The Molecular Fingerprint of High Grade Serous Ovarian Cancer Reflects Its Fallopian Tube Origin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
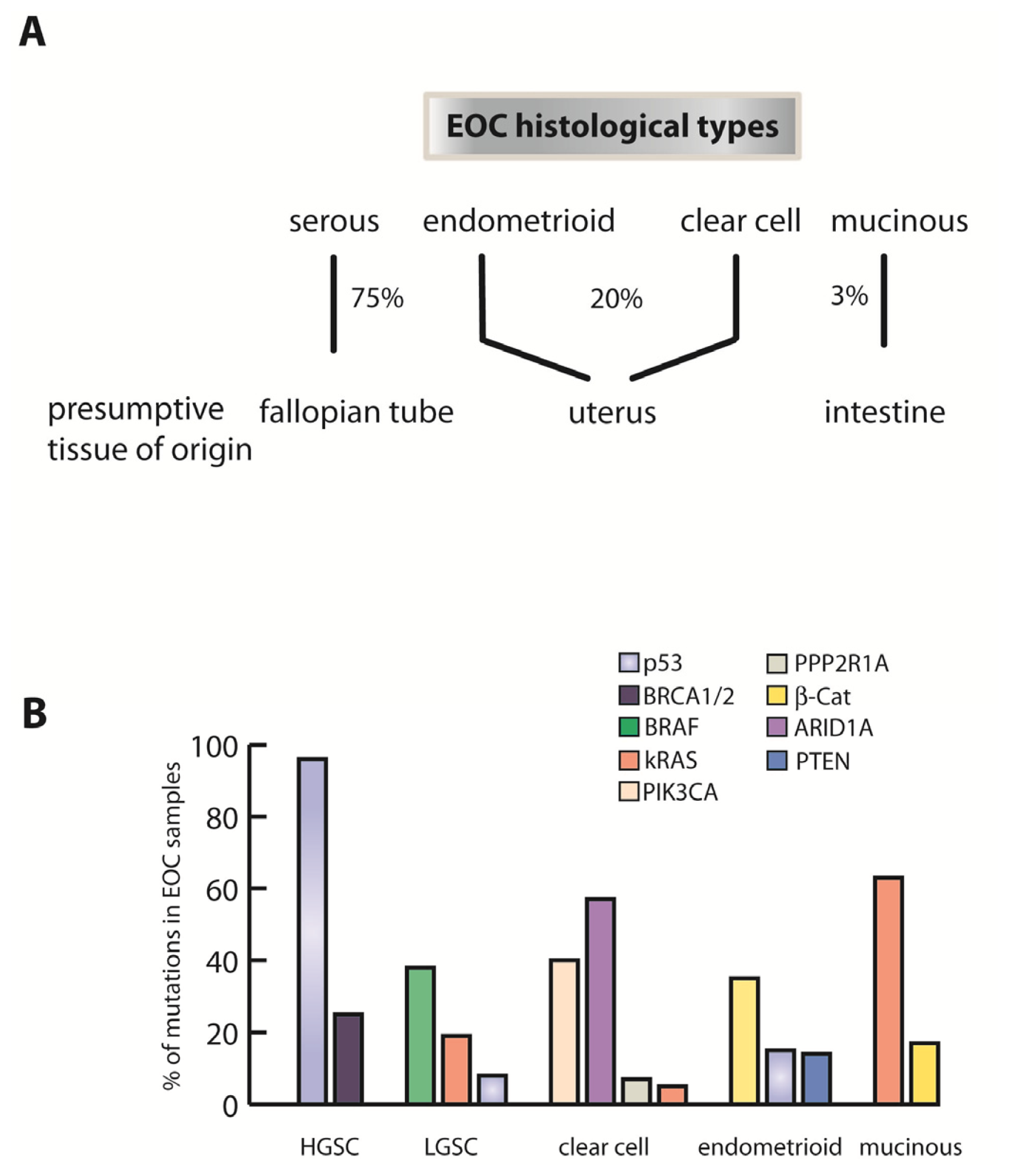
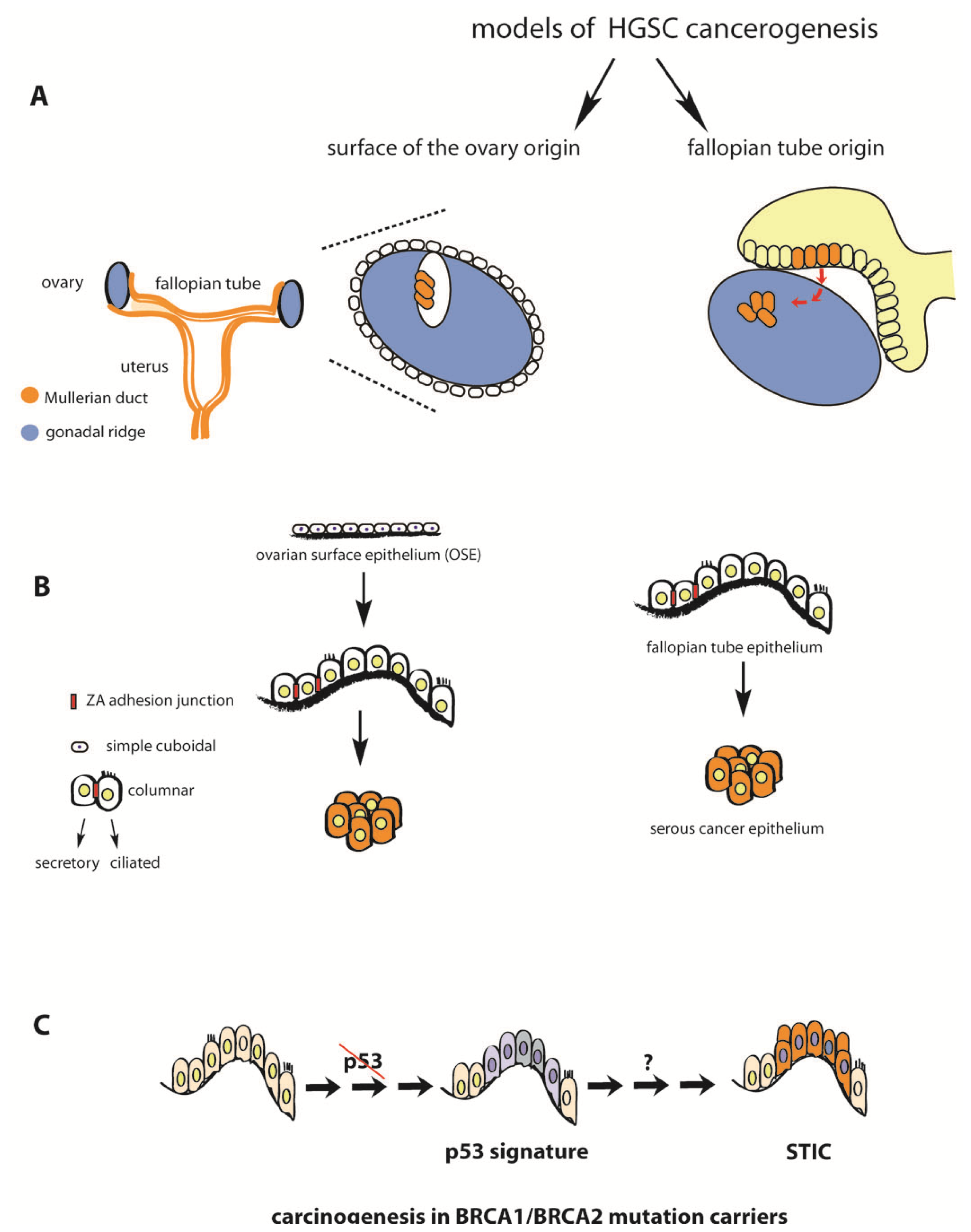
2. Fallopian Tube as Extra-Ovarian Tissue of Origin of Serous Ovarian Cancer
3. Serous Tubal Intraepithelial Carcinoma (STIC) in Asymptomatic BRCA Mutation Carriers and Sporadic HGSC Cases
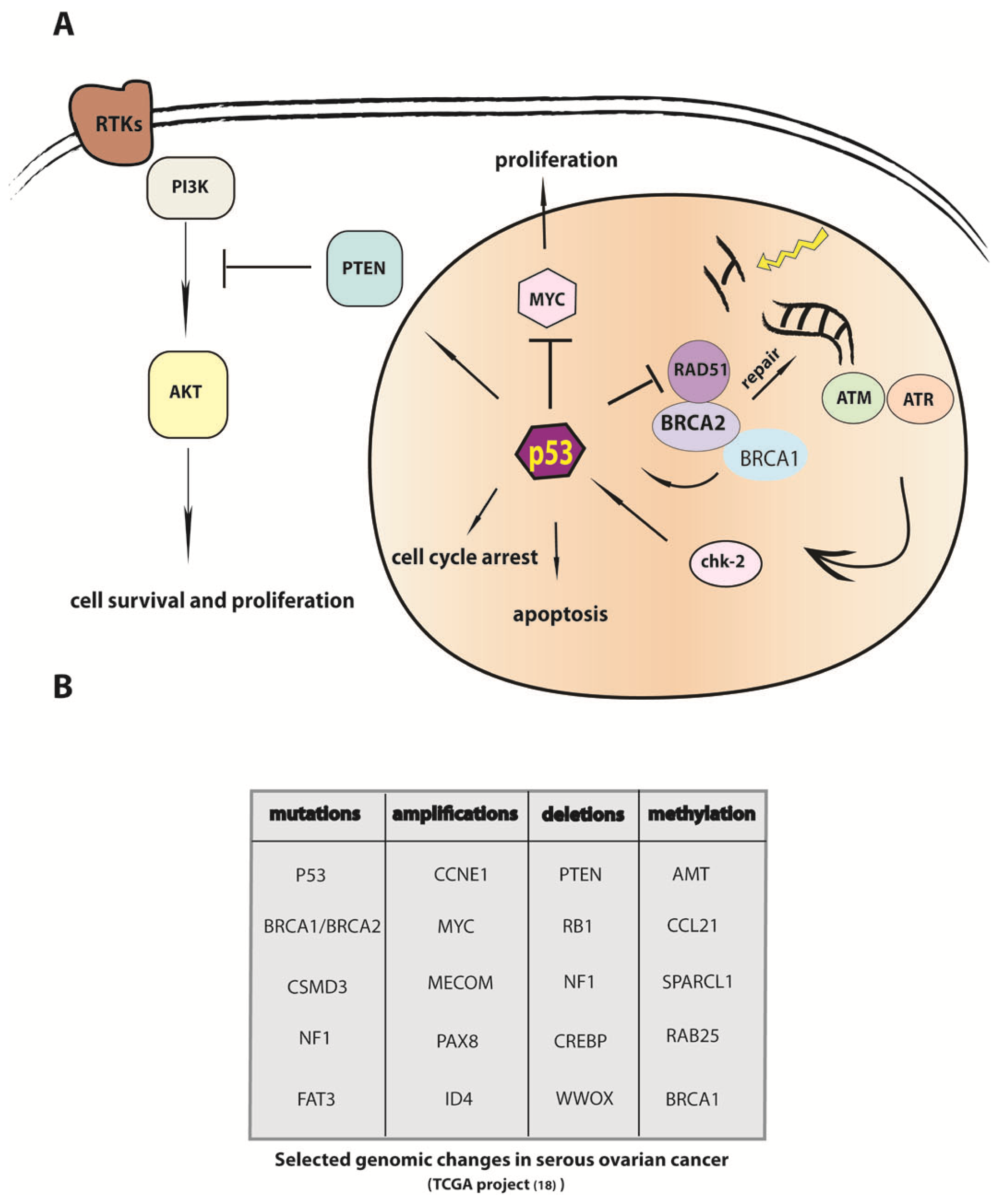
4. P53 Inactivation Is a Necessary but not Sufficient Step in the Etiology of HGSC
5. Modulation of DNA Repair in Carcinogenesis and Mechanisms of Chemoresistance
6. Fallopian Tube Epithelium in BRCA1/2 Mutation Carriers: Searching for the Molecular Fingerprint of Carcinogenesis
7. The Tumorigenic Effects of Oxidative Stress and Inflammation in the Fallopian Tube
8. The Molecular Basis for Malignant Transformation in Fallopian Tube Epithelium: Analysis of Cellular Pathways
9. The Genomic Sequence of Ovarian Carcinoma and Implications for Understanding the Etiology of the Cancer Cell
10. Regulation of Epithelial-Mesenchymal (EMT) Transition and Dissemination of HGSC
11. Notch Paracrine Signaling, Stemness, and Tumor Progression
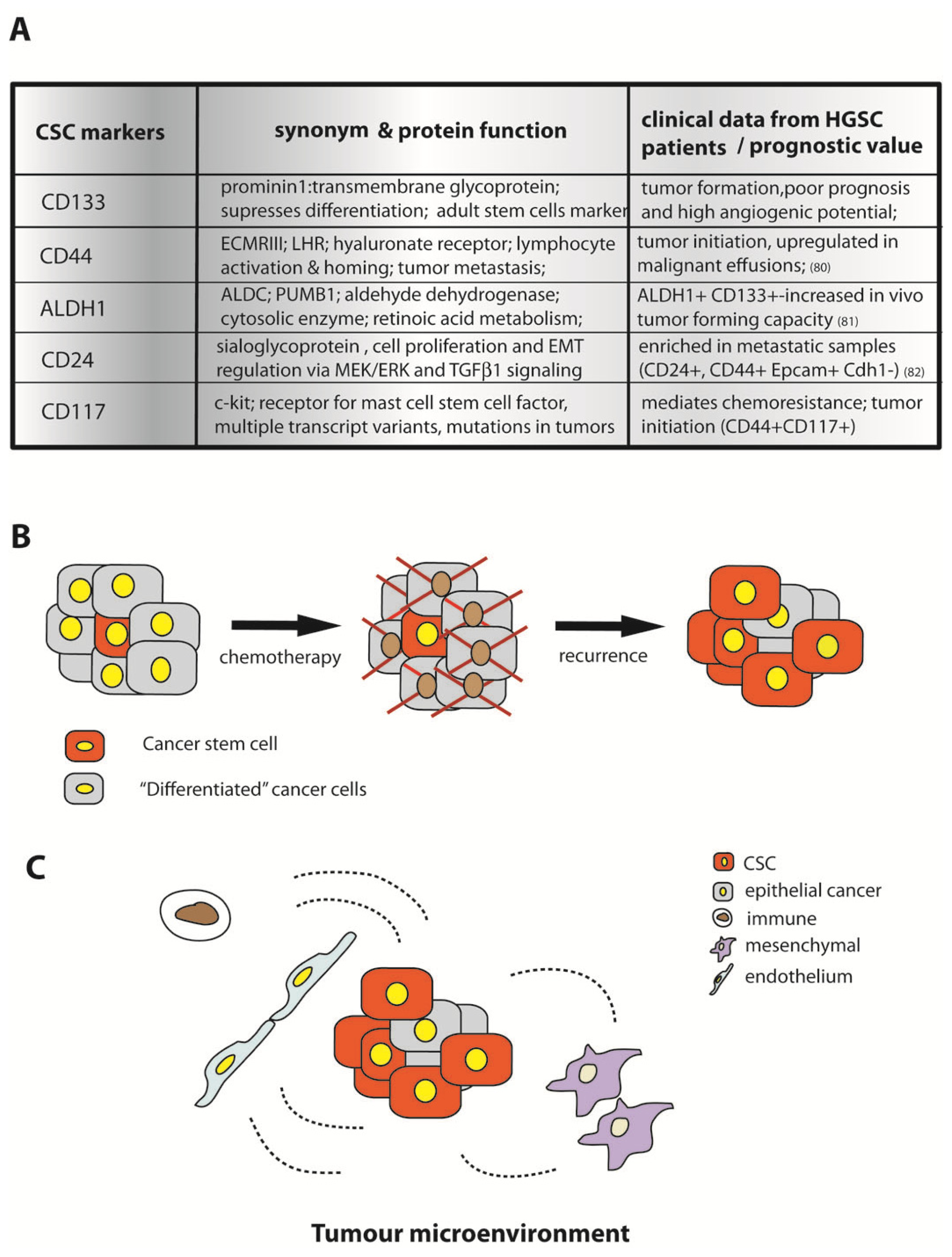
12. Cancer Stem Cells in Proliferation and Survival of HGSC
13. Conclusions
Acknowledgments
Conflict of Interest
References
- Coleman, M.P.; Forman, D.; Bryant, H.; Butler, J.; Rachet, B.; Maringe, C.; Nur, U.; Tracey, E.; Coory, M.; Hatcher, J.; et al. Cancer survival in Australia, Canada, Denmark, Norway, Sweden, and the UK, 1995–2007 (the International Cancer Benchmarking Partnership): An analysis of population-based cancer registry data. Lancet 2011, 377, 127–138. [Google Scholar]
- Baldwin, L.A.; Huang, B.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; DeSimone, C.P.; Ueland, F.R.; van Nagell, J.R.; Seamon, L.G. Ten-year relative survival for epithelial ovarian cancer. Obstet Gynecol 2012, 120, 612–618. [Google Scholar]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar]
- Abe, A.; Minaguchi, T.; Ochi, H.; Onuki, M.; Okada, S.; Matsumoto, K.; Satoh, T.; Oki, A.; Yoshikawa, H. PIK3CA overexpression is a possible prognostic factor for favorable survival in ovarian clear cell carcinoma. Hum. Pathol 2012, 44, 199–207. [Google Scholar]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar]
- Oliva, E.; Sarrio, D.; Brachtel, E.F.; Sanchez-Estevez, C.; Soslow, R.A.; Moreno-Bueno, G.; Palacios, J. High frequency of beta-catenin mutations in borderline endometrioid tumours of the ovary. J. Pathol 2006, 208, 708–713. [Google Scholar]
