Molecular Analysis of RNA-RNA Interactions between 5’ and 3’ Untranslated Regions during the Initiation of Translation of a Cardiovirulent and a Live-Attenuated Coxsackievirus B3 Strains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
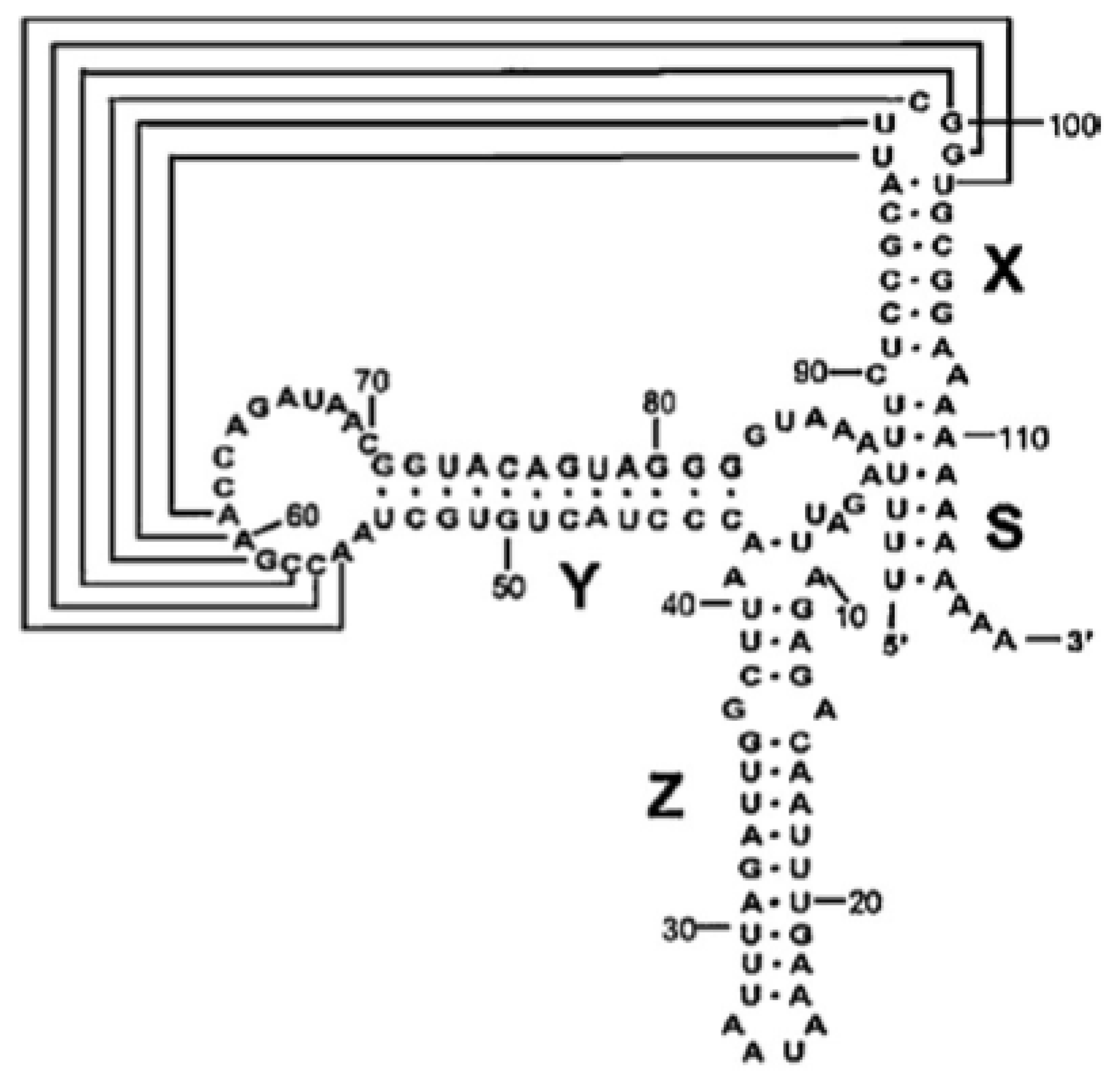
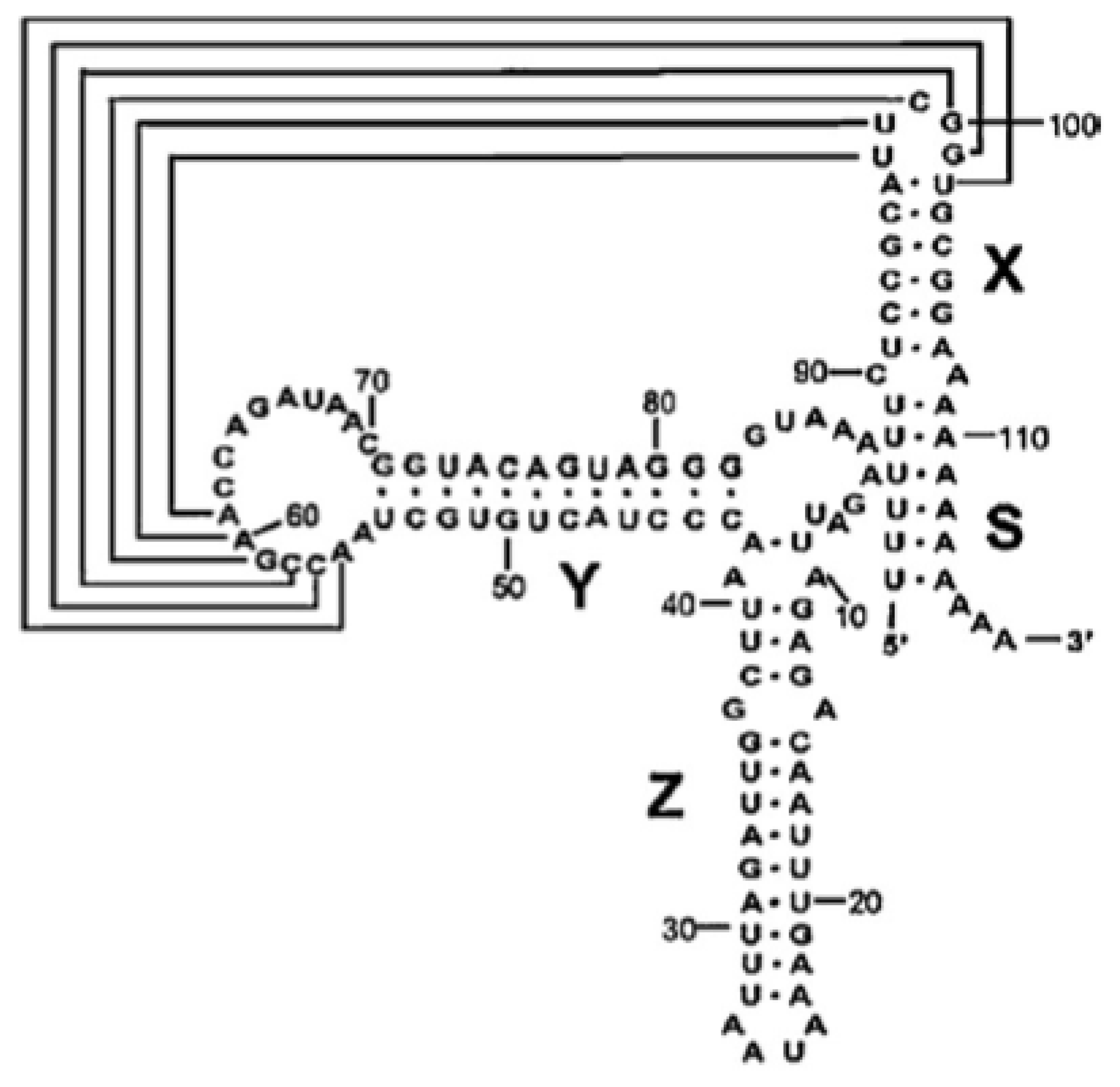
2.1. Cloning of the CVB3 5′ and 3′ Untranslated Regions
2.2. In Vitro Transcription
2.3. RNA and Fluorescent Labeling
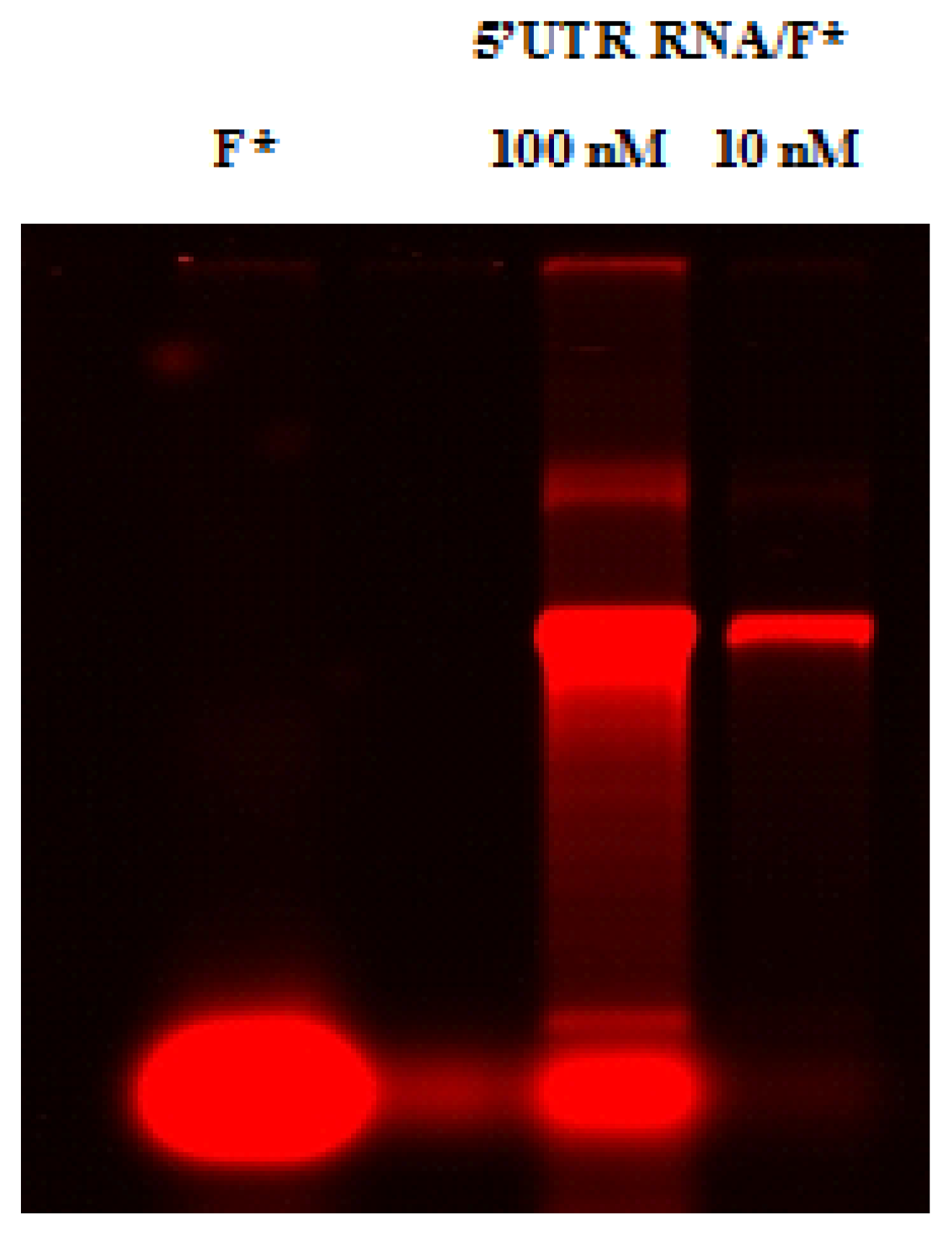
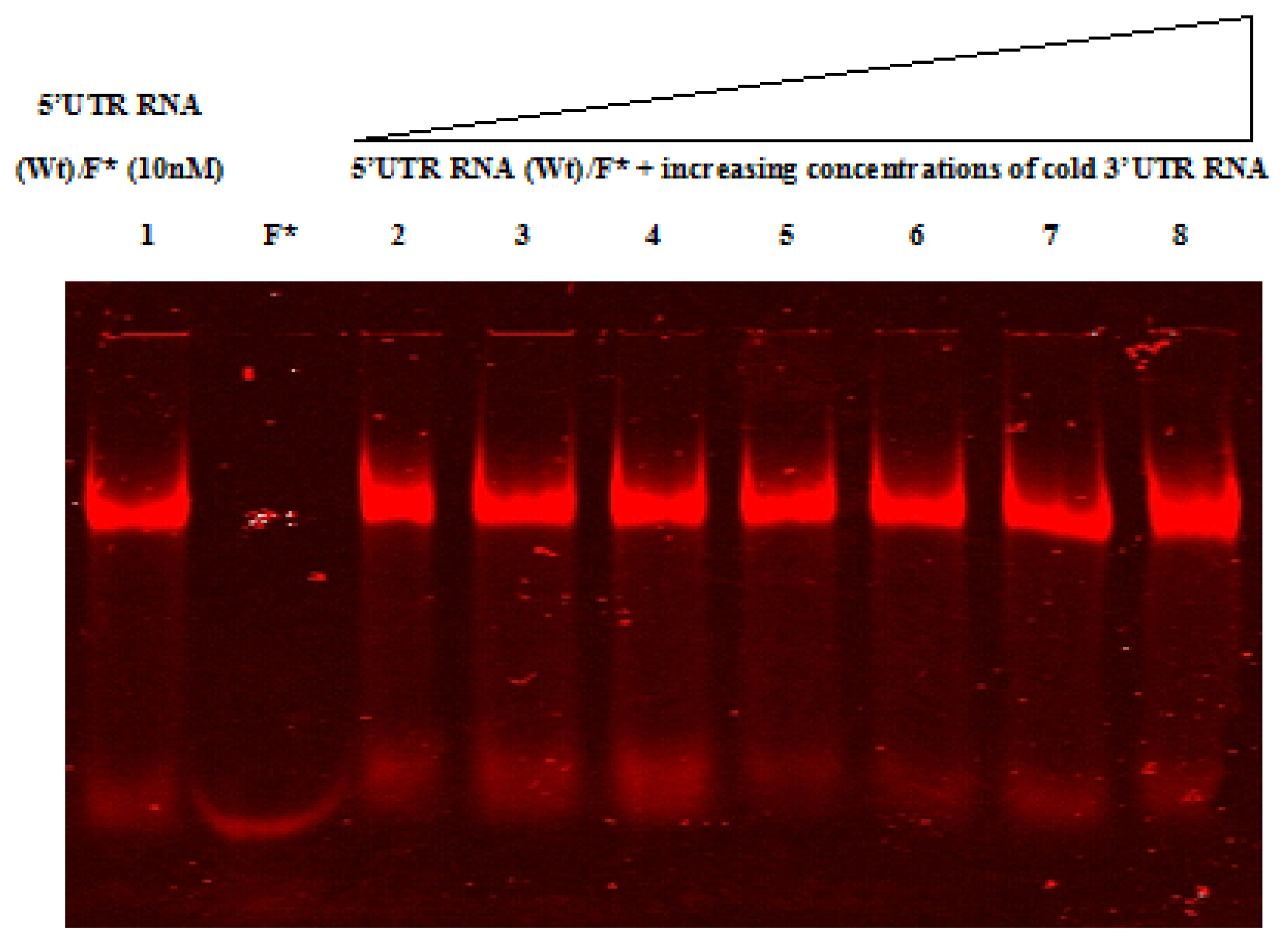
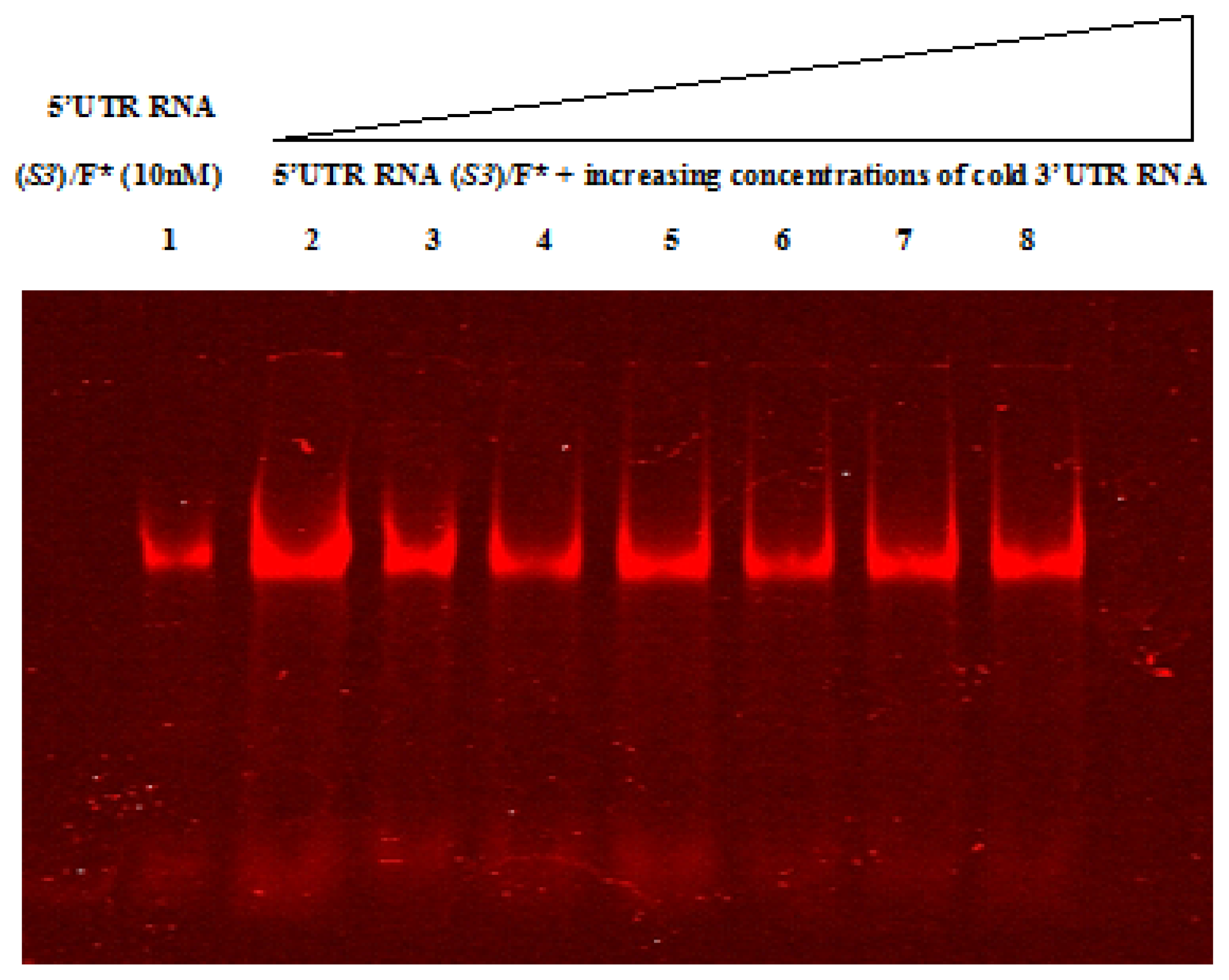
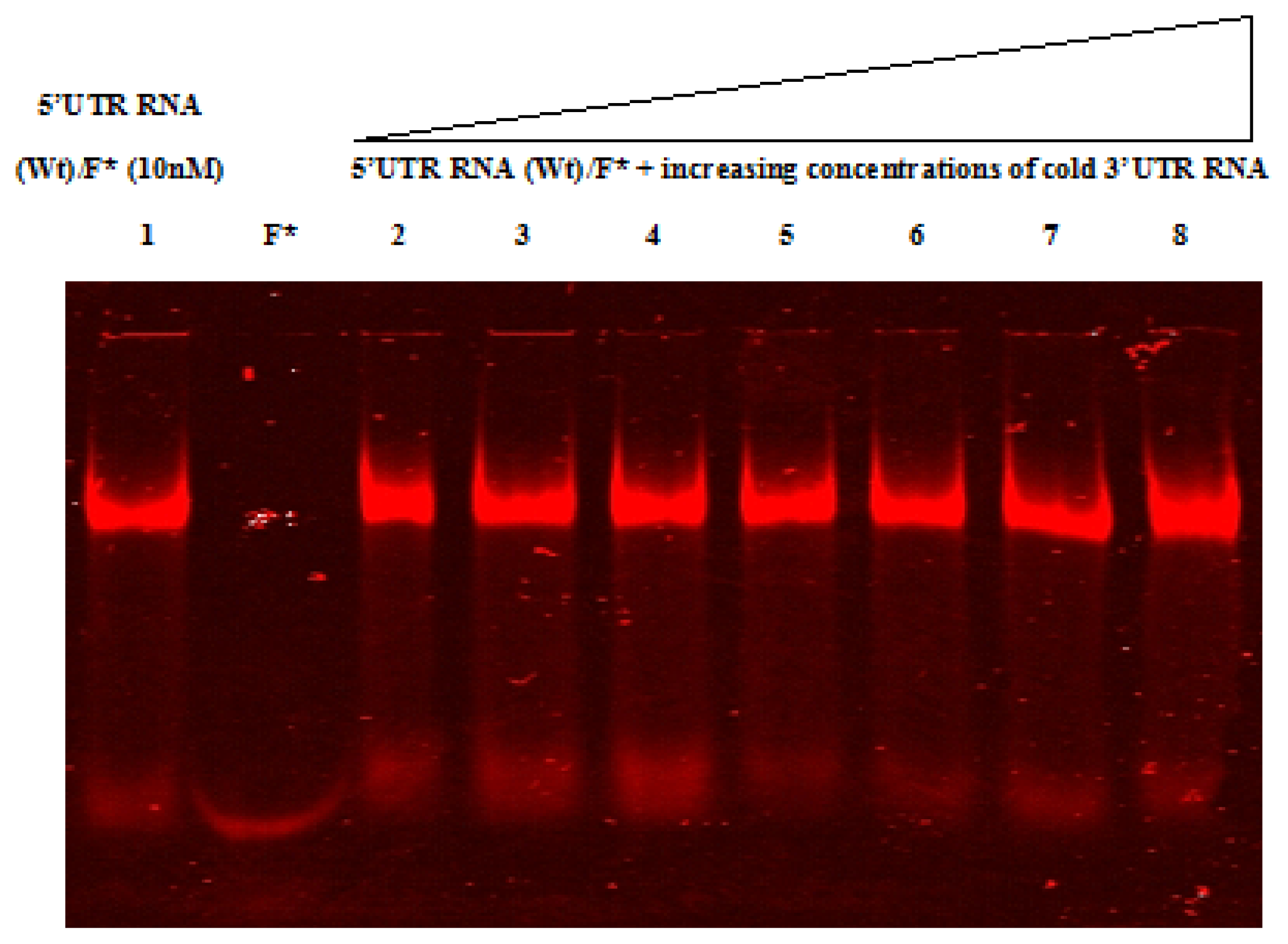
2.4. Native Gel Shift Assay Analysis
3. Discussion
4. Experimental Section
4.1. Virus
4.2. Plasmids
4.3. Bacterial Strains
4.4. Viral RNA Extraction
4.5. Reverse Transcription (RT)
4.6. Amplification of CVB3 Wild-type and Sabin3-Like 5′UTRs
4.7. Synthesis of the CVB3 3′UTR by Primers Hybridization and Extension
4.7.1. Primers Design
4.7.2. Synthesis of the CVB3 3′UTR
4.8. Cloning of the 5′ and 3′UTRs into the pUC19 Vector
4.9. Screening of Transformed Clones
4.10. RNA and In Vitro Transcription
4.11. Periodate Oxidation and Fluorophore Coupling
4.12. RNA-RNA Gel Shift Assay
5. Conclusion
Acknowledgments
Conflict of Interest
References
- Baboonian, C.; Davies, M.J.; Booth, J.C.; McKenna, W.J. Coxsackie B viruses and human heartn disease. Curr. Top. Microbiol. Immunol 1997, 223, 31–52. [Google Scholar]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol 2008, 3, 127–155. [Google Scholar]
- Dunn, J.J.; Chapman, N.M.; Tracy, S.; Romero, J.R. Natural genetics of cardiovirulence in coxsackievirus B3 clinical isolates: Localization to the 5′ nontranslated region. J. Virol 2000, 74, 4787–4794. [Google Scholar]
- Verma, B.; Bhattacharyya, S.; Das, S. Polypyrimidine tract binding protein interacts with coxsackievirus B3 RNA and influences its translation. J. Gen. Virol 2010, 91, 1245–1255. [Google Scholar]
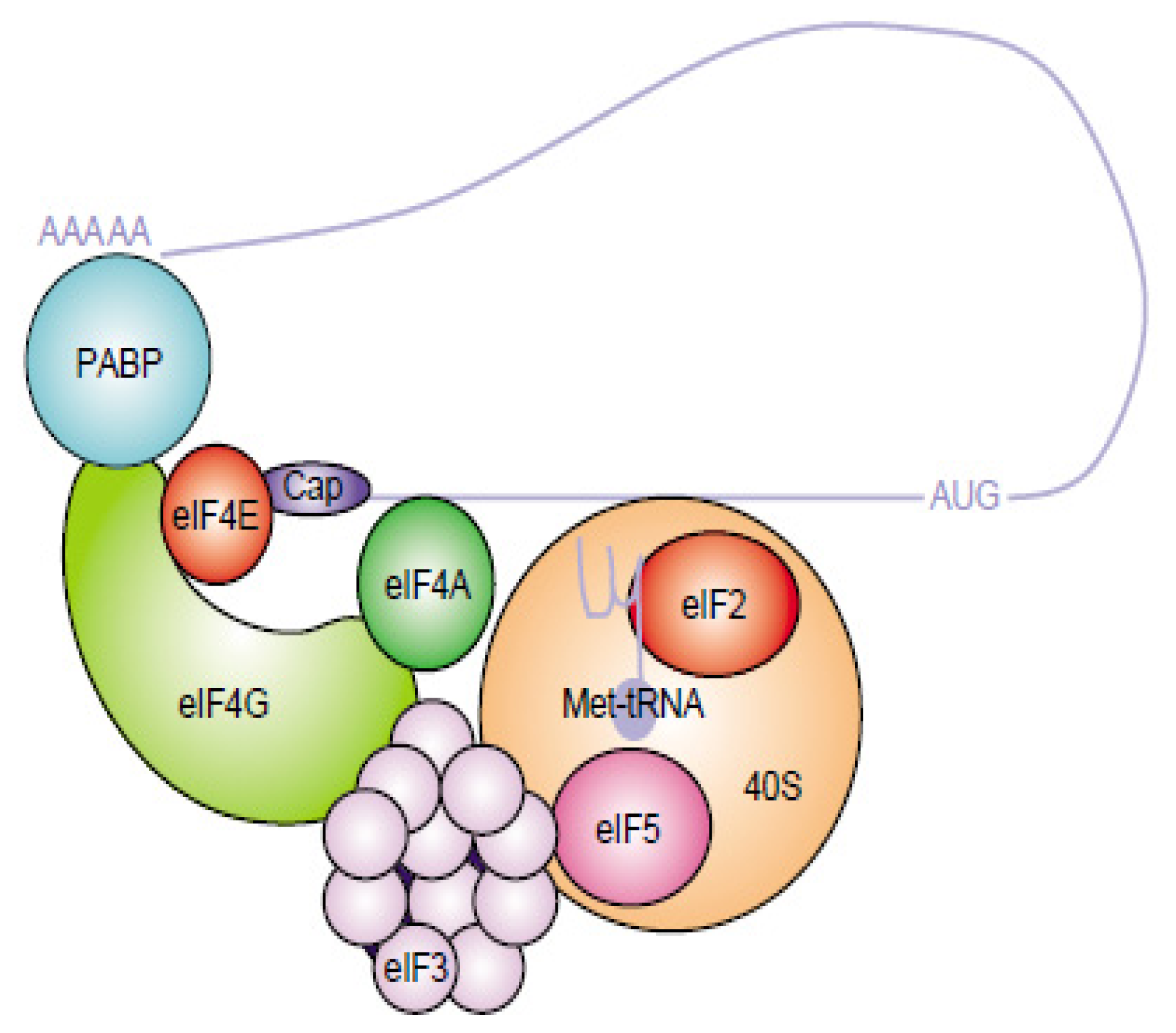
