Inhibition of Poly(ADP-Ribose) Polymerase Enhances Radiochemosensitivity in Cancers Proficient in DNA Double-Strand Break Repair

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
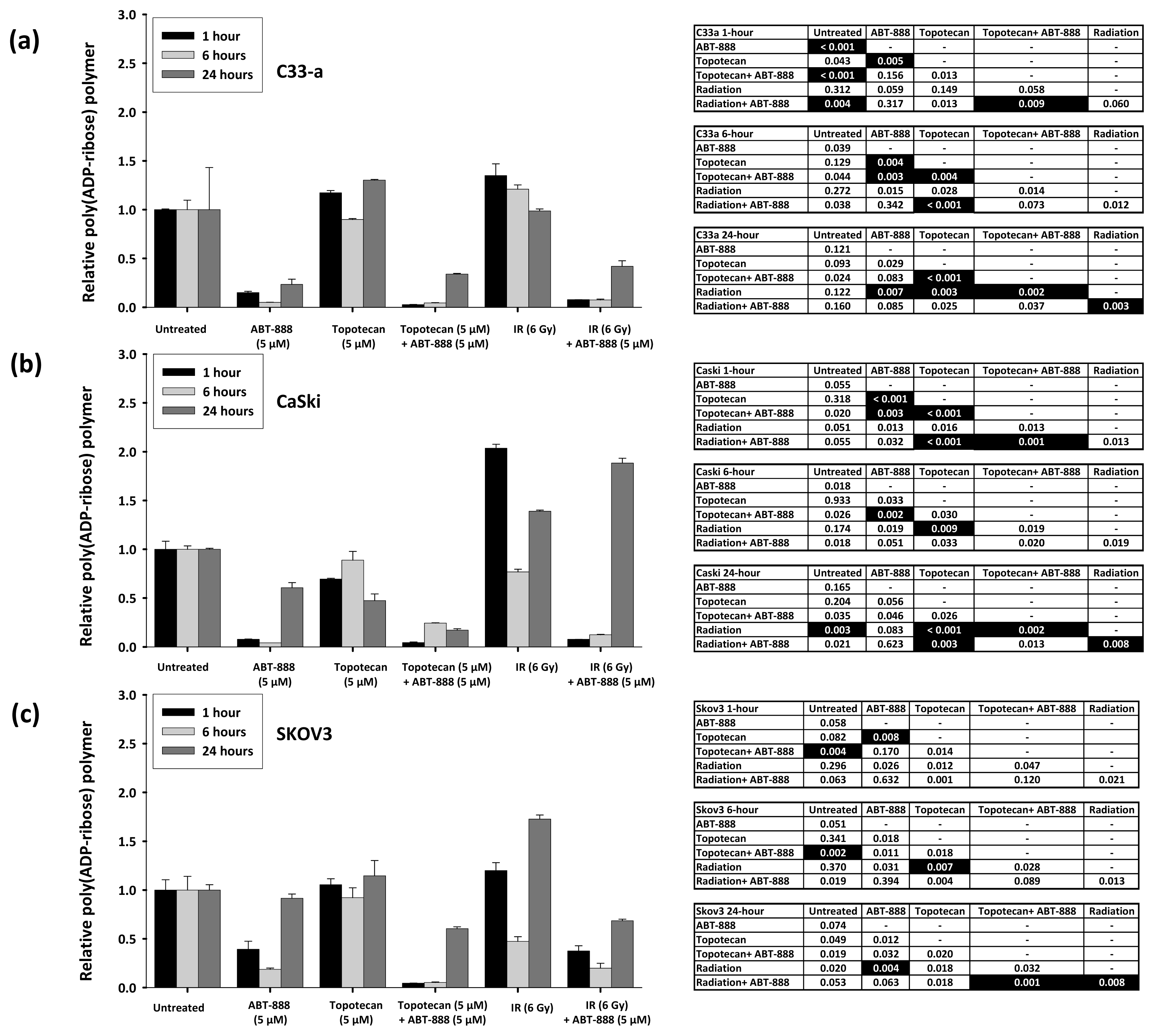
2.1. Poly(ADP-Ribose) Polymer Formation after Ionizing Radiation, Topotecan, and ABT-888 Exposure
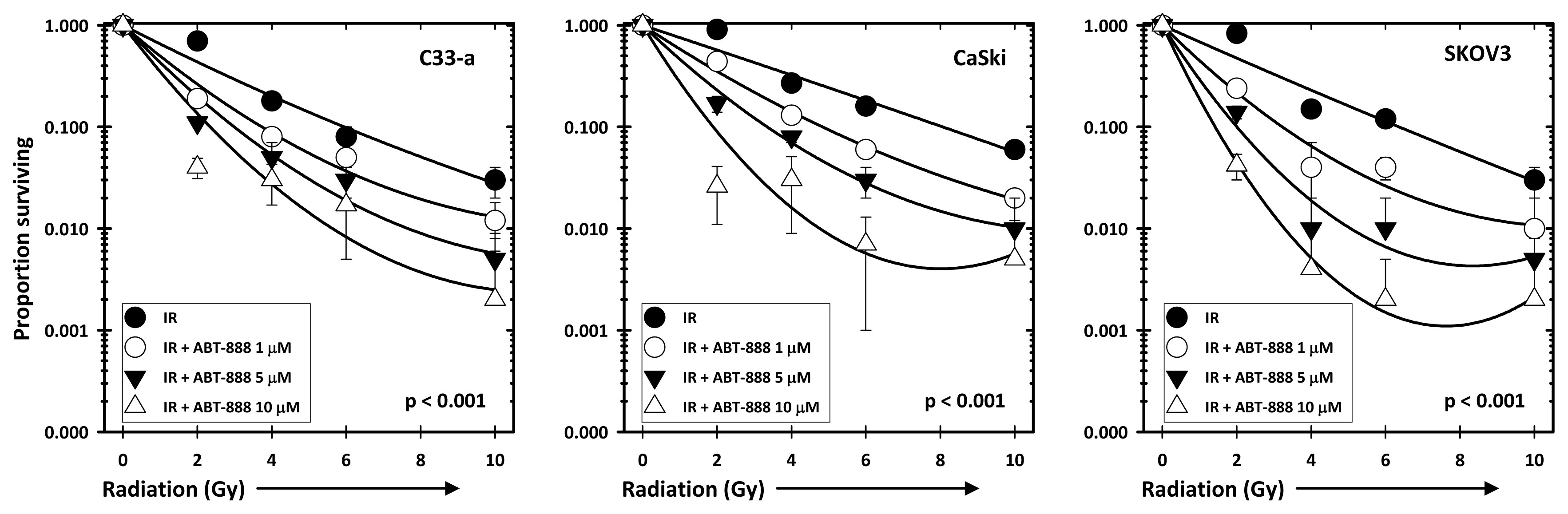
2.2. Radiochemosensitivity Is Enhanced by PARP Inhibition in Cells Proficient in DNA Damage Repair
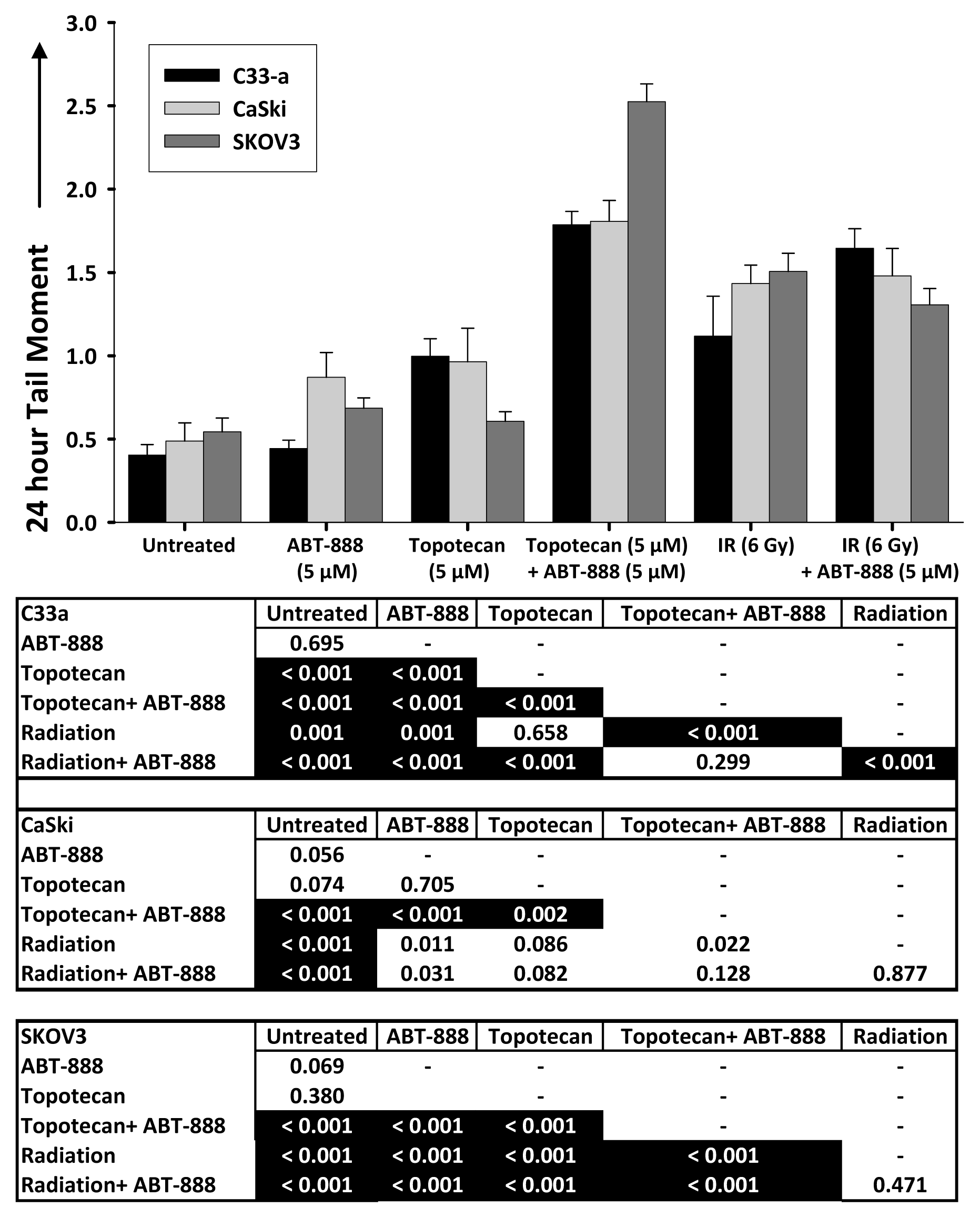
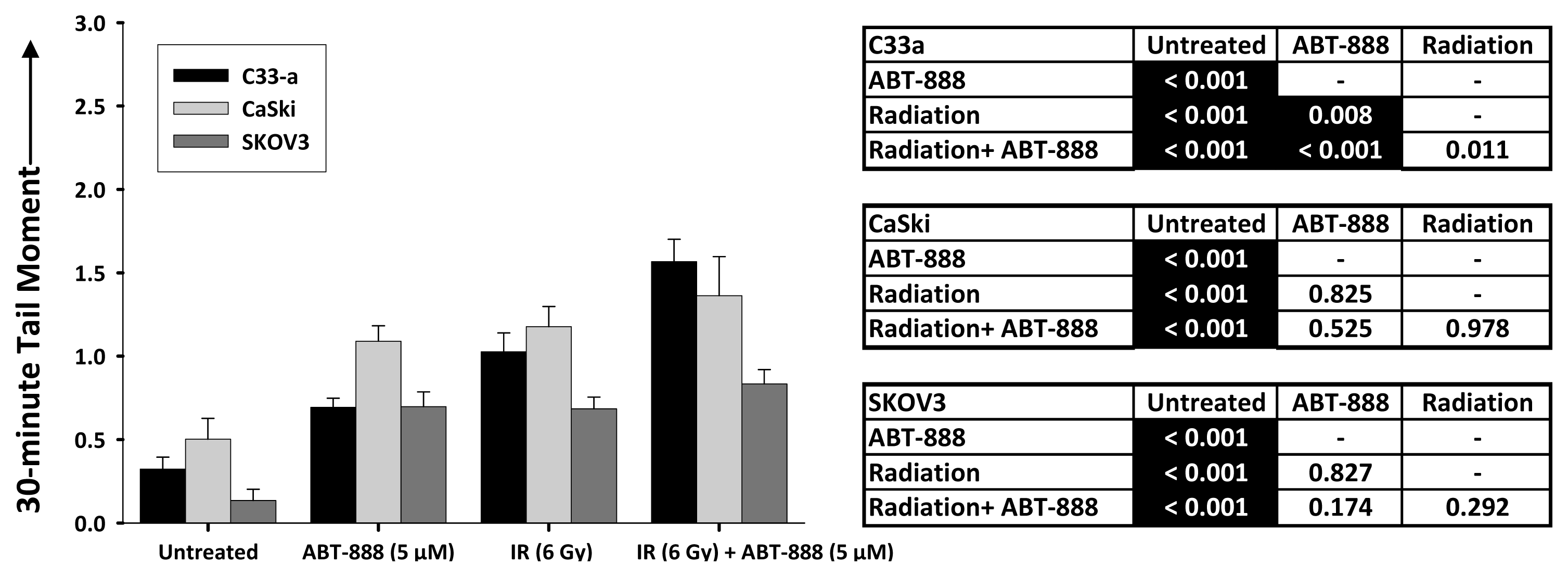
2.3. DNA Double-Strand Break Resolution after Radiation or Topotecan Is Delayed by PARP Inhibition
3. Experimental Section
3.1. Cell Cultures and Chemicals
3.2. Radiation and Drug Treatments
3.3. Clonogenic Survival Assays
3.4. Poly(ADP-Ribose) Polymerase Activity Assay
3.5. DNA Damage (γH2AX) Cytometry
3.6. Neutral Single Cell Electrophoresis (Comet) Assays
3.7. Statistical Methods
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Staker, B.; Hjerrild, K.; Feese, M.; Behnke, C.; Burgin, A.J.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar]