- Hunter, S.M.; Gorringe, K.L.; Christie, M.; Rowley, S.M.; Bowtell, D.D.; Campbell, I.G. Pre-invasive ovarian mucinous tumors are characterized by CDKN2A and RAS pathway aberrations. Clin. Cancer Res 2012, 18, 5267–5277. [Google Scholar]
- Shih Ie, M.; Panuganti, P.K.; Kuo, K.T.; Mao, T.L.; Kuhn, E.; Jones, S.; Velculescu, V.E.; Kurman, R.J.; Wang, T.L. Somatic mutations of PPP2R1A in ovarian and uterine carcinomas. Am. J. Pathol 2011, 178, 1442–1447. [Google Scholar]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet 2001, 68, 700–710. [Google Scholar]
- Tone, A.A.; Virtanen, C.; Shaw, P.; Brown, T.J. Prolonged postovulatory proinflammatory signaling in the fallopian tube epithelium may be mediated through a BRCA1/DAB2 axis. Clin. Cancer Res 2012, 18, 4334–4344. [Google Scholar]
- Cuatrecasas, M.; Villanueva, A.; Matias-Guiu, X.; Prat, J. K-ras mutations in mucinous ovarian tumors: A clinicopathologic and molecular study of 95 cases. Cancer 1997, 79, 1581–1586. [Google Scholar]
- Marquez, R.T.; Baggerly, K.A.; Patterson, A.P.; Liu, J.; Broaddus, R.; Frumovitz, M.; Atkinson, E.N.; Smith, D.I.; Hartmann, L.; Fishman, D.; et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin. Cancer Res 2005, 11, 6116–6126. [Google Scholar]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar]
- Mittag, J.; Winterhager, E.; Bauer, K.; Grummer, R. Congenital hypothyroid female pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology 2007, 148, 719–725. [Google Scholar]
- Bowen, N.J.; Logani, S.; Dickerson, E.B.; Kapa, L.B.; Akhtar, M.; Benigno, B.B.; McDonald, J.F. Emerging roles for PAX8 in ovarian cancer and endosalpingeal development. Gynecol. Oncol 2007, 104, 331–337. [Google Scholar]
- Sundfeldt, K.; Piontkewitz, Y.; Ivarsson, K.; Nilsson, O.; Hellberg, P.; Brannstrom, M.; Janson, P.O.; Enerback, S.; Hedin, L. E-cadherin expression in human epithelial ovarian cancer and normal ovary. Int. J. Cancer 1997, 74, 275–280. [Google Scholar]
- Kabawat, S.E.; Bast, R.C., Jr; Bhan, A.K.; Welch, W.R.; Knapp, R.C.; Colvin, R.B. Tissue distribution of a coelomic-epithelium-related antigen recognized by the monoclonal antibody OC125. Int. J. Gynecol. Pathol. 1983, 2, 275–285. [Google Scholar]
- Li, J.; Abushahin, N.; Pang, S.; Xiang, L.; Chambers, S.K.; Fadare, O.; Kong, B.; Zheng, W. Tubal origin of “ovarian” low-grade serous carcinoma. Mod. Pathol 2011, 24, 1488–1499. [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.[Green Version]
