Transgenerational, Dynamic Methylation of Stomata Genes in Response to Low Relative Humidity

Abstract
:1. Introduction
2. Results
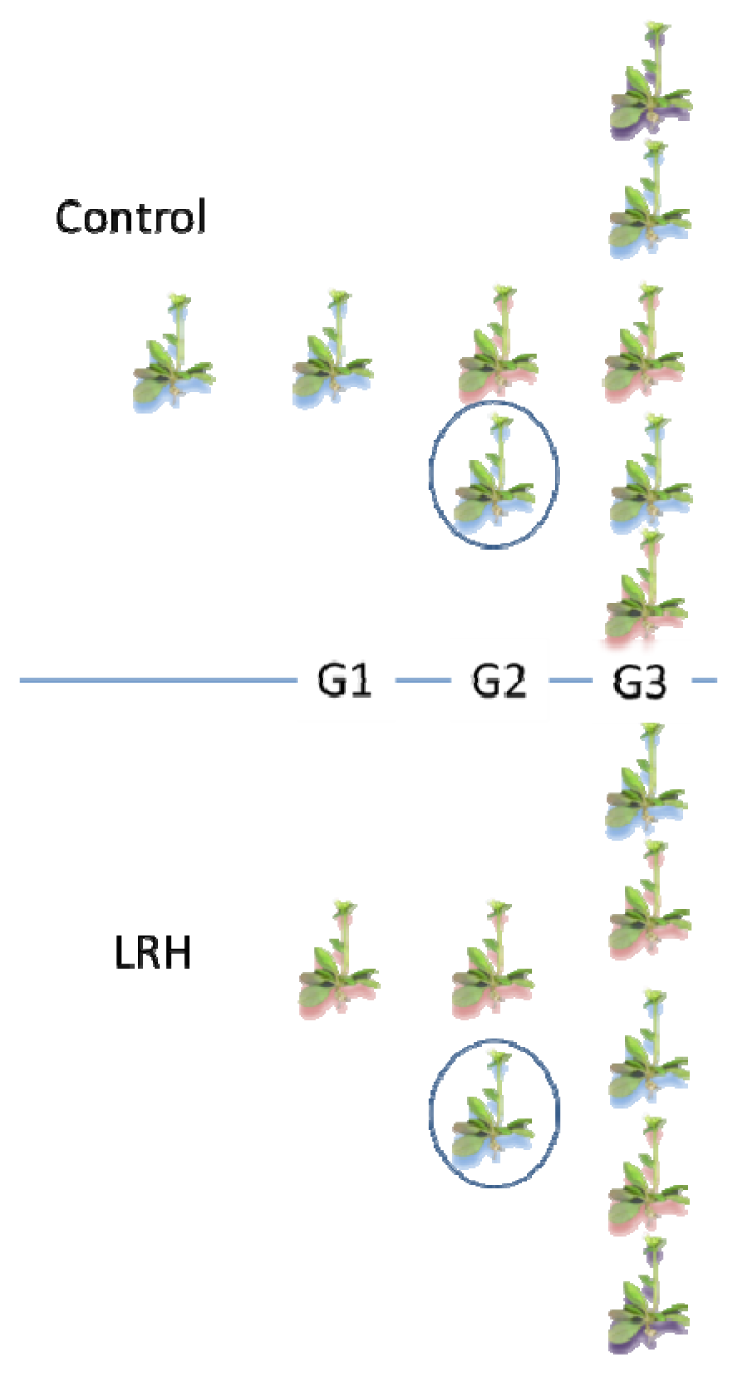
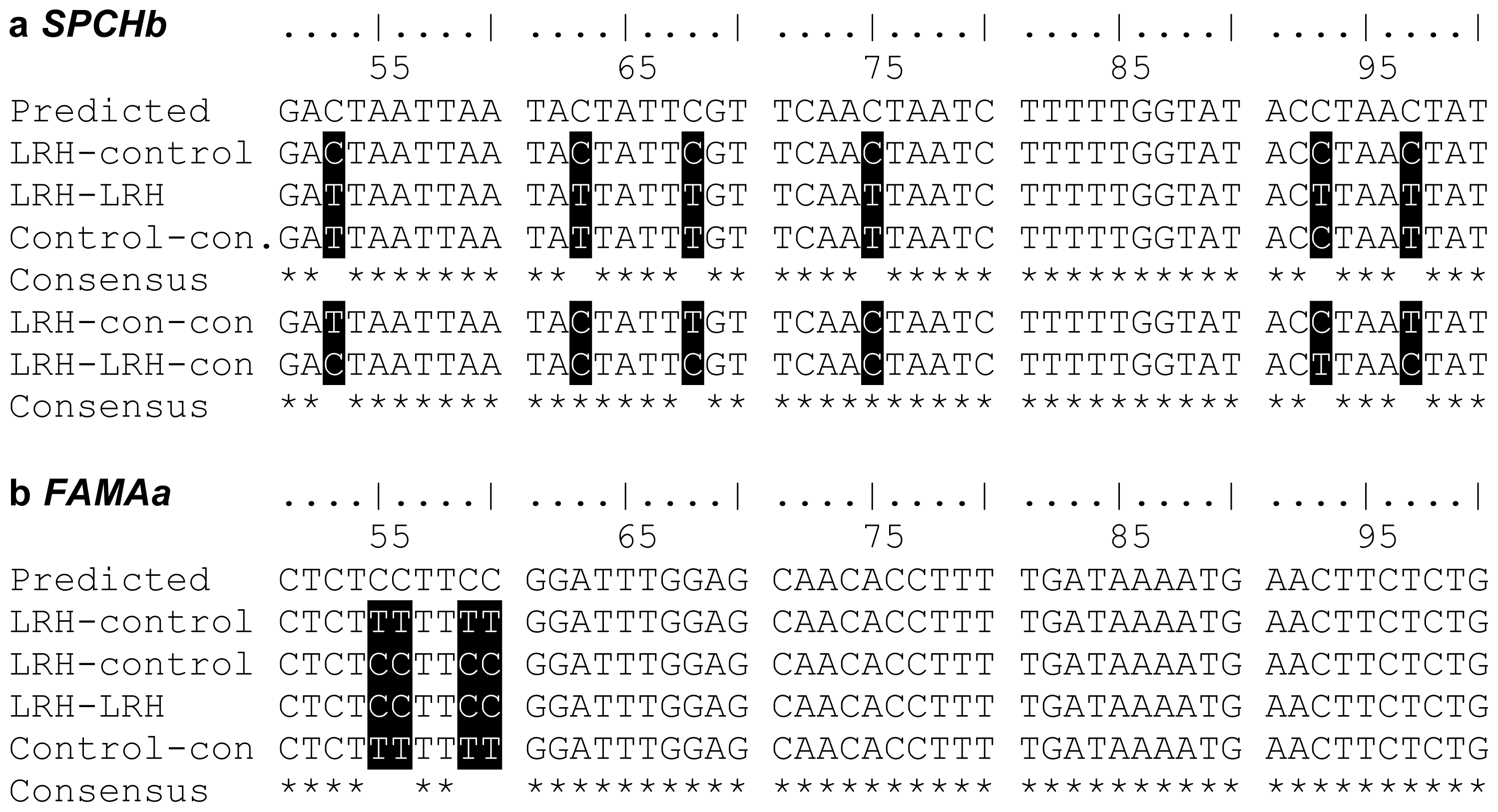
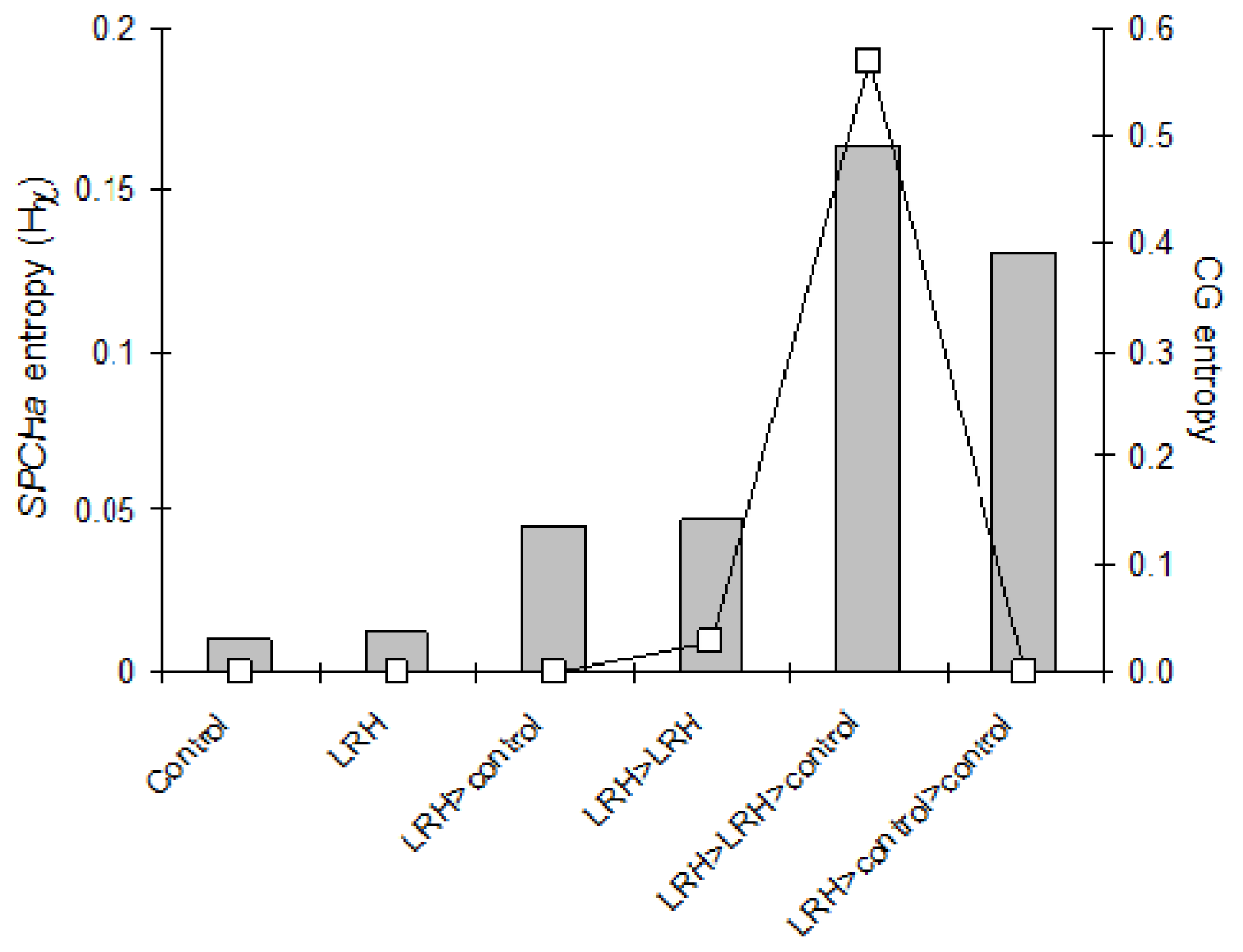
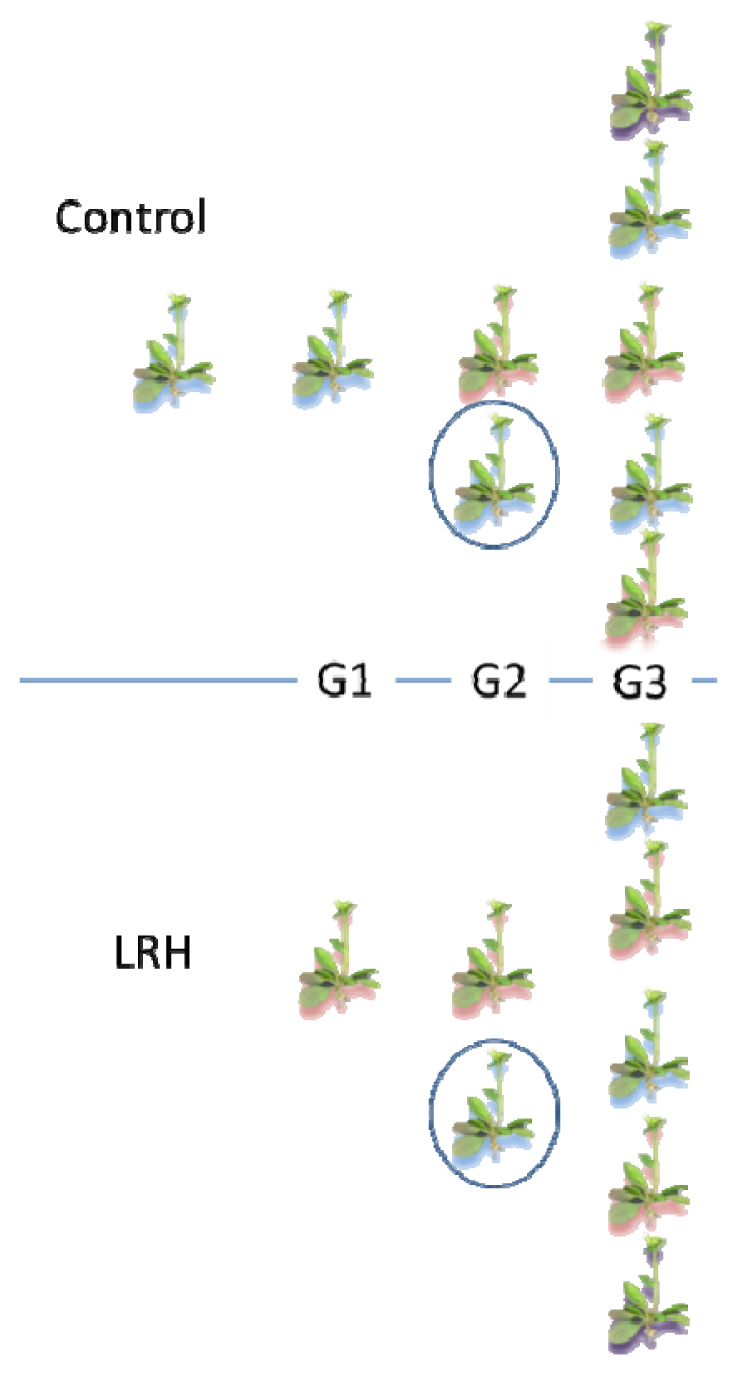
2.1. Inheritance of Stress-Induced DNA Methylation
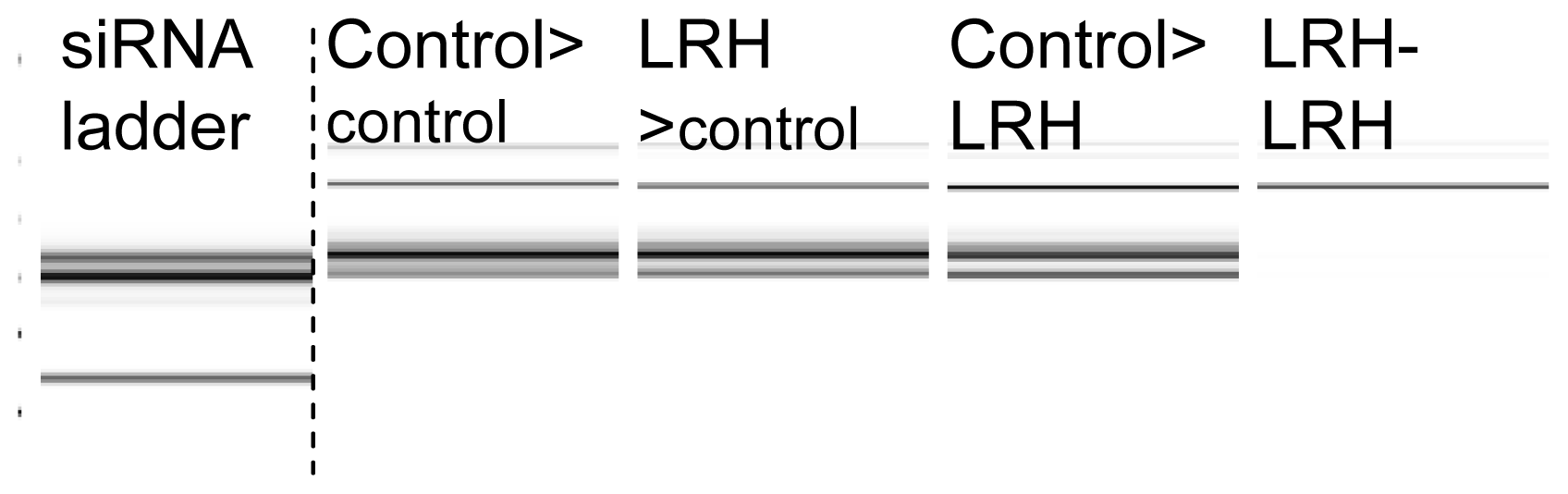
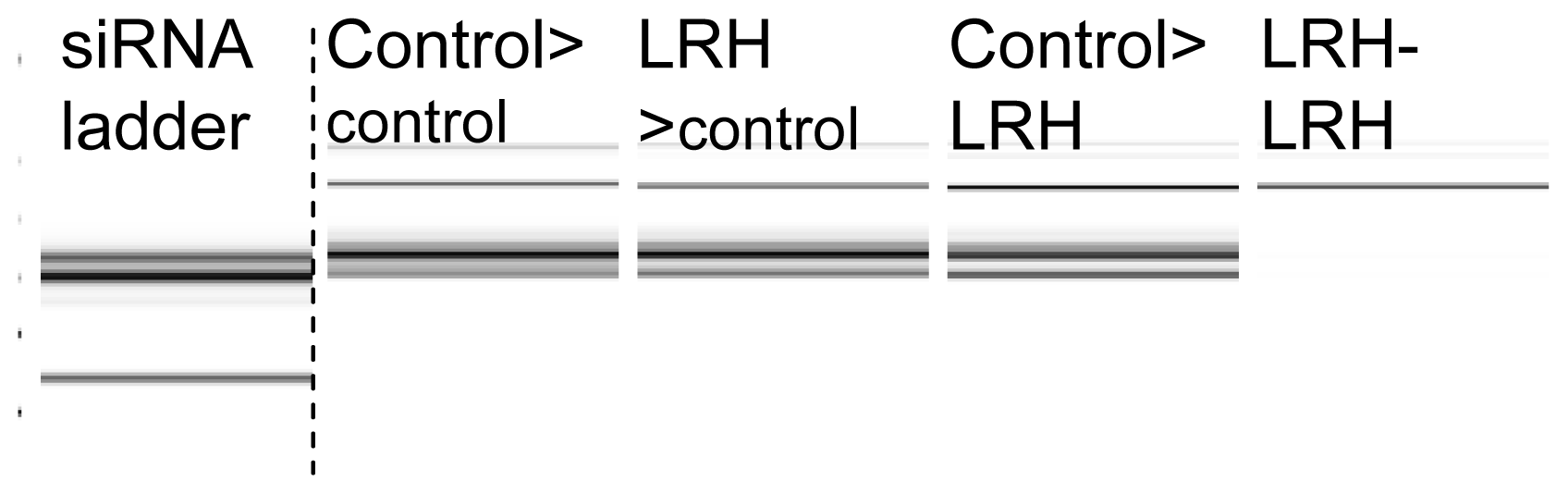
2.2. Gene Expression and the Abundance of siRNAs
2.3. sRNAs Are Lost When the Progeny of Stressed Parents Are Exposed to Repeated Stress
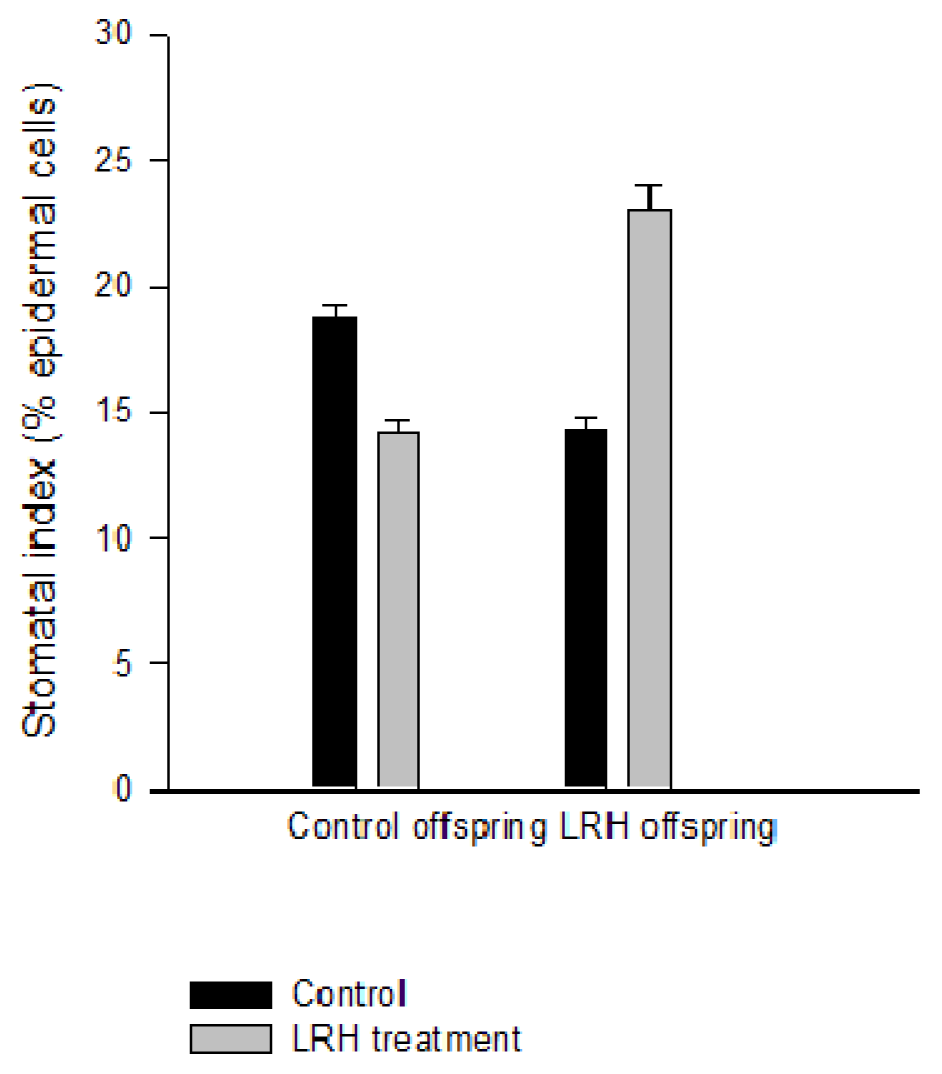
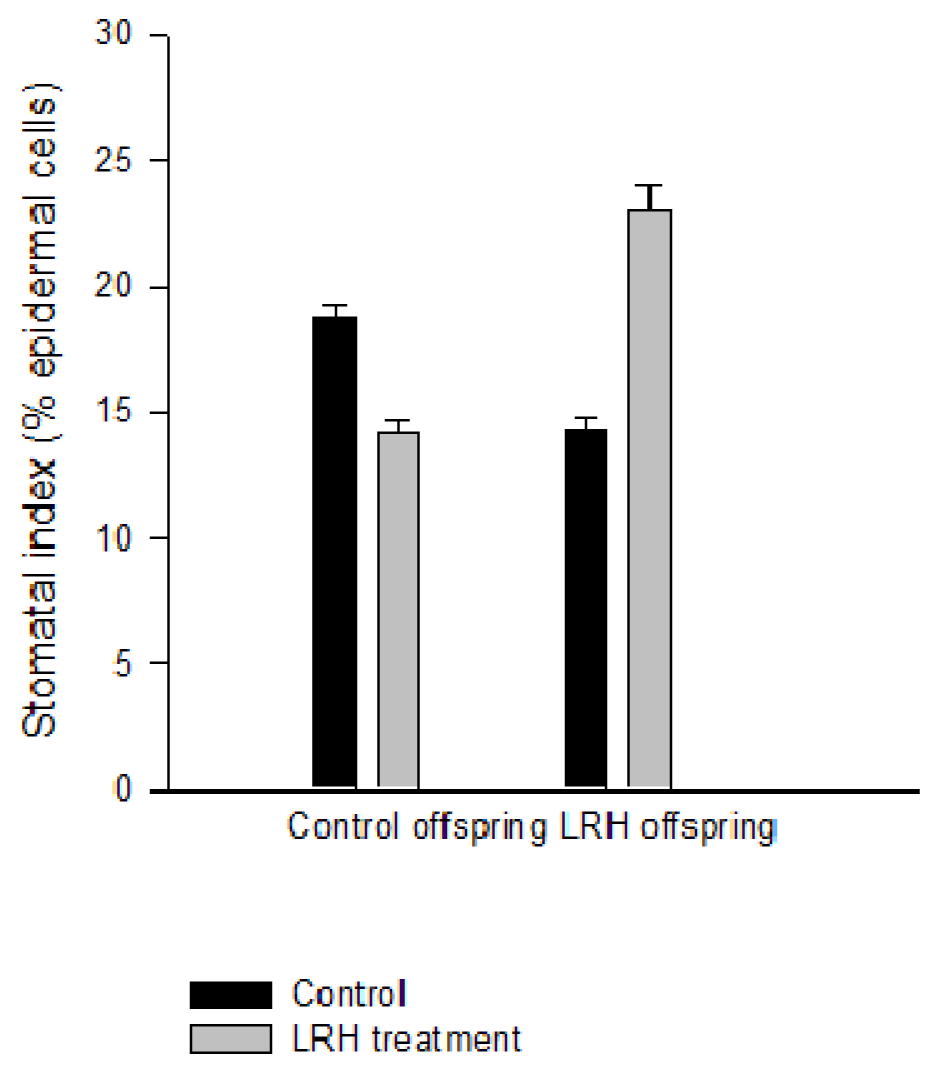
2.4. Stomatal Phenotype under LRH Stress Is Heritable
3. Discussion
4. Experimental Section
4.1. Plants and Growth Environment
4.2. DNA Methylation Analyses
4.3. RNA Analyses
4.4. Primer and Probe Designs
4.5. Stomatal Analyses
4.6. Statistical Analyses
5. Conclusions
Supplementary Information
ijms-14-06674-s001.docxAcknowledgments
Conflict of Interest
References
- Karl, T.R.; Trenberth, K.E. Modern global climate change. Science 2003, 302, 1719–1723. [Google Scholar]
