Dual Role of MicroRNAs in NAFLD


Abstract
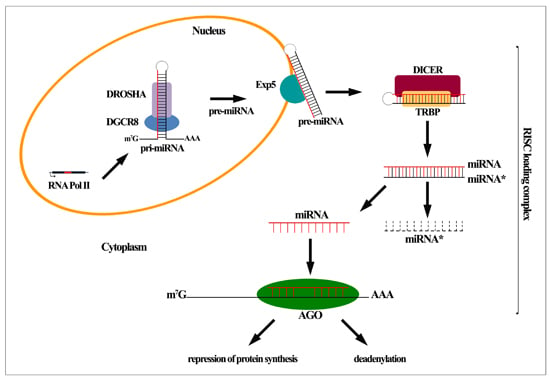
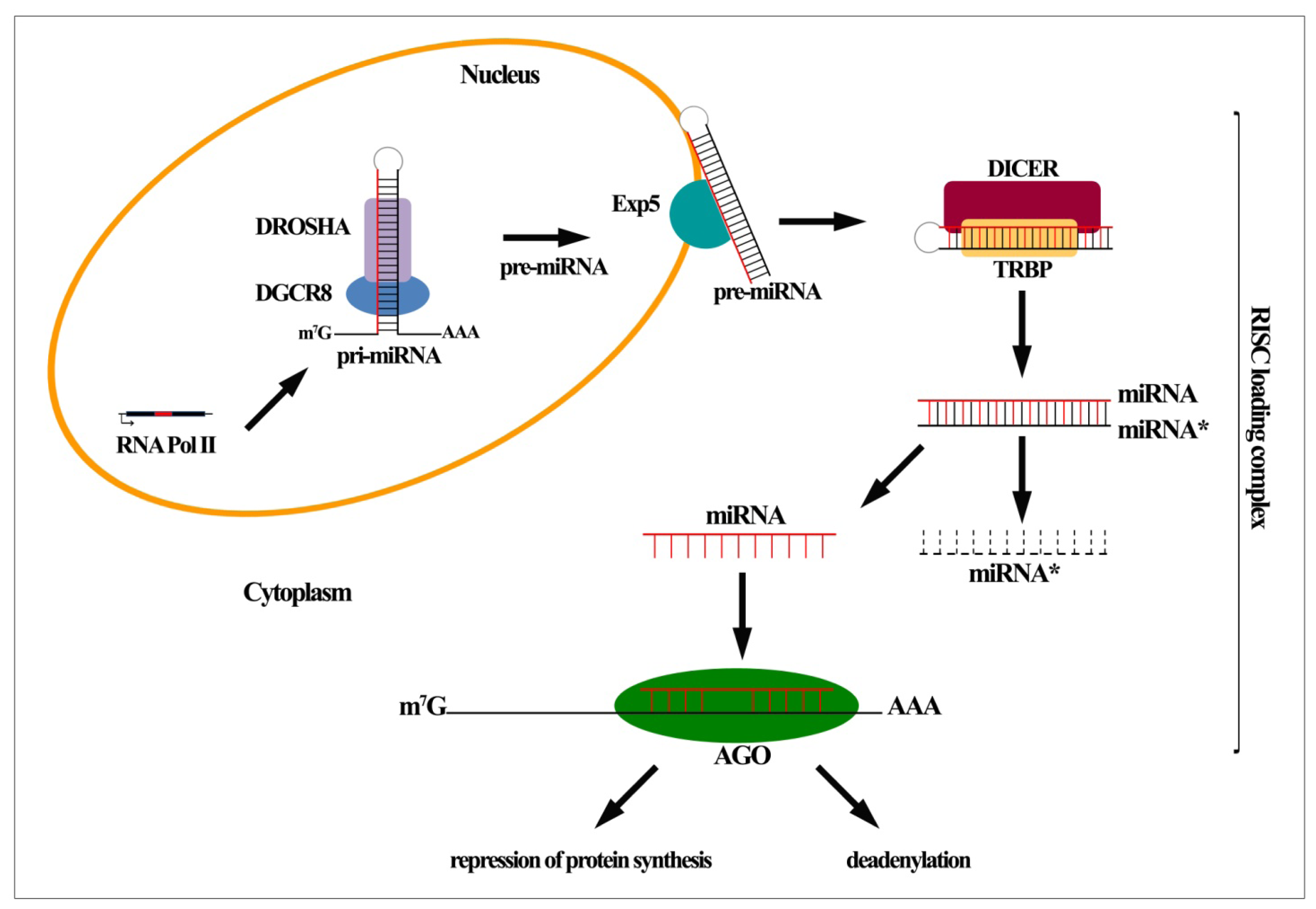
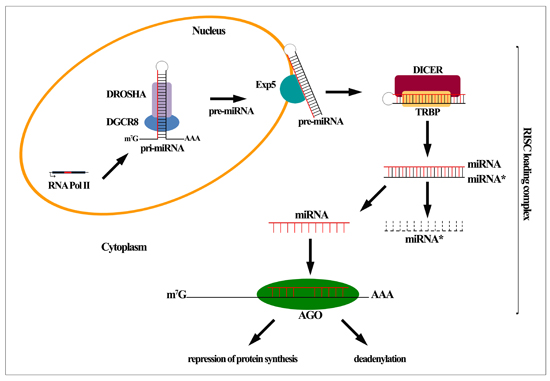
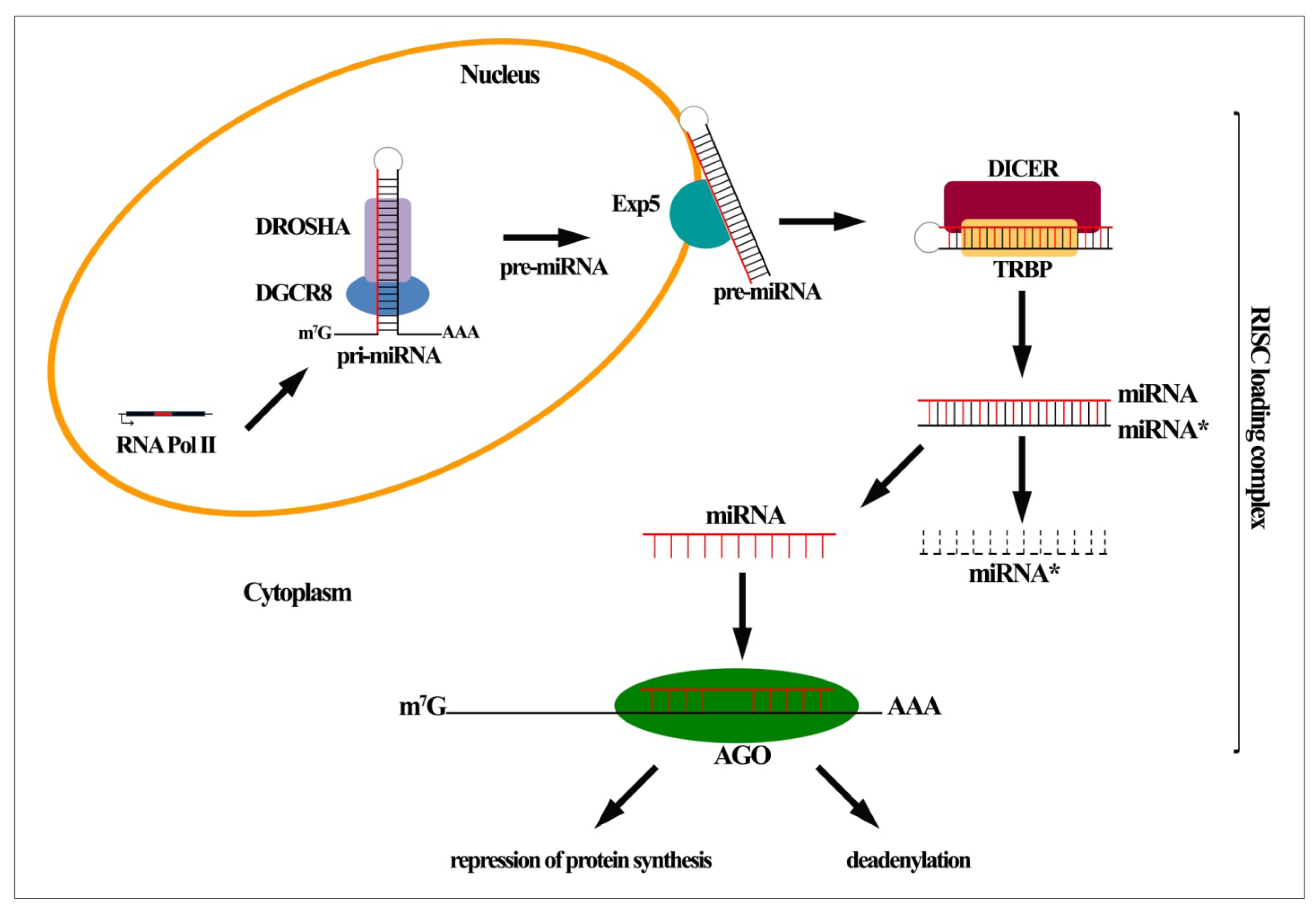
:
1. Introduction
2. MiRNA Biogenesis and Function
3. MiRNA Regulation, Activity and Decay
4. MiRNA Analysis
5. MiRNAs: Metabolic Functions and Their Implication in NAFLD
6. MiRNAs and Their Implication in Hepatic Fibrosis
7. Concluding Remarks
Conflicts of Interest
References
- Leel, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementary to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet 2012, 13, 271–282. [Google Scholar]
- Lakner, A.M.; Bonkovsky, H.L.; Schrum, L.W. MicroRNAs: Fad or future of liver disease. World J. Gastroenterol 2011, 17, 2536–2542. [Google Scholar]
- Siomi, H.; Siomi, M.C. On the road to reading the RNA-interference cose. Nature 2009, 457, 396–404. [Google Scholar]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet 2010, 11, 597–610. [Google Scholar]
- Kerr, T.A.; Korenblat, K.M.; Davidson, N.O. MicroRNAs and liver disease. Transl. Res 2011, 157, 241–252. [Google Scholar]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar]
- Miyoshi, K.; Miyoshi, T.; Siomi, H. Many ways to generate microRNA-like small RNAs: Non-canonical pathways for microRNA production. Mol. Genet. Genomics 2010, 284, 95–103. [Google Scholar]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and function. Thromb. Haemost 2012, 107, 605–610. [Google Scholar]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.A. ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar]
- Sato, F.; Tsuchiya, S.; Meltzer, S.J.; Shimizu, K. MicroRNAs and epigenetics. FEBS J 2011, 278, 1598–1609. [Google Scholar]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar]
- Sakamoto, S.; Aoki, K.; Higuchi, T.; Todaka, H.; Morisawa, K.; Tamaki, N.; Hatano, E.; Fukushima, A.; Taniguchi, T.; Agata, Y. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol. Cell. Biol 2009, 29, 3754–3769. [Google Scholar]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol 2006, 13, 13–21. [Google Scholar]
