Synthesis and Structural Characterization of Substituted 2-Phenacylbenzoxazoles

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
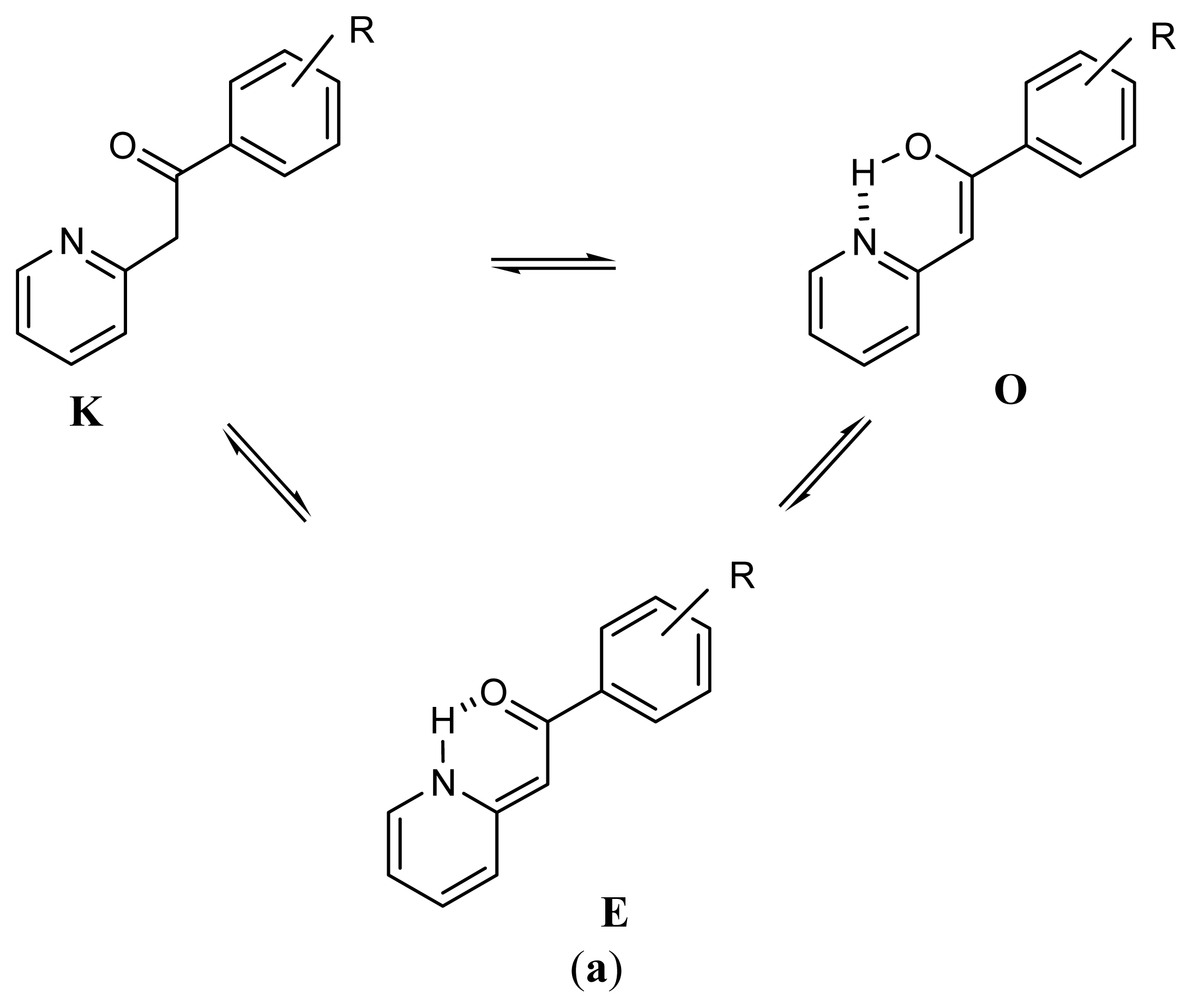
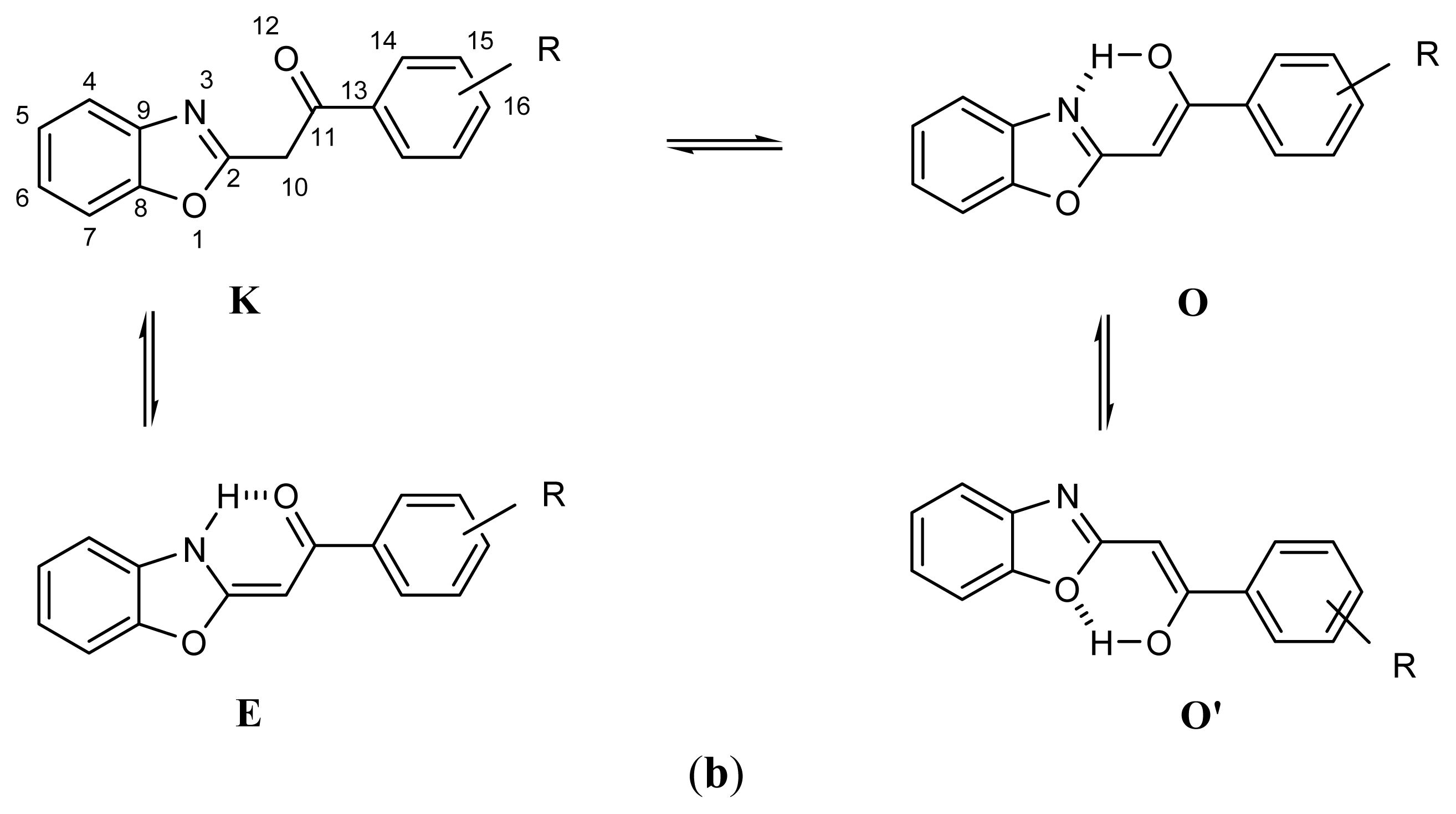
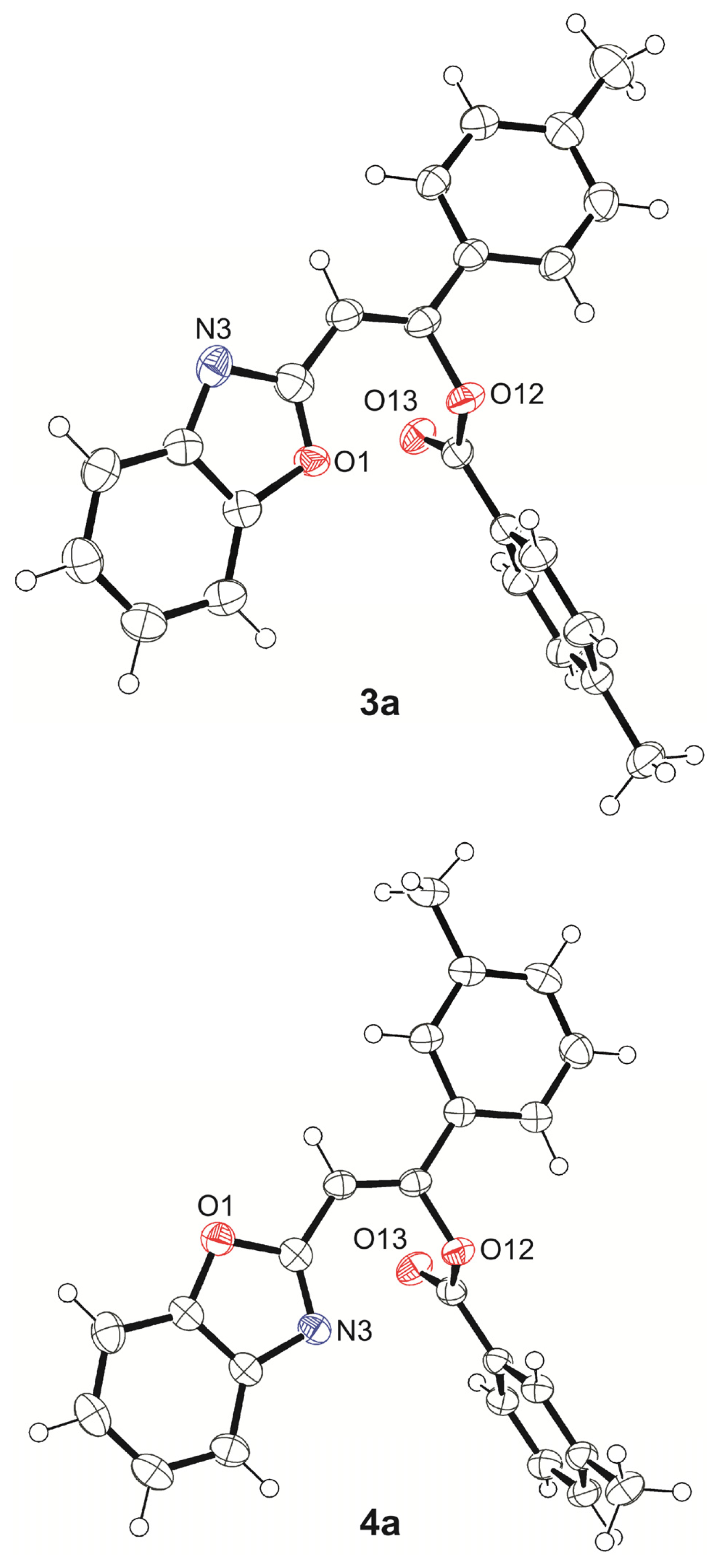
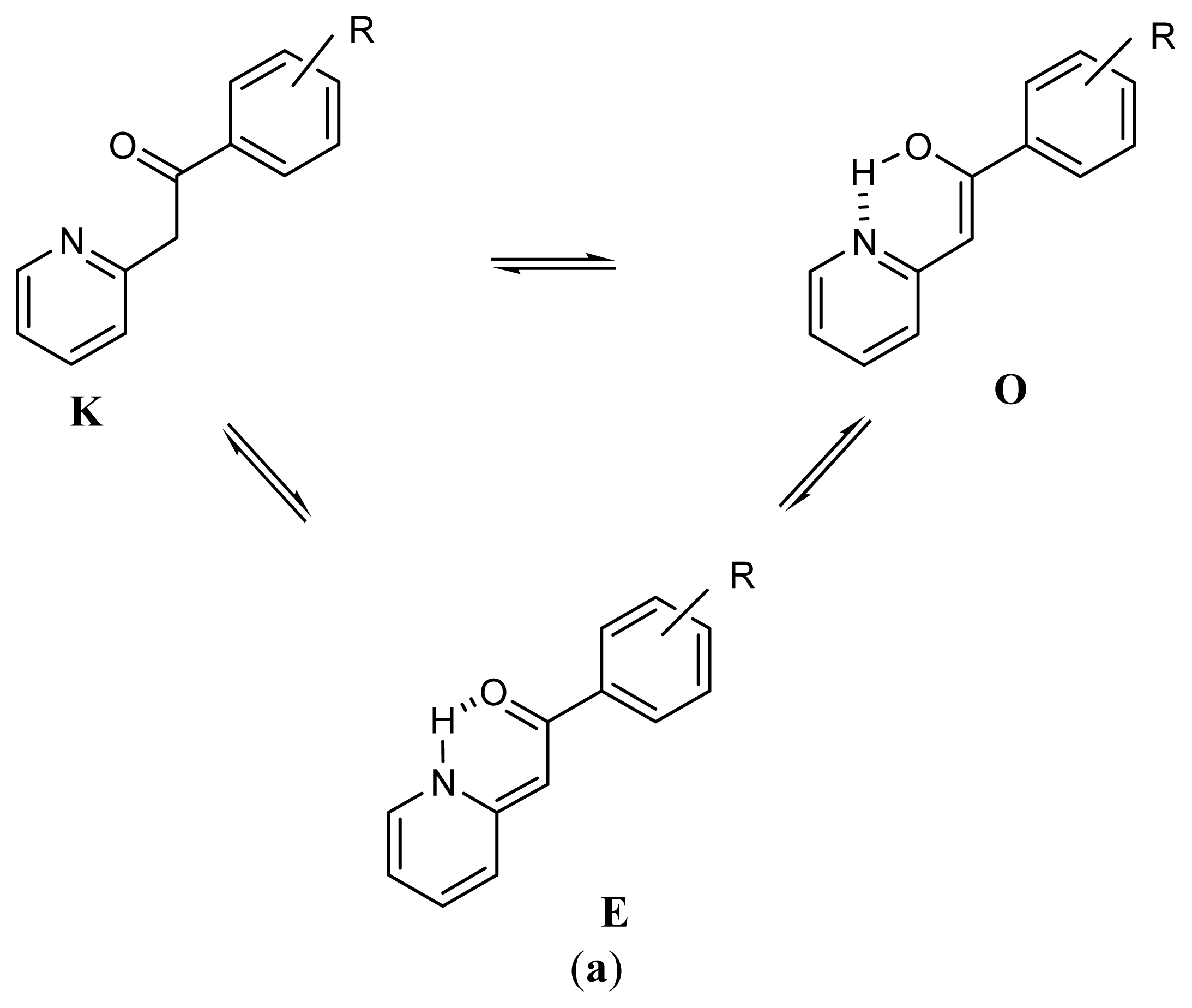
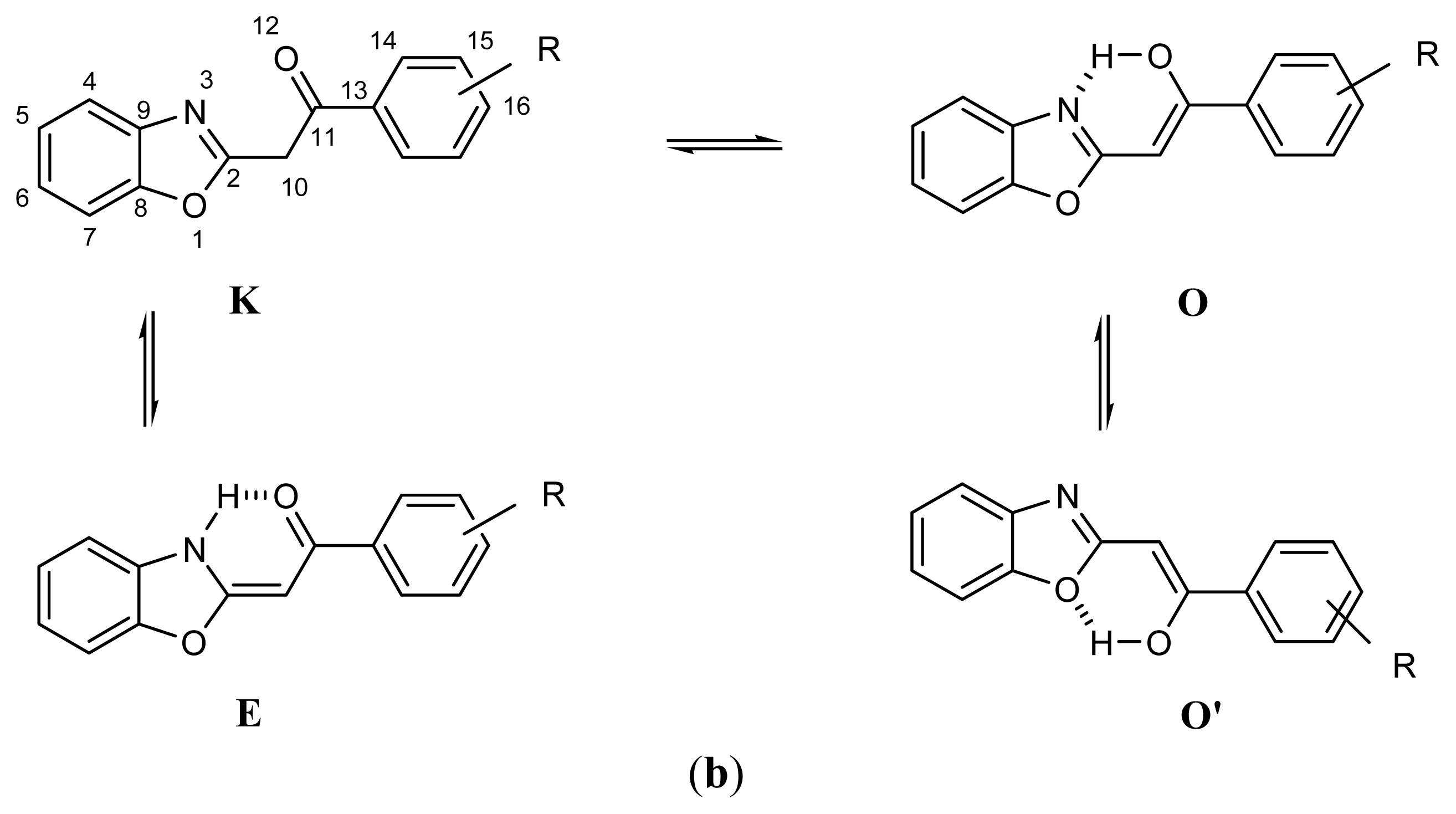
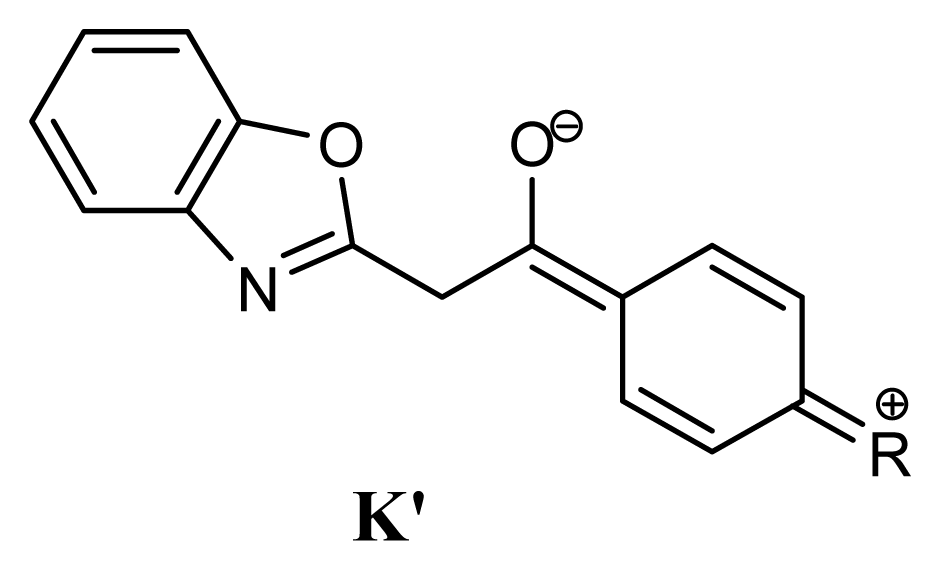
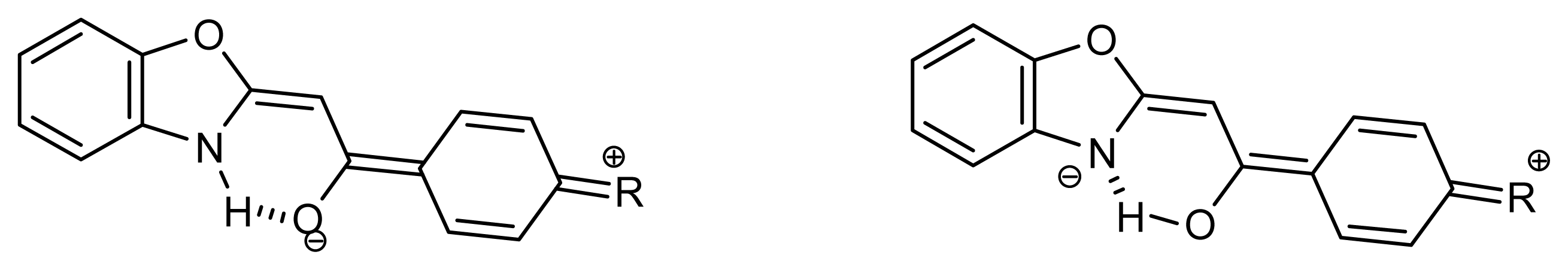
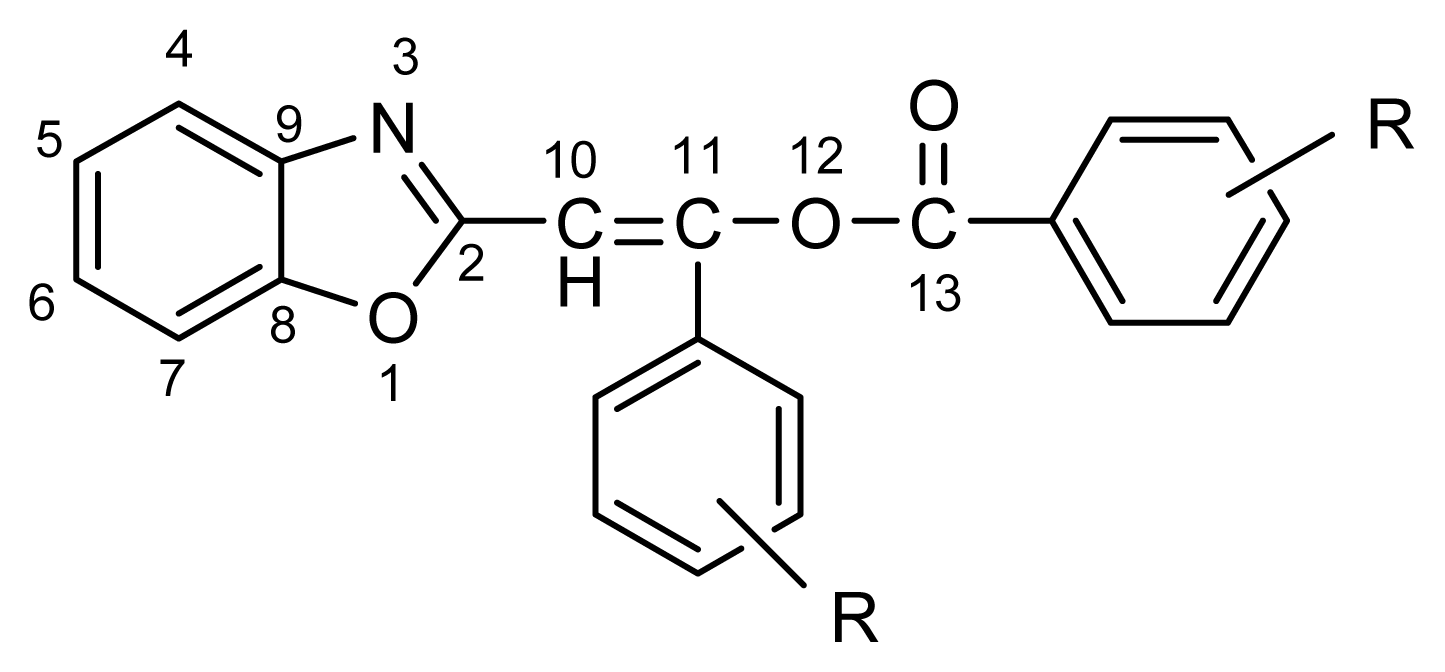
2.1. Synthesis and Identification of Tautomers
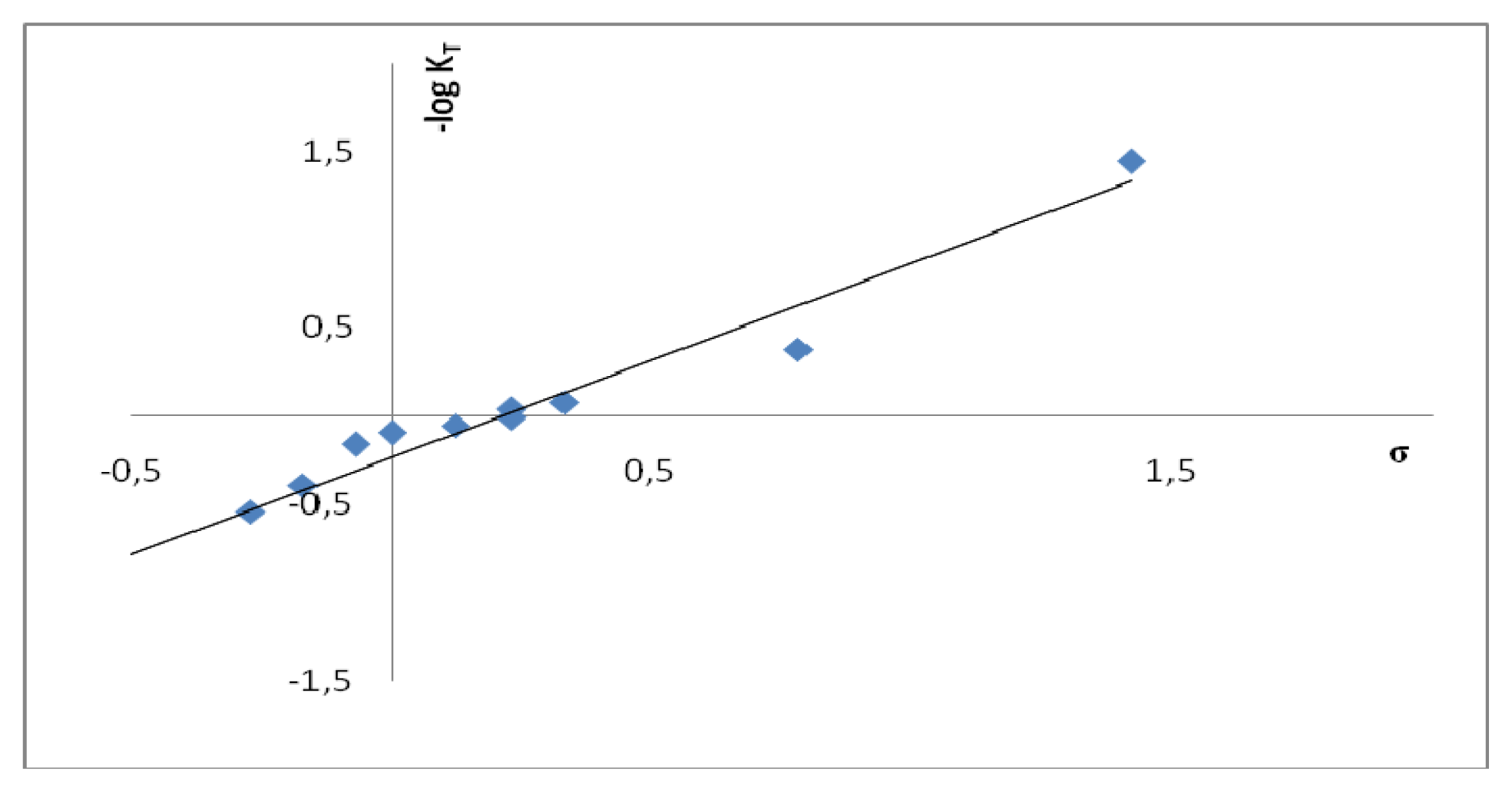