- Seneviratne, S.I.; Lüthi, D.; Litschi, M.; Schär, C. Land-atmosphere coupling and climate change in Europe. Nature 2006, 443, 205–209. [Google Scholar]
- Scoville, A.G.; Barnett, L.L.; Bodbyl-Roels, S.; Kelly, J.K.; Hileman, L.C. Differential regulation of a MYB transcription factor is correlated with transgenerational epigenetic inheritance of trichome density in Mimulus guttatus. New Phytol 2011, 191, 251–263. [Google Scholar]
- Boyko, A.; Kovalchuk, I. Genome instability and epigenetic modification—Heritable responses to environmental stress? Curr. Opin. Plant Biol 2011, 14, 260–266. [Google Scholar]
- Johnsen, Ø.; Dæhlen, O.G.; Østreng, G.; Skrøppa, T. Daylength and temperature during seed production interactively affect adaptive performance of Picea abies progenies. New Phytol. 2005, 168, 589–596. [Google Scholar]
- Blödner, C.; Goebel, C.; Feussner, I.; Gatz, C.; Polle, A. Warm and cold parental reproductive environments affect seed properties, fitness and cold responsiveness in Arabidopsis thaliana progenies. Plant Cell Environ 2007, 30, 165–175. [Google Scholar]
- Galloway, L.F.; Etterson, J.R. Transgenerational plasticity is adaptive in the wild. Science 2007, 318, 1134–1136. [Google Scholar]