- Tomaselli, S.; Panera, N.; Gallo, A.; Alisi, A. Circulating miRNA profiling to identify biomarkers of dysmetabolism. Biomark. Med 2012, 6, 729–742. [Google Scholar]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective blockade of microRNA processing by Lin28. Science 2008, 320, 97–100. [Google Scholar]
- Lehrbach, N.J.; Miska, E.A. Regulation of pre-miRNA Processing. Adv. Exp. Med. Biol 2010, 700, 67–75. [Google Scholar]
- Heo, I.; Joo, C.; Kim, Y.K.; Ha, M.; Yoon, M.J.; Cho, J.; Yeom, K.H.; Han, J.; Kim, V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009, 138, 696–708. [Google Scholar]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar]
- Diederichs, S.; Haber, D.A. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 2007, 131, 1097–1108. [Google Scholar]
- Melo, S.A.; Moutinho, C.; Ropero, S.; Calin, G.A.; Rossi, S.; Spizzo, R.; Fernandez, A.F.; Davalos, V.; Villanueva, A.; Montoya, G.; et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010, 18, 303–315. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet 2011, 12, 861–874. [Google Scholar]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet 2012, 13, 358–369. [Google Scholar]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol 2012, 13, 239–250. [Google Scholar]
- Lynn, F.C. Meta-regulation: MicroRNA regulation of glucose and lipid metabolism. Trends Endocrinol. Metab 2009, 20, 452–459. [Google Scholar]
- Krützfeldt, J.; Stoffel, M. MicroRNAs: A new class of regulatory genes affecting metabolism. Cell Metab 2006, 4, 9–12. [Google Scholar]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 195–203. [Google Scholar]
- Hu, J.; Xu, Y.; Hao, J.; Wang, S.; Li, C.; Meng, S. MiR-122 in hepatic function and liver diseases. Protein Cell 2012, 3, 364–371. [Google Scholar]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol 2012, 9, 137–142. [Google Scholar]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 2006, 3, 87–98. [Google Scholar]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in nonhuman primates. Nature 2008, 452, 896–899. [Google Scholar]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig 2012, 122, 2871–2883. [Google Scholar]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig 2012, 122, 2884–2897. [Google Scholar]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar]
