Protein-Phospholipid Interactions in Nonclassical Protein Secretion: Problem and Methods of Study

{kind=link}
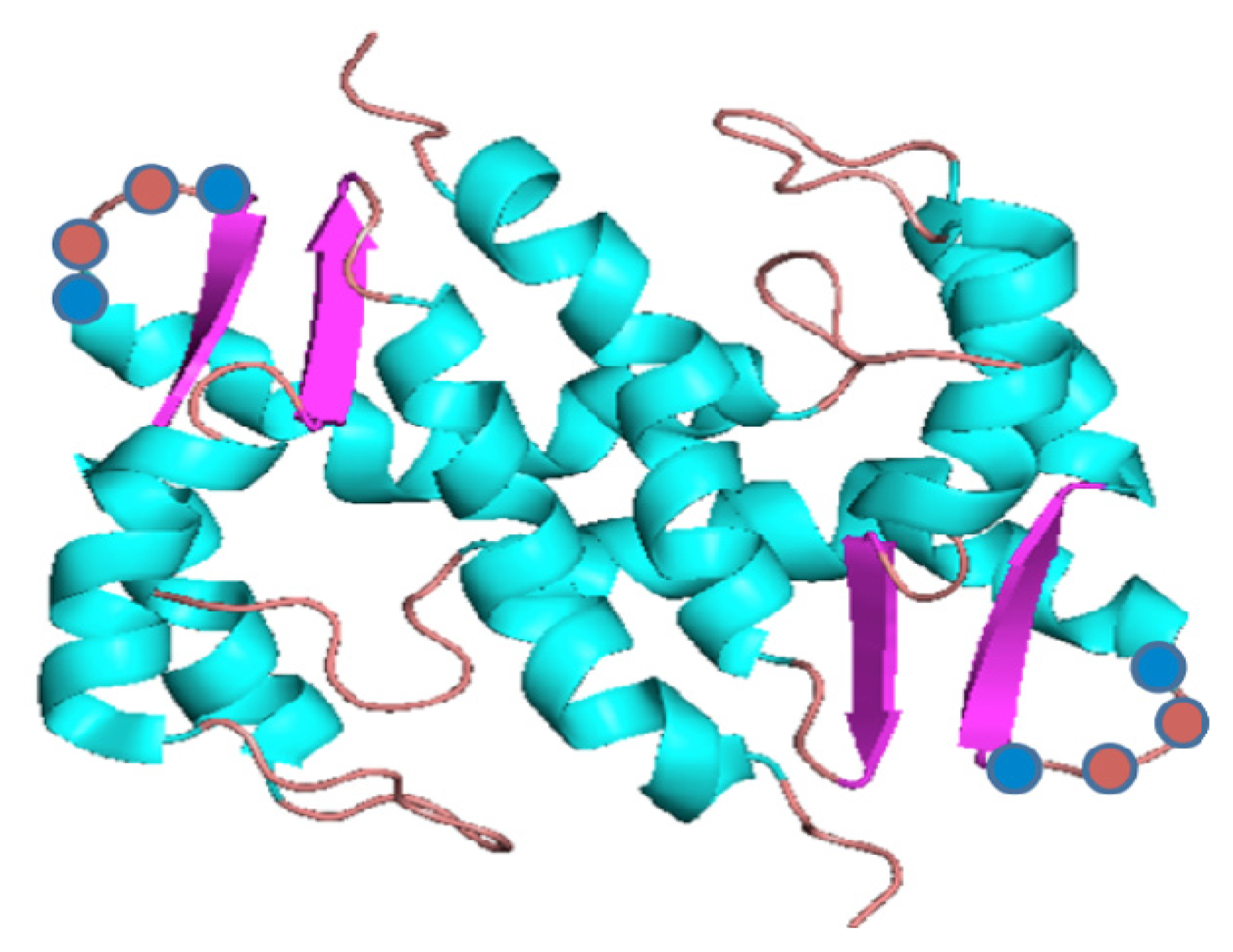{kind=link}
{kind=link}