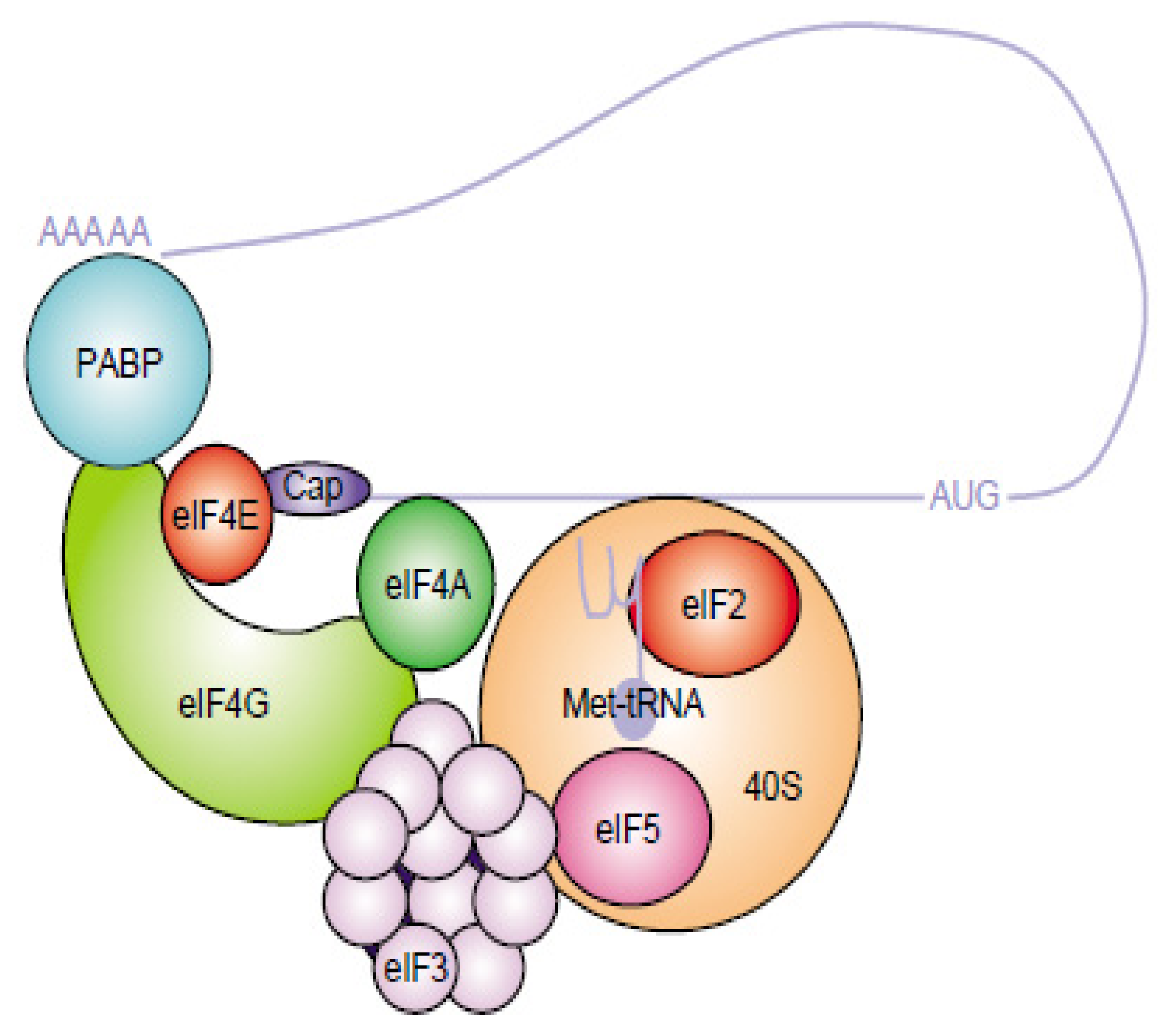
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol 2010, 11, 113–127. [Google Scholar]
- Vallejos, M.; Deforges, J.; Plank, T.D.M.; Letelier, A.; Ramdohr, P.; Abraham, C.G.; Valiente-Echeverrìa, F.; Kieft, J.S.; Sargueil, B.; López-Lastra, M. Activity of the human immunodeficiency virus type 1 cell cycle-dependent internal ribosomal entry site is modulated by IRES trans-acting factors. Nucleic Acid Res 2011, 39, 6186–6200. [Google Scholar]
- Lopez-Lastra, M.; Rivas, A.; Barria, M.I. Protein synthesis in eukaryotes: The growing biological relevance of cap-independent translation initiation. Biol. Res 2005, 38, 121–146. [Google Scholar]
- Fitzgerald, K.D.; Semler, B.L. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 2009, 1789, 518–528. [Google Scholar]
- Flanegan, J.B.; Pettersson, R.F.; Ambros, V.; Hewlett, N.J.; Baltimore, D. Covalent linkage of a protein to a defined nucleotide sequence at the 5′-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl. Acad. Sci. USA 1977, 74, 961–965. [Google Scholar]
- Lee, Y.F.; Nomoto, A.; Detjen, B.M.; Wimmer, E. A protein covalently linked to poliovirus genome RNA. Proc. Natl. Acad. Sci. USA 1977, 74, 59–63. [Google Scholar]
- Etchison, D.; Milburn, S.C.; Edery, I.; Sonenberg, N.; Hershey, J.W. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220.000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem 1982, 257, 14806–14810. [Google Scholar]
- Krausslich, H.G.; Nicklin, M.J.; Toyoda, H.; Etchison, D.; Wimmer, E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol 1987, 61, 2711–2718. [Google Scholar]
- Jang, S.K.; Krausslich, H.G.; Nicklin, M.J.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol 1988, 62, 2636–2643. [Google Scholar]
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from Poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar]