- Alisi, A.; da Sacco, L.; Bruscalupi, G.; Piemonte, F.; Panera, N.; de Vito, R.; Leoni, S.; Bottazzo, G.F.; Masotti, A.; Nobili, V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab. Investig. 2011, 91, 283–293. [Google Scholar]
- Iliopoulos, D.; Drosatos, K.; Hiyama, Y.; Goldberg, I.J.; Zannis, V.I. MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J. Lipid Res 2010, 51, 1513–1523. [Google Scholar]
- Zheng, L.; Lv, G.C.; Sheng, J.; Yang, Y.D. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-alpha expression, a novel mechanism for the pathogenesis of NAFLD. J. Gastroenterol. Hepatol 2010, 25, 156–163. [Google Scholar]
- Ahn, J.; Lee, H.; Chung, C.H.; Ha, T. High fat diet induced downregulation of microRNA-467b increased lipoprotein lipase in hepatic steatosis. Biochem. Biophys. Res. Commun 2011, 414, 664–669. [Google Scholar]
- Hoekstra, M.; van der Sluis, R.J.; Kuiper, J.; van Berkel, T.J. Nonalcoholic fatty liver disease is associated with an altered hepatocyte microRNA profile in LDL receptor knockout mice. J. Nutr. Biochem 2012, 23, 622–628. [Google Scholar]
- Sacco, J.; Adeli, K. MicroRNAs: Emerging roles in lipid and lipoprotein metabolism. Curr. Opin. Lipidol 2012, 23, 220–225. [Google Scholar]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar]
- Gerin, I.; Clerbaux, L.A.; Haumont, O.; Lanthier, N.; Das, A.K.; Burant, C.F.; Leclercq, I.A.; Macdougald, O.A.; Bommer, G.T. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J. Biol. Chem 2010, 285, 33652–33661. [Google Scholar]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 2012, 15, 665–674. [Google Scholar]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS One 2011, 6, e23937. [Google Scholar]