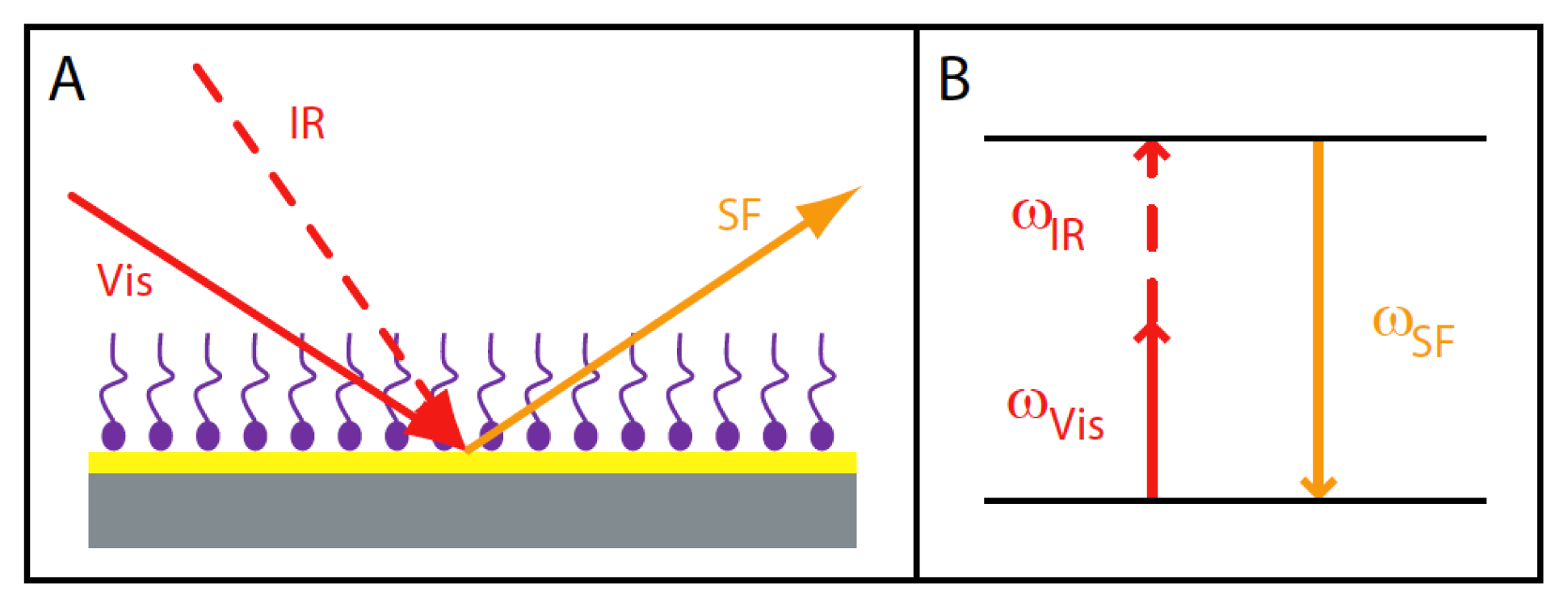{kind=link}
{kind=link}
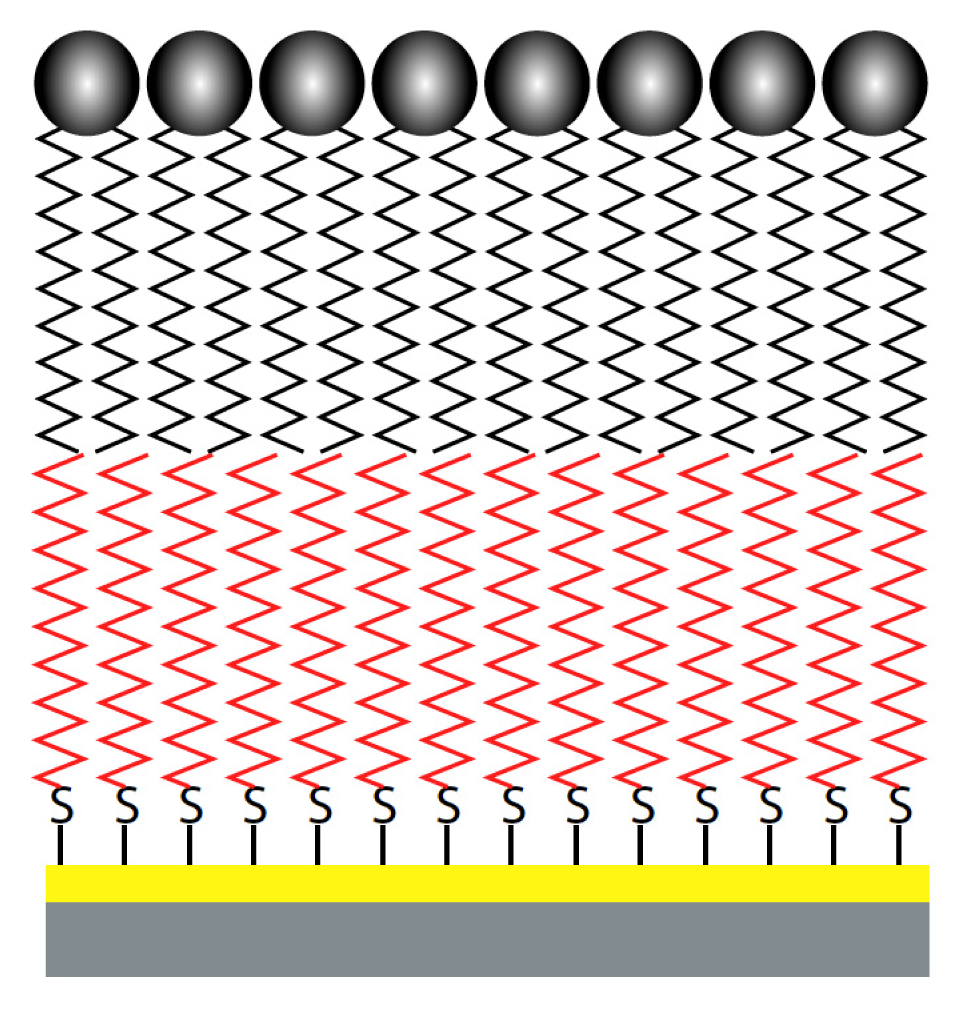{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Determinants