- Kolupaeva, V.G.; Hellen, C.U.; Shatsky, I.N. Structural analysis of the interaction of the pyrimidine tract-binding protein with the internal ribosomal entry site of encephalomyocarditis virus and foot-and-mouth disease virus RNAs. RNA 1996, 2, 1199–1212. [Google Scholar]
- Vagner, S.; Galy, B.; Pyronnet, S. Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. EMBO Rep 2001, 2, 893–898. [Google Scholar]
- Zell, R.; Ihle, Y.; Seitz, S.; Gündel, U.; Wutzler, P.; Gürlach, M. Poly (rC)-binding protein 2 interacts with the oligo (rC) tract of coxsackievirus B3. Biochem. Biophys. Res. Commun 2008, 366, 917–921. [Google Scholar]
- Yang, D.; Wilson, J.E.; Anderson, D.R.; Bohunek, L.; Cordeiro, C.; Kandol, R.; Mcmanus, B.M. In vitro mutational and inhibitory analysis of the cis-acting translational elements within the 5′ untranslated region of coxsackievirus B3: Potential targets for antiviral action of antisense oligomers. Virology 1997, 228, 63–73. [Google Scholar]
- Bailey, J.M.; Tapprich, W.E. Structure of the 5′-nontranslated region of the coxsackievirus B3 Genome: Chemical modification and comparative sequence analysis. J. Virol 2007, 81, 650–668. [Google Scholar]
- Ray, P.S.; Das, S. La autoantigen is required for the internal ribosome entry site-mediated translation of Coxsackievirus B3 RNA. Nucleic Acid Res 2002, 30, 4500–4508. [Google Scholar]
- Chen, T.C.; Weng, K.F.; Chang, S.C.; Lin, J.Y.; Huang, P.N.; Shih, S.R. Development of antiviral agents for enteroviruses. J. Antimicrob. Chemother 2008, 62, 1169–1173. [Google Scholar]
- Bhattacharyya, S.; Das, S. Mapping of secondary structure of the spacer region within the 5′-untranslated region of the coxsackievirus B3 RNA: Possible role of an apical GAGA loop in binding La protein and influencing internal initiation of translation. Virus Res 2005, 108, 89–100. [Google Scholar]
- Ben M’hadheb-Gharbi, M.; Paulous, S.; Aouni, M.; Kean, K.M.; Gharbi, J. The substitution U475→ C with Sabin3-like mutation within the IRES attenuate coxsackievirus B3 cardiovirulence. Mol. Biotechnol 2007, 36, 52–60. [Google Scholar]