2.2. Substituent Effect on the Tautomeric Equilibrium
2.3. Quantum-Chemical Calculations
3. Conclusions
4. Experimental and Computational
4.1. X-Ray Crystallography
4.2. NMR Spectroscopy
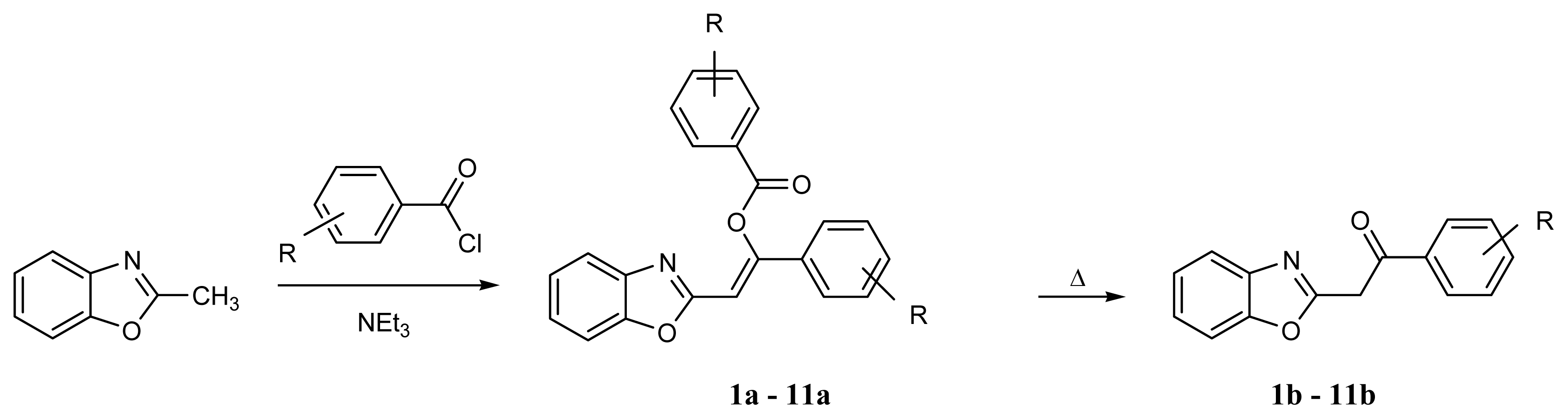
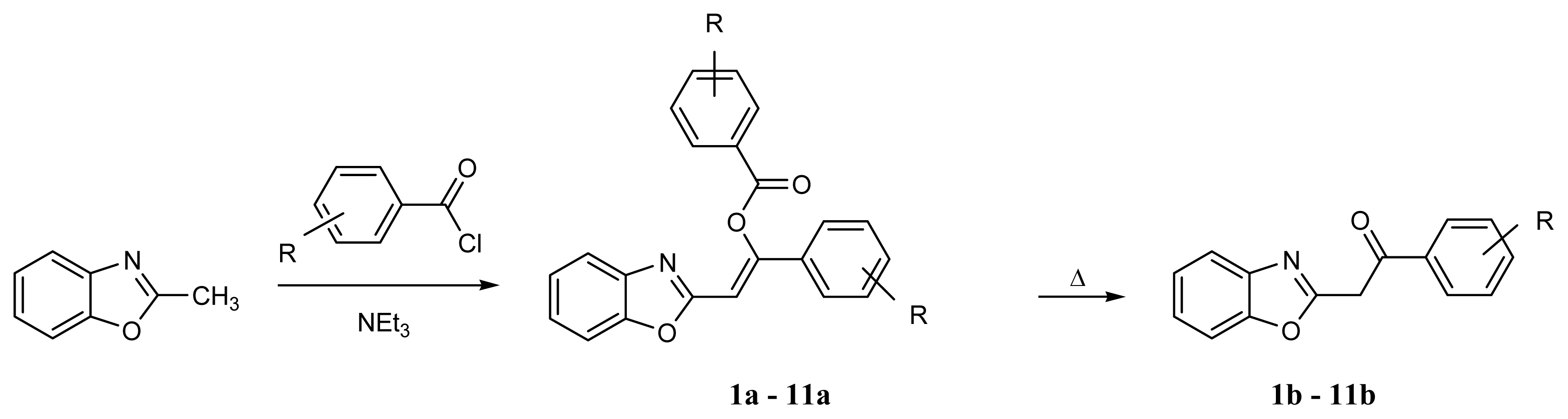
4.3. Syntheses of 2-Phenacylbenzoxazoles
4.3.1. Method A
4.3.2. Method B
- 3a: C23H19NO3 (357.39): calcd. C 77.29, H 5.36, N 3.92; found C 77.04, H 5.47, N 4.08.
- 4a: C23H19NO3 (357.39): calcd. C 77.29, H 5.36, N 3.92; found C 77.09, H 5.11, N 4.06.
- 6a: C23H19NO5 (389.39): calcd. C 70.94, H 4.92, N 3.60; found C 70.69, H 4.68, N 3.69.