- Eklund, H.; Uhlin, U.; Farnegardh, M.; Logan, D.T.; Nordlund, P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol 2001, 77, 177–268. [Google Scholar]
- Kunos, C.; Chiu, S.; Pink, J.; Kinsella, T. Modulating radiation resistance by inhibiting ribonucleotide reductase in cancers with virally or mutationally silenced p53 protein. Radiat. Res 2009, 172, 666–676. [Google Scholar]
- Kunos, C.A.; Radivoyevitch, T.; Pink, J.; Chiu, S.M.; Stefan, T.; Jacobberger, J.; Kinsella, T.J. Ribonucleotide reductase inhibition enhances chemoradiosensitivity of human cervical cancers. Radiat. Res 2010, 174, 574–581. [Google Scholar]
- Kunos, C.; Ferris, G.; Pyatka, N.; Pink, J.; Radivoyevitch, T. Deoxynucleoside salvage facilitates DNA repair during ribonucleotide reductase blockade in human cervical cancers. Radiat. Res 2011, 176, 425–433. [Google Scholar]
- Kunos, C.; Radivoyevitch, T. Molecular strategies of deoxynucleotide triphosphate supply inhibition used in the treatment of gynecologic malignancies. Gynecol. Obstet 2011, S4, 1–5. [Google Scholar]
- D’Armours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J 1999, 342, 249–268. [Google Scholar]