- Verma, B.; Ponnuswamy, A.; Gnanasundram, S.V.; Das, S. Cryptic AUG is important for 48S ribosomal assembly during internal initiation of translation of coxsackievirus B3 RNA. J. Gen. Virol 2011, 92, 1–10. [Google Scholar]
- López de Quinto, S.; Martínez-Salas, E. Conserved structural motifs located in distal loops of aphthovirus internal ribosome entry site domain 3 are required for internal initiation of translation. J. Virol 1997, 71, 4171–4175. [Google Scholar]
- Malnou, C.E.; Pöyry, T.A.; Jackson, R.J.; Kean, K.M. Poliovirus internal ribosome entry segment structure alterations that specifically affect function in neuronal cells: Molecular genetic analysis. J. Virol 2002, 76, 10617–10626. [Google Scholar]
- Bhattacharyya, S.; Verma, B.; Pandey, G.; Das, S. The structure and function of a cis-acting element located upstream of the IRES that influences Coxsackievirus B3 RNA translation. Virology 2008, 377, 345–354. [Google Scholar]
- Kok, C.C.; Phuektes, P.; Bek, E.; McMinn, P.C. Modification of the untranslated regions of human enterovirus 71 impairs growth in a cell-specific manner. J. Virol 2012, 86, 542–552. [Google Scholar]
- Klump, W.M.; Bergmann, I.; Muller, B.C.; Ameis, D.; Kandolf, R. Complete nucleotide sequence of infectious coxsackievirus B3 cDNA: Two initial 5′ uridine residues are regained during plus-strand RNA synthesis. J. Virol 1990, 64, 1573–1583. [Google Scholar]
- Melchers, W.J.; Hoenderop, J.G.; Bruins Slot, H.J.; Pleij, C.W.; Pilipenko, E.V. Kissing of the two predominant hairpin loops in the coxsackie B virus 3′ untranslated region is the essential structural feature of the origin of replication required for negative-strand RNA synthesis. J. Virol 1997, 71, 686–696. [Google Scholar]
- Ye, X.; Liu, Z.; Hemida, M.G.; Yang, D. Targeted delivery of mutant tolerant anti-coxsackievirus artificial micrornas using folate conjugated bacteriophage Phi29 pRNA. PLoS One 2011, 6, e21215. [Google Scholar]
