6: 11424-11437. https://doi.org/10.3390/ijms140611424