- Chalmers, A. Poly(ADP-ribose) polymerase-1 and ionizing radiation: Sensor, signaller, and therapeutic target. Clin. Oncol 2004, 16, 29–39. [Google Scholar]
- Ame, J.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar]
- Chambon, P.; Weill, J.; Mandel, P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun 1963, 11, 39–43. [Google Scholar]
- Dantzer, F.; de la Rubia, G.; Menissier-de Murcia, J.; Hostomsky, Z.; De Murcia, G.; Schreiber, V. Base excision repair is inhibited in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol 1998, 18, 3563–3571. [Google Scholar]
- Mackey, Z.B.; Niedergang, C.; Murcia, J.M.; Leppard, J.; Au, K.; Chen, J.; de Murcia, G.; Tomkinson, A.E. DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly(ADP-ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J. Biol. Chem 1999, 374, 21679–21687. [Google Scholar]
- Fernet, M.; Ponette, V.; Deniaud-Alexandre, E.; Ménissier-De Murcia, J.; de Murcia, G.; Giocanti, N.; Megnin-Chanet, F.; Favaudon, V. Poly(ADP-ribose) polymerase, a major determinant of early cell response to ionizing radiation. Int. J. Radiat. Oncol. Biol 2000, 76, 1621–1629. [Google Scholar]