- 7a: C21H13Cl2NO3 (398.23): calcd. C 63.33, H 3.29, N 3.52; found C 63.12, H 3.47, N 3.39.
- 8a: C21H13Br2NO3 (487.15): calcd. C 51.77, H 2.69, N 2.88; found C 51.85, H 2.82, N 3.01.
- 10a: C21H13N3O7 (419.33): calcd. C 60.15, H 3.12, N 10.02; found C 59.90, H 3.01, N 9.81.
- 1b: C17H16N2O2 (280.32): calcd. C 72.83, H 5.75, N 10.00; found C 72.91, H 5.55, N 9.79.
- 9b: C15H10FNO2 (255.24): calcd. C 70.58, H 3.95, N 5.49; found C 70.73, H 4.06, N 5.70.
- 11b: C15H9N3O6 (327.24): calcd. C 55.05, H 2.77, N 12.84; found C 54.95, H 3.01, N 12.62.
4.4. Quantum-Chemical Calculations
Supplementary Information
ijms-14-04444-s001.docAcknowledgments
Conflict of Interest
References
- Kolehmainen, E.; Ośmiałowski, B.; Krygowski, T.M.; Kauppinen, R.; Nissinen, M.; Gawinecki, R. Substituent and temperature controlled tautomerism: multinuclear magnetic resonance, X-ray, and theoretical studies on 2-phenacylquinolines. J. Chem. Soc. Perkin Trans 2000, 2, 1259–1266. [Google Scholar]
- Kolehmainen, E.; Ośmiałowski, B.; Nissinen, M.; Kauppinen, R.; Gawinecki, R. Substituent and temperature controlled tautomerism of 