- Powell, C.B.; Chen, L.M.; McLennan, J.; Crawford, B.; Zaloudek, C.; Rabban, J.T.; Moore, D.H.; Ziegler, J. Risk-reducing salpingo-oophorectomy (RRSO) in BRCA mutation carriers: Experience with a consecutive series of 111 patients using a standardized surgical-pathological protocol. Int. J. Gynecol. Cancer 2011, 21, 846–851. [Google Scholar]
- Mingels, M.J.; Roelofsen, T.; van der Laak, J.A.; de Hullu, J.A.; van Ham, M.A.; Massuger, L.F.; Bulten, J.; Bol, M. Tubal epithelial lesions in salpingo-oophorectomy specimens of BRCA-mutation carriers and controls. Gynecol. Oncol 2012, 127, 88–93. [Google Scholar]
- Callahan, M.J.; Crum, C.P.; Medeiros, F.; Kindelberger, D.W.; Elvin, J.A.; Garber, J.E.; Feltmate, C.M.; Berkowitz, R.S.; Muto, M.G. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J. Clin. Oncol 2007, 25, 3985–3990. [Google Scholar]
- Olivier, R.I.; van Beurden, M.; Lubsen, M.A.; Rookus, M.A.; Mooij, T.M.; van de Vijver, M.J.; van’t Veer, L.J. Clinical outcome of prophylactic oophorectomy in BRCA1/BRCA2 mutation carriers and events during follow-up. Br. J. Cancer 2004, 90, 1492–1497. [Google Scholar]
- Finch, A.; Beiner, M.; Lubinski, J.; Lynch, H.T.; Moller, P.; Rosen, B.; Murphy, J.; Ghadirian, P.; Friedman, E.; Foulkes, W.D.; et al. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 Mutation. J. Am. Med. Assoc 2006, 296, 185–192. [Google Scholar]
- Leeper, K.; Garcia, R.; Swisher, E.; Goff, B.; Greer, B.; Paley, P. Pathologic findings in prophylactic oophorectomy specimens in high-risk women. Gynecol. Oncol 2002, 87, 52–56. [Google Scholar]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.-M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma–evidence supporting the clonal relationship of the two lesions. J. Pathol 2012, 226, 421–426. [Google Scholar]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih Ie, M.; Vang, R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am. J. Surg. Pathol 2010, 34, 1407–1416. [Google Scholar]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol 2007, 31, 161–169. [Google Scholar]
- Carlson, J.W.; Miron, A.; Jarboe, E.A.; Parast, M.M.; Hirsch, M.S.; Lee, Y.; Muto, M.G.; Kindelberger, D.; Crum, C.P. Serous tubal intraepithelial carcinoma: Its potential role in primary peritoneal serous carcinoma and serous cancer prevention. J. Clin. Oncol 2008, 26, 4160–4165. [Google Scholar]
- Pothuri, B.; Leitao, M.M.; Levine, D.A.; Viale, A.; Olshen, A.B.; Arroyo, C.; Bogomolniy, F.; Olvera, N.; Lin, O.; Soslow, R.A.; et al. Genetic analysis of the early natural history of epithelial ovarian carcinoma. PLoS One 2010, 5, e10358. [Google Scholar]