- Amé, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; de Murcia, G. PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem 1999, 274, 17860–17868. [Google Scholar]
- Schreiber, V.; Amé, J.C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision repair in association with PARP-1 and XRCC1. J. Biol. Chem 2002, 277, 23028–23036. [Google Scholar]
- Donawho, C.; Luo, Y.; Luo, Y.; Penning, T.; Bauch, J.; Bouska, J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT; -888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. In Clin. Cancer Res; 2007; Volume 13, pp. 2728–2737. [Google Scholar]
- Fukushima, M.; Kuzuya, K.; Ota, K.; Ikai, K. Poly(ADP-ribose) synthesis in human cervical cancer cell-diagnostic cytological usefulness. Cancer Lett 1981, 14, 227–236. [Google Scholar]
- Zhang, Y.; Regairaz, M.; Seiler, J.; Agama, K.; Doroshow, J.; Pommier, Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res 2011, 39, 3607–3620. [Google Scholar]
- Crook, T.; Wrede, D.; Vousden, K. p53 point mutation in HPV negative human cervical carcinoma cell lines. Oncogene 1991, 6, 873–875. [Google Scholar]
- Patillo, R.; Hussa, R.; Story, M.; Ruckert, A.; Shalaby, M.; Mattingly, R. Tumor antigen and human chorionic gonadotropin in CaSki cells: A new epidermoid cervical cancer cell line. Science 1977, 196, 1456–1458. [Google Scholar]
- Delaney, C.A.; Wang, L.Z.; Kyle, S.; White, A.W.; Calvert, A.H.; Curtin, N.J.; Durkacz, B.W.; Hostomsky, Z.; Newell, D.R. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin. Cancer Res 2000, 6, 2860–2867. [Google Scholar]
- Löser, D.; Shibata, A.; Shibata, A.; Woodbine, L.; Jeggo, P.; Chalmers, A. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol. Cancer Ther 2010, 9, 1775–1787. [Google Scholar]
- Krishnakumar, R.; Kraus, W. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar]
- Hakansson, P.; Hofer, A.; Thelander, L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem 2006, 281, 7834–7841. [Google Scholar]
- Liu, S.; Coackley, C.; Krause, M.; Jalali, F.; Chan, N.; Bristow, R. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol 2008, 88, 258–268. [Google Scholar]
- Banath, J.; Macphail, S.; Olive, P. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res 2004, 64, 7144–7149. [Google Scholar]