- Daxinger, L.; Whitelaw, E. Transgenerational epigenetic inheritance: More questions than answers. Genome Res 2010, 20, 1623–1628. [Google Scholar]
- Molinier, J.; Ries, G.; Zipfel, C.; Hohn, B. Transgeneration memory of stress in plants. Nature 2006, 442, 1046–1049. [Google Scholar]
- Verhoeven, K.J.F.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol 2010, 185, 1108–1118. [Google Scholar]
- Jullien, P.E.; Berger, F. DNA methylation reprogramming during plant sexual production? Trends Genet 2010, 26, 394–399. [Google Scholar]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdžić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar]
- Pecinka, A.; Rosa, M.; Schikora, A.; Berlinger, M.; Hirt, H.; Luschnig, C.; Scheid, O.M. Transgenerational stress memory is not a general response in Arabidopsis. PLoS One 2009, 4, e5202. [Google Scholar]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins, F., Jr; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS One 2010, 5, e9514. [Google Scholar]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 2009, 5, e1000530. [Google Scholar]
- Mathieu, O.; Reinders, J.; Čaikovski, M.; Smathajitt, C.; Paszkowski, J. Transgenerational stability of the Arabidopsis epigeneome is coordinated by CG methylation. Cell 2007, 130, 851–862. [Google Scholar]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F.; et al. A role for RNAi in the selective correction of DNA methylation defects. Science 2009, 323, 1600–1604. [Google Scholar]
- Soppe, W.J.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J.M. The late flowering phenotype of fwa mutants is caused by gain-of function epigenetic alleles of a homeodomain gene. Mol. Cell 2000, 6, 791–802. [Google Scholar]
- McCue, A.D.; Nuthikattu, S.; Reeder, S.H.; Slotkin, R.K. Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet 2012, 8, e1002474. [Google Scholar]
- Lang-Mladek, C.; Popova, O.; Kiok, K.; Berlinger, M.; Rakic, B.; Aufsatz, W.; Jonak, C.; Hauser, M.-T.; Luschnig, C. Transgenerational inheritance and resetting of stress-induced loss of epigenetic gene silencing in Arabidopsis. Mol. Plant 2010, 3, 594–602. [Google Scholar]
- Pecinka, A.; Dinh, H.Q.; Baubec, T.; Rosa, M.; Lettner, N.; Scheid, O.M. Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 2010, 22, 1–12. [Google Scholar]