- Dobrikova, E.Y.; Grisham, R.N.; Kaizer, C.; Lin, J.; Gromeier, M. Competitive translation efficiency and the picornavirus type I internal ribosome entry site facilitated by viral cis and trans factors. J. Virol 2006, 80, 3310–3321. [Google Scholar]
- Jacobson, S.J.; Konings, D.A.; Sarnow, P. Biochemical and genetic evidence for a pseudoknot structure at the 3′ terminus of the poliovirus RNA genome and its role in viral RNA amplification. J. Virol 1993, 67, 2961–2971. [Google Scholar]
- Jacobson, A.; Peltz, S.W. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem 1996, 65, 693–739. [Google Scholar]
- Cheung, P.; Lim, T.; Yuan, J.; Zhang, M.; Chau, D.; McManus, B.; Yang, D. Specific interaction of HeLa cell proteins with Coxsackievirus B3 3′UTR: La autoantigen binds the 3′ and 5′UTRs independently of the poly (A) tail. Cell Microbiol 2007, 9, 1705–1715. [Google Scholar]
- Merkle, I.; van Ooij, M.J.M.; van Kuppeveld, F.J.M.; Glaudemans, D.H.R.F.; Galama, J.M.D.; Henke, A.; Zell, R.; Melchers, W.J.G. Biological significance of a human enterovirus B-specific RNA element in the 3′ nontranslated region. J. Virol 2002, 76, 9900–9909. [Google Scholar]
- Dobrikova, E.; Florez, P.; Bradrick, S.; Gromeier, M. Activity of a type 1 picornavirus internal ribosomal entry site is determined by sequences within the 3′ nontranslated region. Proc. Natl. Acad. Sci. USA 2003, 100, 15125–15130. [Google Scholar]
- Todd, S.; Towner, J.S.; Brown, D.M.; Semler, B.L. Replication competent picornaviruses with complete genomic RNA 3′ noncoding region deletions. J. Virol 1997, 71, 8868–8874. [Google Scholar]
- Piron, M.; Vende, P.; Cohen, J.; Poncet, D. Rotavirus RNA binding protein NSP3 interacts with eIF4GI and evicts the poly (A) binding protein from eIF4F. EMBO J 1998, 17, 5811–5821. [Google Scholar]