- Radivoyevitch, T.; Taverna, P.; Schupp, J.; Kinsella, T.J. The linear-quadratic log-survival radiation dose response model: Confidence ellipses, drug-drug interactions, and brachytherapeutic gains. Med. Hypotheses Res 2004, 1, 23–28. [Google Scholar]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph Stat 1996, 5, 299–314. [Google Scholar]
- Patel, A.G.; Flatten, K.S.; Schneider, P.A.; Dai, NT; McDonald, J.S.; Poirier, G.G.; Kaufmann, S.H. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012, 287, 4198–4210. [Google Scholar]
- D’Onofrio, G.; Tramontano, F.; Dorio, A.S.; Muzi, A.; Maselli, V.; Fulgione, D.; Graziani, G.; Malanga, M.; Quesada, P. Poly(ADP-ribose) polymerase signaling of topoisomerase 1-dependent DNA damage in carcinoma cells. Biochem. Pharmacol 2011, 81, 194–202. [Google Scholar]
- Kummar, S.; Chen, A.; Ji, J.; Zhang, Y.; Reid, J.M.; Ames, M.; Jia, L.; Weil, M.; Speranza, G.; Murgo, A.J.; et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res 2011, 71, 5626–5634. [Google Scholar]

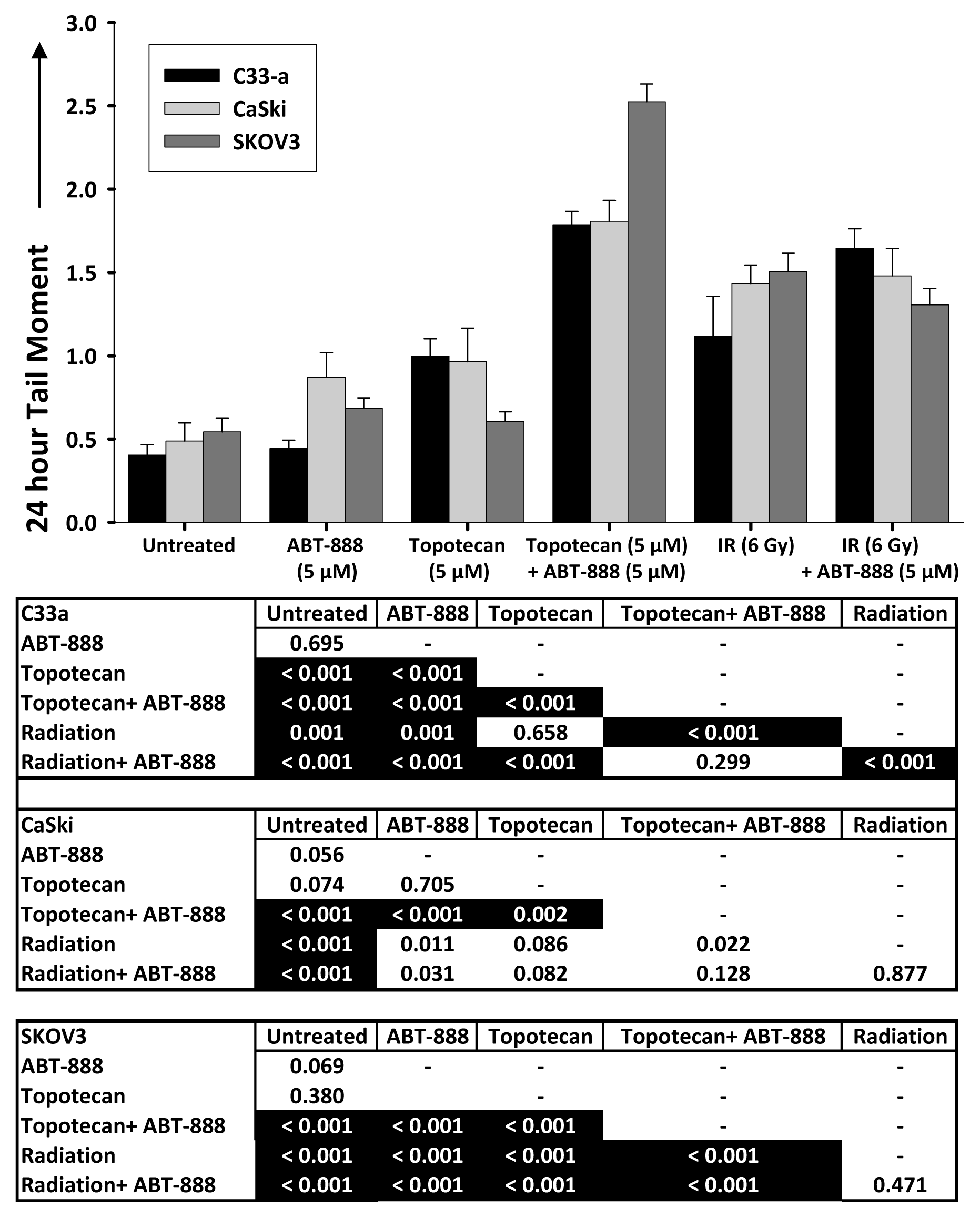
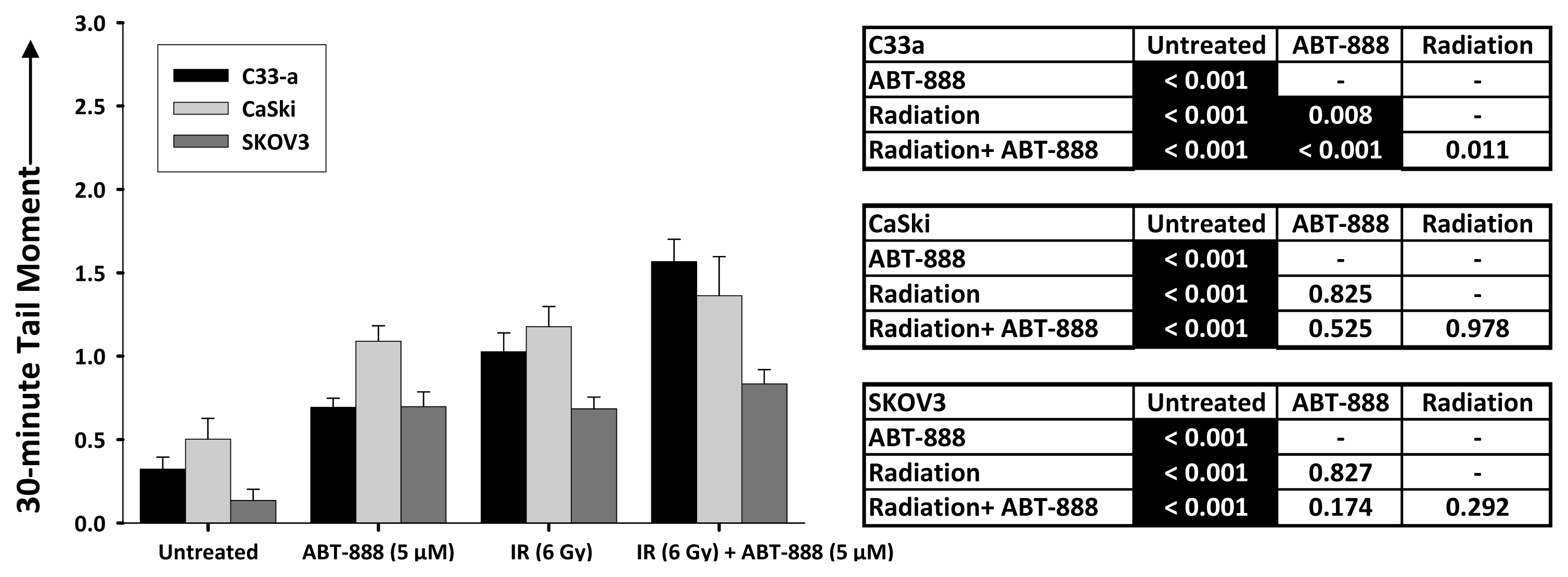
Supplementary Files