- Tittel-Elmer, M.; Bucher, E.; Broger, L.; Mathieu, O.; Paszkowski, J.; Vaillant, I. Stress-induced activation of heterochromatic transcription. PLoS Genet 2010, 6, e1001175. [Google Scholar]
- Tricker, P.J.; Gibbings, J.G.; Rodríguez López, C.M.; Hadley, P.; Wilkinson, M.J. Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot 2012, 63, 3799–3813. [Google Scholar]
- De Block, M.; Van Lijsebettens, M. Energy efficiency and energy homeostasis as genetic and epigenetic components of plant performance and crop productivity. Curr. Opin. Plant Biol 2011, 14, 275–282. [Google Scholar]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol 2011, 14, 267–274. [Google Scholar]
- Reinders, J.; Wulff, B.B.H.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev 2009, 23, 939–950. [Google Scholar]
- Roux, F.; Tatché, M.C.; Edelist, C.; Wardenaar, R.; Guerche, P.; Hospital, F.; Colot, V.; Jansen, R.C.; Johannes, F. Genome-wide epigenetic perturbation jump starts patterns of heritable variation found in nature. Genetics 2011. [Google Scholar] [CrossRef]
- Saze, H.; Scheid, O.M.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar]
- Akimoto, K.; Katakami, H.; Kim, H.-J.; Ogawa, E.; Sano, C.M.; Wada, Y.; Sano, H. Epigenetic inheritance in rice plants. Ann. Bot 2007, 100, 205–217. [Google Scholar]
- Gluckman, P.D.; Hanson, M.A.; Spencer, H.G. Predictive adaptive responses and human evolution. Trends Ecol. Evol 2005, 20, 527–533. [Google Scholar]
- Abrash, E.B.; Bergmann, D.C. Asymmetric cell divisions: A view from plant development. Dev. Cell 2009, 16, 783–796. [Google Scholar]
- Coupe, S.A.; Palmer, B.G.; Lake, J.A.; Overy, S.A.; Oxborough, K.; Woodward, F.I.; Gray, J.E.; Quick, W.P. Systemic signalling of environmental cues in Arabidopsis leaves. J. Exp. Bot 2006, 57, 329–341. [Google Scholar]
- Lake, J.A.; Quick, W.P.; Beerling, D.J.; Woodward, F.I. Signals from mature to new leaves. Nature 2001, 411, 154. [Google Scholar]
- Lake, J.A.; Woodward, F.I. Response of stomatal numbers to CO2 and humidity: Control by transpiration rate and abscisic acid. New Phytol 2008, 179, 397–404. [Google Scholar]
- Hollister, J.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 2009, 19, 1419–1428. [Google Scholar]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Görlach, J. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser 1999, 41, 95–98. [Google Scholar]