- Tryndyak, V.P.; Latendresse, J.R.; Montgomery, B.; Ross, S.A.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Plasma microRNAs are sensitive indicators of inter-strain differences in the severity of liver injury induced in mice by a choline- and folate-deficient diet. Toxicol. Appl. Pharm 2012, 262, 52–59. [Google Scholar]
- Castro, R.E.; Ferreira, D.M.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol 2013, 58, 119–125. [Google Scholar]
- Jin, X.; Ye, Y.F.; Chen, S.H.; Yu, C.H.; Liu, J.; Li, Y.M. MicroRNA expression pattern in different stages of nonalcoholic fatty liver disease. Dig. Liver Dis 2009, 41, 289–297. [Google Scholar]
- Pogribny, I.P.; Starlard-Davenport, A.; Tryndyak, V.P.; Han, T.; Ross, S.A.; Rusyn, I.; Beland, F.A. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab. Investig 2010, 90, 1437–1446. [Google Scholar]
- Milić, S.; Stimac, D. Nonalcoholic fatty liver disease/steatohepatitis: Epidemiology, pathogenesis, clinical presentation and treatment. Dig. Dis 2012, 30, 158–162. [Google Scholar]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol 2011, 25, 195–206. [Google Scholar]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev 2008, 88, 125–172. [Google Scholar]
- Murakami, Y.; Toyoda, H.; Tanaka, M.; Kuroda, M.; Harada, Y.; Matsuda, F.; Tajima, A.; Kosaka, N.; Ochiya, T.; Shimotohno, K. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One 2011, 6, e16081. [Google Scholar]
- Li, W.Q.; Chen, C.; Xu, M.D.; Guo, J.; Li, Y.M.; Xia, Q.M.; Liu, H.M.; He, J.; Yu, H.Y.; Zhu, L. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J 2011, 278, 1522–1532. [Google Scholar]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar]
- Iizuka, M.; Ogawa, T.; Enomoto, M.; Motoyama, H.; Yoshizato, K.; Ikeda, K.; Kawada, N. Induction of microRNA-214-5p in human and rodent liver fibrosis. Fibrogenesis Tissue Repair 2012, 5. [Google Scholar] [CrossRef]
- Guo, C.J.; Pan, Q.; Li, D.G.; Sun, H.; Liu, B.W. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. J. Hepatol 2009, 50, 766–778. [Google Scholar]
- Chen, C.; Wu, C.Q.; Zhang, Z.Q.; Yao, D.K.; Zhu, L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp. Cell Res 2011, 317, 1714–1725. [Google Scholar]