- Gilbert, L.; Basso, O.; Sampalis, J.; Karp, I.; Martins, C.; Feng, J.; Piedimonte, S.; Quintal, L.; Ramanakumar, A.V.; Takefman, J.; et al. Assessment of symptomatic women for early diagnosis of ovarian cancer: Results from the prospective DOvE pilot project. Lancet Oncol 2012, 13, 285–291. [Google Scholar]
- Piek, J.M.; van Diest, P.J.; Zweemer, R.P.; Jansen, J.W.; Poort-Keesom, R.J.; Menko, F.H.; Gille, J.J.; Jongsma, A.P.; Pals, G.; Kenemans, P.; et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol 2001, 195, 451–456. [Google Scholar]
- Bernardini, M.Q.; Baba, T.; Lee, P.S.; Barnett, J.C.; Sfakianos, G.P.; Secord, A.A.; Murphy, S.K.; Iversen, E.; Marks, J.R.; Berchuck, A. Expression signatures of TP53 mutations in serous ovarian cancers. BMC Cancer 2010, 10, 237, :1–237:10.. [Google Scholar]
- Jimenez, G.S.; Khan, S.H.; Stommel, J.M.; Wahl, G.M. p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 1999, 18, 7656–7665. [Google Scholar]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol 2007, 211, 26–35. [Google Scholar]
- Xian, W.; Miron, A.; Roh, M.; Semmel, D.R.; Yassin, Y.; Garber, J.; Oliva, E.; Goodman, A.; Mehra, K.; Berkowitz, R.S.; et al. The Li-Fraumeni syndrome (LFS): A model for the initiation of p53 signatures in the distal Fallopian tube. J. Pathol 2010, 220, 17–23. [Google Scholar]
- Van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet 2001, 2, 196–206. [Google Scholar]
- Richardson, C.; Stark, J.M.; Ommundsen, M.; Jasin, M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004, 23, 546–553. [Google Scholar]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep 2006, 7, 219–224. [Google Scholar] [Green Version]
- Ludwig, T.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Targeted mutations of breast cancer susceptibility gene homologs in mice: Lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 1997, 11, 1226–1241. [Google Scholar]
- Hakem, R.; de la Pompa, J.L.; Sirard, C.; Mo, R.; Woo, M.; Hakem, A.; Wakeham, A.; Potter, J.; Reitmair, A.; Billia, F.; et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996, 85, 1009–1023. [Google Scholar]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. J. Am. Med. Assoc 2012, 307, 382–390. [Google Scholar]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: Sensitivity to PARP inhibitors, platinum, and survival. Cancer Res 2012, 72, 5675–5682. [Google Scholar]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol 2011, 29, 3008–3015. [Google Scholar]