2-phenacylpyridine: hydrogen bond as a configurational lock of (Z) 2-(2-hydroxy-2-phenylvinyl)pyridine. J. Chem. Soc. Perkin Trans 2000, 2, 2185–2191. [Google Scholar]
- Gawinecki, R.; Kolehmainen, E.; Loghmani-Khouzani, H.; Ośmiałowski, B.; Lovász, T.; Rosa, P. Effect of π-electron delocalization on tautomeric equilibria. Benzoannulated annulated 2-phenacylpyridines. Eur. J. Org. Chem 2006, 12, 2817–2824. [Google Scholar]
- Sun, J.; Yan, Ch.-G.; Han, Y. KF-Al2O3 catalyzed the condensations of 2-methylbenzoxazole and pyrazol-5-one with aromatic aldehydes. Synth. Commun. 2001, 31, 151–154. [Google Scholar]
- Lazukina, L.A.; Kukhar, V.P. Reactions of trichloromethyldialkylamines with 2-methylbenzothiazole, 2-methylbenzoxazole, and their salts. Chem. Heterocycl. Comp 1974, 10, 668–670. [Google Scholar]
- Stepanov, F.N.; Davydowa, S.L. Heterocyclic derivatives of methylketones. Zh. Obshch. Khim 1958, 28, 891–896. [Google Scholar]
- Babichev, F.S.; Volovenko, Y.M. Acylalation of 2-methylbenzazoles with use of esters of carboxylic acids. Ukr. Khim. Zh 1977, 43, 163–165. [Google Scholar]Sov. Prog. Chem. (Engl. Transl.) 1977, 43, 49–50.