- Vende, P.; Piron, M.; Castagnè, N.; Poncet, D. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′end. J. Virol 2000, 74, 7064–7071. [Google Scholar]
- Herold, J.; Andino, R. Poliovirus RNA replication requires genome circularization through a protein–protein bridge. Mol. Cell 2001, 7, 581–591. [Google Scholar]
- Isken, O.; Grassmann, C.W.; Sarisky, R.T.; Kann, M.; Zhang, S.; Grosse, F.; Kao, P.N.; Behrens, S.E. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J 2003, 22, 5655–5665. [Google Scholar]
- Isken, O.; Grassmann, C.W.; Yu, H.; Behrens, S.E. Complex signals in the genomic 3′ nontranslated region of bovine viral diarrhea virus coordinate translation and replication of the viral RNA. RNA 2004, 10, 1637–1652. [Google Scholar]
- Serrano, P.; Pulido, M.R.; Saiz, M.; Martinez-Salas, E. The 3′ end of the foot-and-mouth disease virus genome establishes two distinct long-range RNA-RNA interactions with the 5′ end region. J. Gen. Virol 2006, 87, 3013–3022. [Google Scholar]
- Ben M’hadheb-Gharbi, M.; Gharbi, J.; Paulous, S.; Brocard, M.; Komaromva, A.; Aouni, M.; Kean, K.M. Effects of the Sabin-like mutations in domain V of the internal ribosome entry segment on translational efficiency of the Coxsackievirus B3. Mol. Gen. Genomics 2006, 276, 402–412. [Google Scholar]
- Souii, A.; Ben M’hadheb-Gharbi, M.; Aouni, M.; Gharbi, J. In vitro molecular characterization of RNA-proteins interactions during initiation of translation of a wild-type and a mutant coxsackievirus B3 RNAs. Mol. Biotechnol 2012. [Google Scholar] [CrossRef]
- Zoll, J.; Heus, H.A.; van Kuppeveld, F.J.M.; Melchers, W.J.G. The structure–function relationship of the enterovirus 3′UTR. Virus Res 2009, 139, 209–216. [Google Scholar]
- Robaglia, C.; Caranta, C. Translation initiation factors: A weak link in plant RNA virus infection. Trends Plant Sci 2006, 11, 40–45. [Google Scholar]
- Romero-Lòpez, C.; Berzal-Herranz, A. A long-range RNA-RNA interaction between the 5′ and 3′ends of the HCV genome. RNA 2009, 15, 1740–1752. [Google Scholar]