- Cohen, J. A power primer. Psychol. Bull 1992, 112, 155–159. [Google Scholar]
- Luna, E.; Ton, J. The epigenetic machinery controlling transgenerational systemic acquired resistance. Plant Signal. Behav 2012, 7, 615–618. [Google Scholar]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methylated bases in 100 bp | SCREAM2 | FAMAa | FAMAb | SPCHa | SPCHb |
|---|---|---|---|---|---|
| LRH (Parent) | 0 | 17 | 14 | 0 | 30.2 |
| LRH-control | 0.1 | 13.5 | 12.9 | 0 | 29.2 |
| Entropy (Hχ) | 0.004 | 0.08 | 0.17 | 0.001 | 0.02 |
| LRH-LRH | 0.13 | 16.6 | 13.5 | 0.03 | 11.4 |
| Entropy (Hχ) | 0.003 | 0.05 | 0.05 | 0.001 | 0.02 |
| Control-control | 0 | 11.5 | 7.2 | 0 | 11.3 |
| Entropy (Hχ) | 0.000 | 0.08 | 0.07 | 0.015 | 0.000 |
| χ2 treatment | ns | <0.001 | <0.001 | ns | <0.001 |
| χ2 inheritance | ns | ns | ns | ns | <0.001 |
| FAMA mRNA | 21–24 nt local siRNAs | SPCH mRNA | 21–24 nt local siRNAs | |
|---|---|---|---|---|
| LRH-control (pg μL−1) | 244 (±52.6) | 378 (±153.9) | 511 (±74.5) | 983 (±108) |
| LRH-LRH (pg μL−1) | 541 (±128) | 203 (±189) | 1732 (±76.4) | 312 (±74.7) |
| Control-control (pg μL−1) | 334 (±54.3) | 394 (±38.1) | 809 (±134) | 665 (±151) |