- Sund, E.H.; Donohue, B.E.; Thomas, T.K. Synthesis of 2-(2-benzoxazolyl)-1-phenylethanone and related ethanones. J. Chem. Eng. Data 1979, 24. [Google Scholar] [CrossRef]
- Stachel, H.-D. Die darstellung einiger heterocyclen aus acylketen-derivaten (benzoxazole, benzothiazole, benzothiodiazine, chinazolone). Über Keten-Derivate, X. Arch. Pharm. (Weinheim) 1963, 296, 337–343. [Google Scholar]
- Huang, Z.-T.; Wang, M.-X. The synthesis and tautomerization of ketene aminals with benzimidazoline ring. Tetrahedron 1992, 48, 2325–2332. [Google Scholar]
- Sakamoto, M.; Abe, M.; Ishii, K. Studies on conjugated nitriles. VI. Reaction of 2-methyl-quinoline and related compounds with acyl cyanides. Chem. Pharm. Bull. (Tokyo) 1991, 39, 277–281. [Google Scholar]
- Nardi, D.; Tajana, A.; Pennini, R. Heterocyclic compounds from 3,3-dimercapto-1-aryl-2-propen- 1-ones. Note 2. Condensation with o-aminophenol and o-aminothiophenol. J. Heterocycl. Chem 1975, 12, 139–142. [Google Scholar]
- More O’Ferrall, R.A.; Murray, B.A. 1H and 13C NMR spectra of α-heterocyclic ketones and assignment of keto, enol and enaminone tautomeric structures. J. Chem. Soc. Perkin Trans 1994, 2, 2461–2470. [Google Scholar]
- Preparation of Omega-Acyl-Azoles. U.S. Patent 2323504, 1939.
- Ciurdaru, Gh.; Ciuciu, M. The acylation of 2-methylbenzazoles. J. Prakt. Chem. 1979, 321, 320–322. [Google Scholar]
- Dzvinchuk, I.B.; Lozinskii, M.O.; Vypirailenko, A.V. C-Mono- and dibenzoylation of 2-methylbenzimidazole with use of benzoyl chloride. Zh. Org. Khim 1994, 30, 909–914. [Google Scholar]
- Rauch, E.B.; Dickinson, P.; Welsh, J.A. Prepartion of 2-benzoylmethylbezoxazoles. U.S. Patent 3375258, 1968. [Google Scholar]
- Dzvinchuk, I.B.; Vypirailenko, A.V.; Lozinskii, M.O. Selective recyclization of 2-aroylmethyl-1H-benzimidazole hydrazones by condensation with dimethylformamide. Chem. Heterocycl. Comp 2001, 37, 1096–1101. [Google Scholar]
- Dzvinchuk, I.B.; Nesterenko, A.M.; Polovinko, V.V.; Ryabitskii, A.B.; Lozinskii, M.O. Synthesis and tautomerism of 2-phenacyl-1H-benzimidazoles and their hydrogen bromide salts. Chem. Heterocycl. Comp 2011, 47, 953–963. [Google Scholar]
- De Silva, H.I.; Chatterjee, S.; Henry, W.P.; Pittman, Ch.U. Synthesis of functionalized fused-ring heterocycles from tautomers of 2-(thiazole, oxazole, benzothaizole, and benzoxazole)-1- phenylethenols and 1,3-diacyl chlorides or N-(chlorocarbonyl) isocyanate. Synthesis 2012, 44, 3453–3464. [Google Scholar]
- Gawinecki, R.; Kolehmainen, E.; Ośmiałowski, B.; Palkovič, P.; Nissinen, M. Synthesis and NMR spectra of 2-methyl-2-quinolin-2-yl-propiophenones. Heterocycl. Commun 1999, 5, 549–554. [Google Scholar]
- Gawinecki, R.; Ośmiałowski, B.; Kolehmainen, E.; Nissinen, M. N-Methyl-1,2-dihydro-2- benzoylmethylenequinolines: Configurational dissimilarity with unmethylated congeners. J. Mol. Struct 2000, 525, 233–239. [Google Scholar]
- Raczyñska, E.D.; Kosiñska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric equilibria in relation to π-electron delocalization. Chem. Rev 2005, 105, 3561–3612. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parametyers. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- Shavitt, I. Molecular Interactions; Scheiner, S., Ed.; Wiley: Chichester, UK, 1997. [Google Scholar]
- Dimitrova, Y.; Peyerimhof, S. Ab initio study of structures of hydrogen-bonded nitric acid complexes. Chem. Phys 2000, 254, 125–134. [Google Scholar]
- Gilli, G.; Belluci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the β-diketone fragment. J. Am. Chem. Soc 1981, 111, 1023–1028. [Google Scholar]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 2. Intercorrelation between crystal structure and spectroscopic parameters in eight intramolecularly hydrogen bonded 1,3-diaryl- 1,3-propanedione enols. J. Am. Chem. Soc 1991, 113, 4917–4915. [Google Scholar]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Covalent nature of the strong homonuclear hydrogen bond. Study of the O–H···O system by crystal structure correlation methods. J. Am. Chem. Soc 1994, 116, 909–915. [Google Scholar]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Resonance-assisted O–H···O hydrogen bonding: Its role in the crystalline self-recognition of β-diketone enols and its structural and IR characterization. Chem. Eur. J 1996, 2, 925–934. [Google Scholar]
- CrysalisPro, version 1.171.36.24; Agilent Technologies: Oxford, UK, 2012.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Giampiero, C.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Crystallogr 2005, 38, 381–388. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr 2008, 64, 112–122. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr 1997, 30, 565. [Google Scholar]