- George, S.H.; Greenaway, J.; Milea, A.; Clary, V.; Shaw, S.; Sharma, M.; Virtanen, C.; Shaw, P.A. Identification of abrogated pathways in fallopian tube epithelium from BRCA1 mutation carriers. J. Pathol 2011, 225, 106–117. [Google Scholar]
- Tsilidis, K.K.; Allen, N.E.; Key, T.J.; Dossus, L.; Lukanova, A.; Bakken, K.; Lund, E.; Fournier, A.; Overvad, K.; Hansen, L.; et al. Oral contraceptive use and reproductive factors and risk of ovarian cancer in the European Prospective Investigation into Cancer and Nutrition. Br. J. Cancer 2011, 105, 1436–1442. [Google Scholar]
- King, S.M.; Hilliard, T.S.; Wu, L.Y.; Jaffe, R.C.; Fazleabas, A.T.; Burdette, J.E. The impact of ovulation on fallopian tube epithelial cells: Evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr. Relat. Cancer 2011, 18, 627–642. [Google Scholar]
- Lin, H.W.; Tu, Y.Y.; Lin, S.Y.; Su, W.J.; Lin, W.L.; Lin, W.Z.; Wu, S.C.; Lai, Y.L. Risk of ovarian cancer in women with pelvic inflammatory disease: A population-based study. Lancet Oncol 2011, 12, 900–904. [Google Scholar]
- Haggerty, C.L.; Gottlieb, S.L.; Taylor, B.D.; Low, N.; Xu, F.; Ness, R.B. Risk of sequelae after Chlamydia trachomatis genital infection in women. J. Infect. Dis 2010, 201, S134–S155. [Google Scholar]
- Piura, B.; Sarov, I.; Sarov, B.; Kleinman, D.; Chaim, W.; Insler, V. Serum IgG and IgA antibodies specific for Chlamydia trachomatis in salpingitis patients as determined by the immunoperoxidase assay. Eur. J. Epidemiol 1985, 1, 110–116. [Google Scholar]
- Cooper, M.D.; Rapp, J.; Jeffery-Wiseman, C.; Barnes, R.C.; Stephens, D.S. Chlamydia trachomatis infection of human fallopian tube organ cultures. J. Gen. Microbiol 1990, 136, 1109–1115. [Google Scholar]
- Hvid, M.; Baczynska, A.; Deleuran, B.; Fedder, J.; Knudsen, H.J.; Christiansen, G.; Birkelund, S. Interleukin-1 is the initiator of Fallopian tube destruction during Chlamydia trachomatis infection. Cell Microbiol 2007, 9, 2795–2803. [Google Scholar]
- Shaw, J.L.; Wills, G.S.; Lee, K.F.; Horner, P.J.; McClure, M.O.; Abrahams, V.M.; Wheelhouse, N.; Jabbour, H.N.; Critchley, H.O.; Entrican, G.; et al. Chlamydia trachomatis infection increases fallopian tube PROKR2 via TLR2 and NFkappaB activation resulting in a microenvironment predisposed to ectopic pregnancy. Am. J. Pathol 2011, 178, 253–260. [Google Scholar]
- Reddy, B.S.; Rastogi, S.; Das, B.; Salhan, S.; Verma, S.; Mittal, A. Cytokine expression pattern in the genital tract of Chlamydia trachomatis positive infertile women-implication for T-cell responses. Clin. Exp. Immunol 2004, 137, 552–558. [Google Scholar]