| Linear regression | FAMAand local siRNAs | SPCHand local siRNAs | ||
| r2 | 0.34 | 0.87 | ||
| p value | 0.875 | 0.013 | ||
| Tukey’s pairwise comparisons | SE difference in means | pvalue | ||
| Target RNA | 104.7 | ns | ||
| Parent | 110.3 | 0.017 | ||
| Treatment | 106.1 | ns | ||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tricker, P.J.; López, C.M.R.; Gibbings, G.; Hadley, P.; Wilkinson, M.J. Transgenerational, Dynamic Methylation of Stomata Genes in Response to Low Relative Humidity. Int. J. Mol. Sci. 2013, 14, 6674-6689. https://doi.org/10.3390/ijms14046674
Tricker PJ, López CMR, Gibbings G, Hadley P, Wilkinson MJ. Transgenerational, Dynamic Methylation of Stomata Genes in Response to Low Relative Humidity. International Journal of Molecular Sciences. 2013; 14(4):6674-6689. https://doi.org/10.3390/ijms14046674
Chicago/Turabian StyleTricker, Penny J., Carlos M. Rodríguez López, George Gibbings, Paul Hadley, and Mike J. Wilkinson. 2013. "Transgenerational, Dynamic Methylation of Stomata Genes in Response to Low Relative Humidity" International Journal of Molecular Sciences 14, no. 4: 6674-6689. https://doi.org/10.3390/ijms14046674