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shunkwiler, L.; Ferris, G.; Kunos, C. Inhibition of Poly(ADP-Ribose) Polymerase Enhances Radiochemosensitivity in Cancers Proficient in DNA Double-Strand Break Repair. Int. J. Mol. Sci. 2013, 14, 3773-3785. https://doi.org/10.3390/ijms14023773
Shunkwiler L, Ferris G, Kunos C. Inhibition of Poly(ADP-Ribose) Polymerase Enhances Radiochemosensitivity in Cancers Proficient in DNA Double-Strand Break Repair. International Journal of Molecular Sciences. 2013; 14(2):3773-3785. https://doi.org/10.3390/ijms14023773
Chicago/Turabian StyleShunkwiler, Lauren, Gina Ferris, and Charles Kunos. 2013. "Inhibition of Poly(ADP-Ribose) Polymerase Enhances Radiochemosensitivity in Cancers Proficient in DNA Double-Strand Break Repair" International Journal of Molecular Sciences 14, no. 2: 3773-3785. https://doi.org/10.3390/ijms14023773
APA StyleShunkwiler, L., Ferris, G., & Kunos, C. (2013). Inhibition of Poly(ADP-Ribose) Polymerase Enhances Radiochemosensitivity in Cancers Proficient in DNA Double-Strand Break Repair. International Journal of Molecular Sciences, 14(2), 3773-3785. https://doi.org/10.3390/ijms14023773