- Sean, P.; Nguyen, J.H.C.; Semler, B.L. Altered interactions between stem-loop IV within the 5′ non coding region of coxsackievirus RNA and poly (rC) binding protein 2: Effects on IRES-mediated translation and viral infectivity. Virology 2009, 389, 45–58. [Google Scholar]
- Song, Y.; Tzima, E.; Ochs, K.; Bassili, G.; Trusheim, H.; Linder, M.; Preissner, K.T.; Niepmann, M. Evidence for an RNA chaperone function of polypyrimidine tract-binding protein in picornavirus translation. RNA 2005, 11, 1809–1824. [Google Scholar]
- Martìnez-Salas, E.; Pineiro, D.; Fernàndez, N. Alternative mechanisms to initiate translation in eukaryotic mRNAs. Comp. Funct. Genomics 2012, 2012. [Google Scholar] [CrossRef]
- Lopez de Quinto, S.; Saiz, M.; de la Morena, D.; Sobrino, F.; Martinez-Salas, E. IRES-driven translation is stimulated separately by the FMDV 3′-NCR and poly (A) sequences. Nucleic Acid Res 2002, 30, 4398–4405. [Google Scholar]
- Alvarez, D.E.; Lodeiro, M.F.; Ludueña, S.J.; Pietrasanta, L.I.; Gamarnik, A.V. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol 2005, 79, 6631–6643. [Google Scholar]
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol 1987, 198, 33–41. [Google Scholar]
- You, S.; Falgout, B.; Markoff, L.; Padmanabhan, R. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5′- and 3′-terminal regions that influence RNA structure. J. Biol. Chem 2001, 276, 15581–15591. [Google Scholar]
- De Nova-Ocampo, M.; Villegas-Sepùlveda, N.; del Angel, R.M. Translation elongation factor-1α, La, and PTB interact with the 3′ untranslated region of dengue 4 virus RNA. Virology 2002, 295, 337–347. [Google Scholar]
- Fabian, M.R.; White, K.A. 5′–3′ RNA-RNA interaction facilitates cap-and poly (A) tail-independent translation of tomato bushy stunt virus mRNA: A potential common mechanism for Tombusviridae. J. Biol. Chem. 2004, 279, 28862–28872. [Google Scholar]