of the Nonclassical Export of FGF1
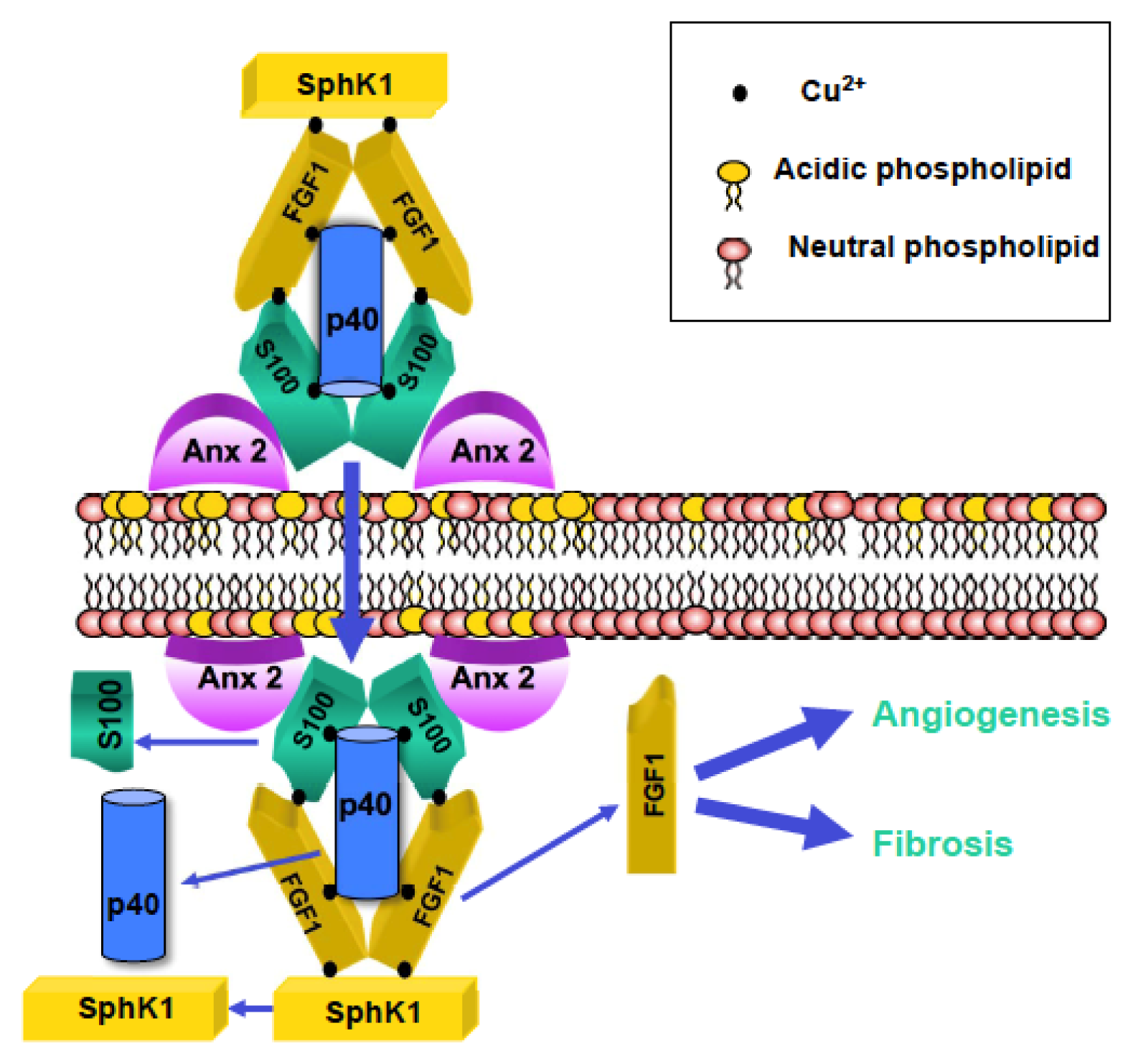
3. Role of Cu2+ in the Organization of the FGF1 MRC
4. Understanding the Formation of FGF1 MRC