- Ji, J.; Zhang, J.; Huang, G.; Qian, J.; Wang, X.; Mei, S. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett 2009, 583, 759–766. [Google Scholar]
- Venugopal, S.K.; Jiang, J.; Kim, T.H.; Li, Y.; Wang, S.S.; Torok, N.J.; Wu, J.; Zern, M.A. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am. J. Physiol. Gastrointest. Liver Physiol 2010, 298, G101–G106. [Google Scholar]
- Ogawa, T.; Iizuka, M.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; Kawada, N. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem. Biophys. Res. Commun 2010, 391, 316–321. [Google Scholar]
- Chakraborty, J.B.; Oakley, F.; Walsh, M.J. Mechanisms and biomarkers of apoptosis in liver disease and fibrosis. Int. J. Hepatol. 2012, 2012. [Google Scholar] [CrossRef]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 425–436. [Google Scholar]
- Guo, C.J.; Pan, Q.; Jiang, B.; Chen, G.Y.; Li, D.G. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 2009, 14, 1331–1340. [Google Scholar]
- Guo, C.J.; Pan, Q.; Cheng, T.; Jiang, B.; Chen, G.Y.; Li, D.G. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J 2009, 276, 5163–5176. [Google Scholar]
- He, Y.; Huang, C.; Sun, X.; Long, X.R.; Lv, X.W.; Li, J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell. Signal 2012, 24, 1923–1930. [Google Scholar]
- Li, J.T.; Liao, Z.X.; Ping, J.; Xu, D.; Wang, H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J. Gastroenterol 2008, 43, 419–428. [Google Scholar]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem 2000, 275, 2247–2250. [Google Scholar]
- Maubach, G.; Lim, M.C.; Chen, J.; Yang, H.; Zhuo, L. miRNA studies in in vitro and in vivo activated hepatic stellate cells. World J. Gastroenterol 2011, 17, 2748–2773. [Google Scholar]
- Shiratori, Y.; Imazeki, F.; Moriyama, M.; Yano, M.; Arakawa, Y.; Yokosuka, O.; Kuroki, T.; Nishiguchi, S.; Sata, M.; Yamada, G.; et al. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Ann. Intern. Med 2000, 132, 517–524. [Google Scholar]