- Kessler, M.; Zielecki, J.; Thieck, O.; Mollenkopf, H.J.; Fotopoulou, C.; Meyer, T.F. Chlamydia trachomatis disturbs epithelial tissue homeostasis in fallopian tubes via paracrine Wnt signaling. Am. J. Pathol 2012, 180, 186–198. [Google Scholar]
- Yu, H.; Schwarzer, K.; Forster, M.; Kniemeyer, O.; Forsbach-Birk, V.; Straube, E.; Rodel, J. Role of high-mobility group box 1 protein and poly(ADP-ribose) polymerase 1 degradation in Chlamydia trachomatis-induced cytopathicity. Infect. Immun 2010, 78, 3288–3297. [Google Scholar]
- Johnson, K.A.; Tan, M.; Sutterlin, C. Centrosome abnormalities during a Chlamydia trachomatis infection are caused by dysregulation of the normal duplication pathway. Cell Microbiol 2009, 11, 1064–1073. [Google Scholar]
- Sharma, M.; Machuy, N.; Bohme, L.; Karunakaran, K.; Maurer, A.P.; Meyer, T.F.; Rudel, T. HIF-1alpha is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell Microbiol 2011, 13, 1573–1385. [Google Scholar]
- Ness, R.B.; Goodman, M.T.; Shen, C.; Brunham, R.C. Serologic evidence of past infection with Chlamydia trachomatis, in relation to ovarian cancer. J. Infect. Dis 2003, 187, 1147–1152. [Google Scholar]
- Ness, R.B.; Shen, C.; Bass, D.; Jackson, C.; Moysich, K.; Edwards, R.; Brunham, R.C. Chlamydia trachomatis serology in women with and without ovarian cancer. Infect. Dis. Obstet. Gynecol 2008, 2008, 219672, :1–219672:5.. [Google Scholar]
- Idahl, A.; Lundin, E.; Jurstrand, M.; Kumlin, U.; Elgh, F.; Ohlson, N.; Ottander, U. Chlamydia trachomatis and Mycoplasma genitalium plasma antibodies in relation to epithelial ovarian tumors. Infect. Dis. Obstet. Gynecol 2011, 2011, 824627, :1–824627:10.. [Google Scholar]
- Collet, T.; Macnaughton, T.; Walsh, T.; Debattista, J.; Timms, P. Identification of novel markers for uncomplicated lower genital tract infections and upper genital tract pathology due to Chlamydia trachomatis. Int. J. Infect. Dis 2011, 15, e257–e266. [Google Scholar]
- Jazaeri, A.A.; Bryant, J.L.; Park, H.; Li, H.; Dahiya, N.; Stoler, M.H.; Ferriss, J.S.; Dutta, A. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia 2011, 13, 899–911. [Google Scholar]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar]
- Kim, J.; Coffey, D.M.; Creighton, C.J.; Yu, Z.; Hawkins, S.M.; Matzuk, M.M. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 3921–3926. [Google Scholar]
- Sangha, N.; Wu, R.; Kuick, R.; Powers, S.; Mu, D.; Fiander, D.; Yuen, K.; Katabuchi, H.; Tashiro, H.; Fearon, E.R.; Cho, K.R. Neurofibromin 1 (NF1) defects are common in human ovarian serous carcinomas and co-occur with TP53 mutations. Neoplasia 2008, 10, 1362–1372. [Google Scholar]