- Bartlett, R.J. Coupled-cluster approach to molecular structure and spectra: A step toward predictive quantum chemistry. J. Phys. Chem 1989, 93, 1697–1708. [Google Scholar]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semidirect algorithms for the MP2 energy and gradient. Chem. Phys. Lett 1990, 166, 281–289. [Google Scholar]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys 1989, 90, 1007–1023. [Google Scholar]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. aug-cc-pVDZ for first row. J. Chem. Phys 1992, 96, 6796–6806. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc: Wallingford, CT, USA; p. 2009.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Yield (%) | mp (°C) |
|---|---|---|
| 3a | 35 | 92–93 |
| 4a | 20 | 91–92 |
| 5a | 52 | 104–106 (109 [15], 97–98 [20]) |
| 6a | 30 | 111–112 |
| 7a | 32 | 106–107 |
| 8a | 75 | 175–177 |
| 10a | 63 | 226–227 |
 | |||||
|---|---|---|---|---|---|
| Compound | H10 | C2 | C10 | C11 | C13 |
| 3a | 6.98 | 164.55 | 102.39 | 154.47 | 160.14 |
| 4a | 7.03 | 164.70 | 103.12 | 154.49 | 159.96 |
| 5a | 7.26 | 164.51 | 103.31 | 154.20 | 159.83 |
| 6a | 7.03 | 164.38 | 103.61 | 153.99 | 159.99 |
| 7a | 7.02 | 163.28 | 104.51 | 152.45 | 160.86 |
| 8a | 7.00 | 163.77 | 103.81 | 152.94 | 161.45 |
| 10a | 7.17 | 162.61 | 106.75 | 151.23 | 158.35 |
| Compound | Method a | Yield (%) | mp (°C) |
|---|---|---|---|
| 1b | A | 73 | 197–198 |
| 2b | A | 62 | 106–108 (107.5–108.5) [8] |
| 3b | B | 65.5 | 96–98 (97.5–98.5) [8] |
| 4b | B | 84 | 66–68 (67–68) [8] |
| 5b | B | 59 | 94–96 (93.5–94.5) [8] (97–98) [12] (88–88.5) [6] (88–88.5) [9] (90–91) [17] (88) [7] 87–89 [20] |
| 6b | B | 61.5 | 58–60 (59.5–60) [8] |
| 7b | B | 87 | 168–170 (168.5–170) [8] |
| 8b | B | 83 | 167–169 (167) [7] |
| 9b | B | 50.5 | 101–102.5 |
| 10b | B | 99 | 233–235 b 248–249 [11] |
| 11b | B | 72 | 156.5–158 |
| Tautomer a | OH b | H10 c | C10 | C11 | N3 |
|---|---|---|---|---|---|
| 1K | - | 4.54 | 39.08 | 190.00 | −135.6 d |
| - | - | 38.76 | 100.72 | - | |
| 1O | e | 6.05 | 80.35 | 166.61 | −168.3 e |
| 2K | - | 4.61 | 39.39 | 190.81 | −134.9 |
| 2O | 12.6 | 6.11 | 82.19 | 166.35 | −166.2 |
| 3K | - | 4.61 | 39.50 | 191.95 | −134.7 |
| 3O | 12.5 | 6.17 | 82.97 | 166.44 | −164.9 |
| - | - | 82.22 | 166.49 | - | |
| 4K | - | 4.62 | 39.58 | 192.56 | −134.6 |
| 4O | 12.4 | 6.20 | 83.61 | 166.48 | −164.2 |
| 5K | - | 4.64 | 39.59 f | 192.36 g | −134.4 |
| 5O | 12.5 | 6.21 | 83.69 h | 166.29 i | −164.0 |
| 6K | - | 4.62 | 39.67 | 192.20 | −134.4 |
| 6O | 12.5 | 6.20 | 83.91 | 166.08 | −164.0 |
| 7K | - | 4.60 | 39.63 | 191.18 | −134.1 |
| 7O | 12.7 | 6.18 | 83.93 | 165.48 | −164.0 |
| 8K | - | 4.59 | 39.61 | 191.39 | −134.3 |
| 8O | 12.7 | 6.19 | 83.97 | 165.46 | −164.0 |
| 9K | - | 4.61 | 39.71 | 191.20 | −134.0 |
| 9O | 12.7 | 6.20 | 84.44 | 165.37 | −163.3 |
| - | - | 83.05 | 162.42 | - | |
| 10K | - | 4.67 | j | j | j |
| 10O | 12.8 | 6.34 | 86.47 | 164.80 | −160.6 |
| - | - | 84.70 | 163.16 | - | |
| 11K | - | 4.74 | j | j | j |
| 11O | k | 6.43 | 86.47 | 165.29 | −163.9 l |
| [K] (%) a | |
|---|---|
| 1 | 94.5 (87.0) b |
| 2 | 77.5 (83.0) c |
| 3 | 71.0 (72.0) c; 56 d,e[10] |
| 4 | 59.0 (57.5) c |
| 5 | 55.5; 50 d,e[10,13]; 51.5 [20]; 20 d,f[12] |
| 6 | 53.0 (50.0) c |
| 7 | 48.0; 33 d,e[10] |
| 8 | 50.5 |
| 9 | 45.5 |
| 10 | 29.5 |
| 11 | 3.5 |
| O12-H12 or N3-H3 | H12···N3 or H12···O1 | H10···H18 | C14C13C11O12 C18C13C11O12 | |
|---|---|---|---|---|
| 1K | - | - | 2.34 2.38 b | −179.97 −0.20 |
| 1Oa | 1.00 | 1.76 | 2.14 | 164.24 −15.76 |
| 1E | 1.03 | 1.79 | 2.04 | 173.70 −5.56 |
| 5K | - | - | 2.34 2.36 b | 179.48 −0.41 |
| 5Oa | 1.00 | 1.76 | 2.20 | 157.31 −22.36 |
| 5O′a | 0.98 | 1.87 | 2.19 | 154.73 −24.91 |
| 5E | 1.04 | 1.80 | 2.14 | 158.62 −20.62 |
| 10K | - | - | 2.35 2.37 b | 179.56 −0.33 |
| 10Oa | 1.00 | 1.75 | 2.14 | 163.19 −16.25 |
| 10E | 1.03 | 1.80 | 2.13 | 160.00 −18.75 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Skotnicka, A.; Kolehmainen, E.; Czeleń, P.; Valkonen, A.; Gawinecki, R. Synthesis and Structural Characterization of Substituted 2-Phenacylbenzoxazoles. Int. J. Mol. Sci. 2013, 14, 4444-4460. https://doi.org/10.3390/ijms14034444
Skotnicka A, Kolehmainen E, Czeleń P, Valkonen A, Gawinecki R. Synthesis and Structural Characterization of Substituted 2-Phenacylbenzoxazoles. International Journal of Molecular Sciences. 2013; 14(3):4444-4460. https://doi.org/10.3390/ijms14034444
Chicago/Turabian StyleSkotnicka, Agnieszka, Erkki Kolehmainen, Przemysław Czeleń, Arto Valkonen, and Ryszard Gawinecki. 2013. "Synthesis and Structural Characterization of Substituted 2-Phenacylbenzoxazoles" International Journal of Molecular Sciences 14, no. 3: 4444-4460. https://doi.org/10.3390/ijms14034444
APA StyleSkotnicka, A., Kolehmainen, E., Czeleń, P., Valkonen, A., & Gawinecki, R. (2013). Synthesis and Structural Characterization of Substituted 2-Phenacylbenzoxazoles. International Journal of Molecular Sciences, 14(3), 4444-4460. https://doi.org/10.3390/ijms14034444