- Poynard, T.; McHutchison, J.; Davis, G.L.; Esteban-Mur, R.; Goodman, Z.; Bedossa, P.; Albrecht, J. Impact of interferon alfa-2b and ribavirin on progression of liver fibrosis in patients with chronic hepatitis C. Hepatology 2000, 32, 1131–1137. [Google Scholar]
- Sekiya, Y.; Ogawa, T.; Iizuka, M.; Yoshizato, K.; Ikeda, K.; Kawada, N. Down-regulation of cyclin E1 expression by microRNA-195 accounts for interferon-β-induced inhibition of hepatic stellate cell proliferation. J. Cell. Physiol 2011, 226, 2535–2542. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar]

{kind=link}
{kind=link}
| Condition | HUMAN | MOUSE/RAT | References | ||
|---|---|---|---|---|---|
| Upregulated miRs | Downregulated miRs | Upregulated miRs | Downregulated miRs | ||
| NAFLD/NASH | 10b, 16, 29c, 33, 34a,122 (circ), 146b | 99b, 122 (liver), 132, 150, 511a | MOUSE: 34a, 155, 200b, 214-5p, 221 RAT: 16, 10b, 29c, 33, 34a, 122, 200a/b, 429 | MOUSE: 29c, 122, 192, 203, 467b, 216, 302a RAT: 27, 29b, 122, 203, 451 | [4,20,41,42,45,46, 52–55,65] |
| FIBROSIS | 34c, 125-5p, 199a/b, 200a/b, 221, 223 | 29a/b/c, 30b/c, 96, 132,193, 341, 183, 877 | MOUSE: mmu-let-7e, 31, 34a, 125-5p,199a-5p, 199b, 200a/b, 497, 802 RAT: 34a/b/c, 214, 221, 146b, 199a-5p, 199a-3p, 223, 324-5p | MOUSE: 29a/b/c RAT: 193, 378, 878 | [4,62–64] |
| Activated HSCs | 214-5p | 29b, 150, 194, | MOUSE: 214-5p RAT: 138, 140, 143, 193, 207, 501, 325-5p, 328, 349, 872, 874 | RAT: 15b, 16, 20b-3p, 27a/b, 29b 92b, 126, 122, 146a, 150, 194, 341, 375 | [65,66,69,70,75] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ceccarelli, S.; Panera, N.; Gnani, D.; Nobili, V. Dual Role of MicroRNAs in NAFLD. Int. J. Mol. Sci. 2013, 14, 8437-8455. https://doi.org/10.3390/ijms14048437
Ceccarelli S, Panera N, Gnani D, Nobili V. Dual Role of MicroRNAs in NAFLD. International Journal of Molecular Sciences. 2013; 14(4):8437-8455. https://doi.org/10.3390/ijms14048437
Chicago/Turabian StyleCeccarelli, Sara, Nadia Panera, Daniela Gnani, and Valerio Nobili. 2013. "Dual Role of MicroRNAs in NAFLD" International Journal of Molecular Sciences 14, no. 4: 8437-8455. https://doi.org/10.3390/ijms14048437