- Ramus, S.J.; Vierkant, R.A.; Johnatty, S.E.; Pike, M.C.; van Den Berg, D.J.; Wu, A.H.; Pearce, C.L.; Menon, U.; Gentry-Maharaj, A.; Gayther, S.A.; et al. Consortium analysis of 7 candidate SNPs for ovarian cancer. Int. J. Cancer 2008, 123, 380–388. [Google Scholar]
- Baker, V.V.; Borst, M.P.; Dixon, D.; Hatch, K.D.; Shingleton, H.M.; Miller, D. c-myc amplification in ovarian cancer. Gynecol. Oncol 1990, 38, 340–342. [Google Scholar]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010, 116, 2621–2634. [Google Scholar]
- Kurose, K.; Zhou, X.P.; Araki, T.; Cannistra, S.A.; Maher, E.R.; Eng, C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am. J. Pathol 2001, 158, 2097–2106. [Google Scholar]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar]
- Teh, M.T.; Wong, S.T.; Neill, G.W.; Ghali, L.R.; Philpott, M.P.; Quinn, A.G. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res 2002, 62, 4773–4780. [Google Scholar]
- Lok, G.T.; Chan, D.W.; Liu, V.W.; Hui, W.W.; Leung, T.H.; Yao, K.M.; Ngan, H.Y. Aberrant activation of ERK/FOXM1 signaling cascade triggers the cell migration/invasion in ovarian cancer cells. PLoS One 2011, 6, e23790. [Google Scholar]
- Gusarova, G.A.; Wang, I.C.; Major, M.L.; Kalinichenko, V.V.; Ackerson, T.; Petrovic, V.; Costa, R.H. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J. Clin. Invest 2007, 117, 99–111. [Google Scholar]
- Cheng, J.C.; Chang, H.M.; Leung, P.C. Egr-1 mediates epidermal growth factor-induced downregulation of E-cadherin expression via Slug in human ovarian cancer cells. Oncogene 2012. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91, :1–91:10.. [Google Scholar]
- Rosano, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res 2011, 17, 2350–2360. [Google Scholar]
- Lee, M.; Nam, E.J.; Kim, S.W.; Kim, S.; Kim, J.H.; Kim, Y.T. Prognostic impact of the cancer stem cell-related marker NANOG in ovarian serous carcinoma. Int. J. Gynecol. Cancer 2012, 22, 1489–1496. [Google Scholar]
- Siu, M.K.; Wong, E.S.; Kong, D.S.; Chan, H.Y.; Jiang, L.; Wong, O.G.; Lam, E.W.; Chan, K.K.; Ngan, H.Y.; Le, X.F.; et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene 2012. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.X.; Xu, M.; Tan, L.; Yang, H.; Permuth-Wey, J.; Kruk, P.A.; Wenham, R.M.; Nicosia, S.V.; Lancaster, J.M.; Sellers, T.A.; et al. MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog. J. Biol. Chem 2012, 287, 34970–34978. [Google Scholar]