- Guo, L.; Allen, E.M.; Miller, W.A. Base-pairing between untranslated regions facilitates translation of uncapped, nonpolyadenylated viral RNA. Mol. Cell 2001, 7, 1103–1109. [Google Scholar]
- Shen, R.; Miller, W.A. The 3′ untranslated region of tobacco necrosis virus RNA contains a barley yellow dwarf virus-like cap-independent translation element. J. Virol 2004, 78, 4655–4664. [Google Scholar]
- Hentze, M.W. eIF4G: A multipurpose ribosome adapter? Science 1997, 275, 500–501. [Google Scholar]
- Song, Y.; Friebe, P.; Tzima, E.; Junemann, C.; Bartenschlager, R.; Niepmann, M. The hepatitis C virus RNA 3′ untranslated region strongly enhances translation directed by the internal ribosome entry site. J. Virol 2006, 80, 11579–11588. [Google Scholar]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by Acid Guanidium thiocyanate-phenol-Chloroform extraction. Anal. Biochem 1987, 162, 156–159. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Souii, A.; Gharbi, J.; M'hadheb-Gharbi, M.B. Molecular Analysis of RNA-RNA Interactions between 5’ and 3’ Untranslated Regions during the Initiation of Translation of a Cardiovirulent and a Live-Attenuated Coxsackievirus B3 Strains. Int. J. Mol. Sci. 2013, 14, 4525-4544. https://doi.org/10.3390/ijms14034525
Souii A, Gharbi J, M'hadheb-Gharbi MB. Molecular Analysis of RNA-RNA Interactions between 5’ and 3’ Untranslated Regions during the Initiation of Translation of a Cardiovirulent and a Live-Attenuated Coxsackievirus B3 Strains. International Journal of Molecular Sciences. 2013; 14(3):4525-4544. https://doi.org/10.3390/ijms14034525
Chicago/Turabian StyleSouii, Amira, Jawhar Gharbi, and Manel Ben M'hadheb-Gharbi. 2013. "Molecular Analysis of RNA-RNA Interactions between 5’ and 3’ Untranslated Regions during the Initiation of Translation of a Cardiovirulent and a Live-Attenuated Coxsackievirus B3 Strains" International Journal of Molecular Sciences 14, no. 3: 4525-4544. https://doi.org/10.3390/ijms14034525