- Jaks, V.; Barker, N.; Kasper, M.; van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgard, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet 2008, 40, 1291–1299. [Google Scholar]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar]
- Paik, D.Y.; Janzen, D.M.; Schafenacker, A.M.; Velasco, V.S.; Shung, M.S.; Cheng, D.; Huang, J.; Witte, O.N.; Memarzadeh, S. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: A site for injury and serous cancer initiation. Stem Cells 2012, 30, 2487–2497. [Google Scholar]
- Park, J.T.; Chen, X.; Trope, C.G.; Davidson, B.; Shih Ie, M.; Wang, T.L. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am. J. Pathol 2010, 177, 1087–1094. [Google Scholar]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68, 4311–4320. [Google Scholar]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res 2011, 71, 3991–4001. [Google Scholar]
- Meirelles, K.; Benedict, L.A.; Dombkowski, D.; Pepin, D.; Preffer, F.I.; Teixeira, J.; Tanwar, P.S.; Young, R.H.; MacLaughlin, D.T.; Donahoe, P.K.; et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc. Natl. Acad. Sci. USA 2012, 109, 2358–2363. [Google Scholar]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar]
- Long, H.; Xie, R.; Xiang, T.; Zhao, Z.; Lin, S.; Liang, Z.; Chen, Z.; Zhu, B. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-kappaB-mediated MMP-9 upregulation. Stem Cells 2012, 30, 2309–2319. [Google Scholar]
- Coffman, L.; Mooney, C.; Lim, J.; Bai, S.; Silva, I.; Gong, Y.; Yang, K.; Buckanovich, R.J. Endothelin receptor-A is required for the recruitment of anti-tumor T cells and modulates chemotherapy induction of cancer stem cells. Cancer Biol. Ther 2012, 14, 184–192. [Google Scholar]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar]



© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kessler, M.; Fotopoulou, C.; Meyer, T. The Molecular Fingerprint of High Grade Serous Ovarian Cancer Reflects Its Fallopian Tube Origin. Int. J. Mol. Sci. 2013, 14, 6571-6596. https://doi.org/10.3390/ijms14046571
Kessler M, Fotopoulou C, Meyer T. The Molecular Fingerprint of High Grade Serous Ovarian Cancer Reflects Its Fallopian Tube Origin. International Journal of Molecular Sciences. 2013; 14(4):6571-6596. https://doi.org/10.3390/ijms14046571
Chicago/Turabian StyleKessler, Mirjana, Christina Fotopoulou, and Thomas Meyer. 2013. "The Molecular Fingerprint of High Grade Serous Ovarian Cancer Reflects Its Fallopian Tube Origin" International Journal of Molecular Sciences 14, no. 4: 6571-6596. https://doi.org/10.3390/ijms14046571