5. Phospholipids and Nonclassically Released Proteins
6. Determining the Lipid Binding Domains of FGF1 MRC Components
7. Methods to Study the Interactions of Nonclassically Released Proteins with Membranes
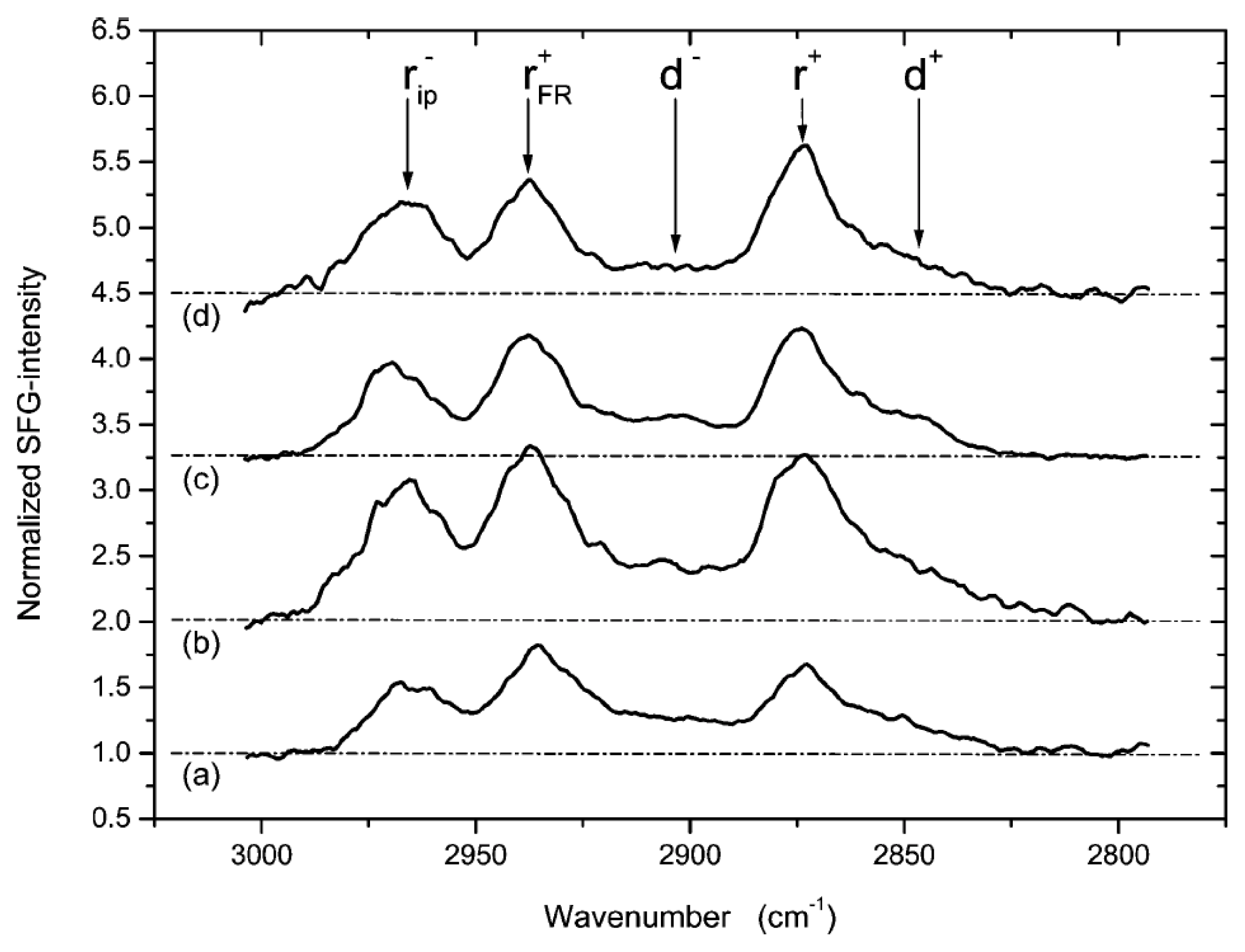
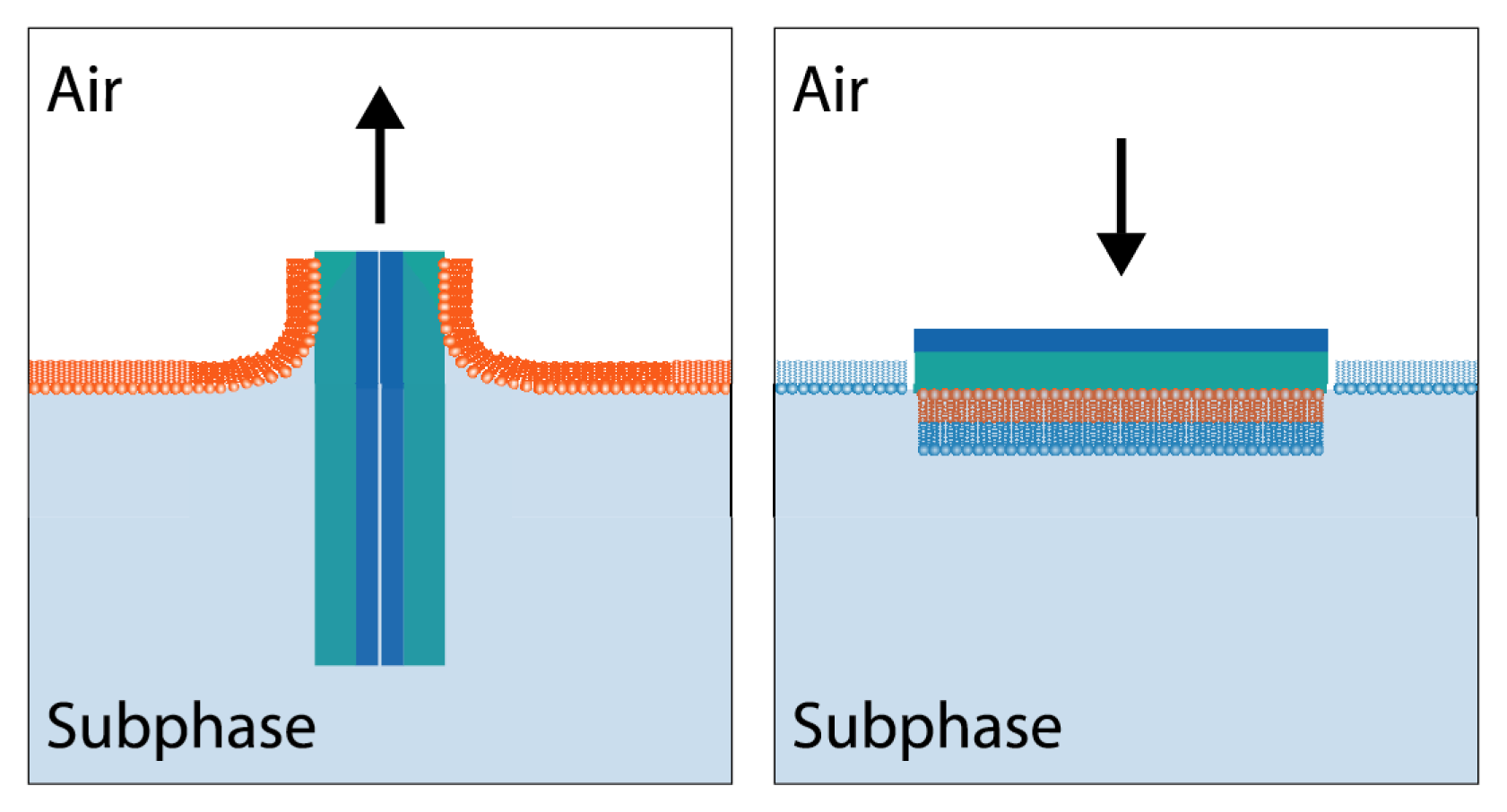
7.1. Sum Frequency Generation Vibrational Spectroscopy
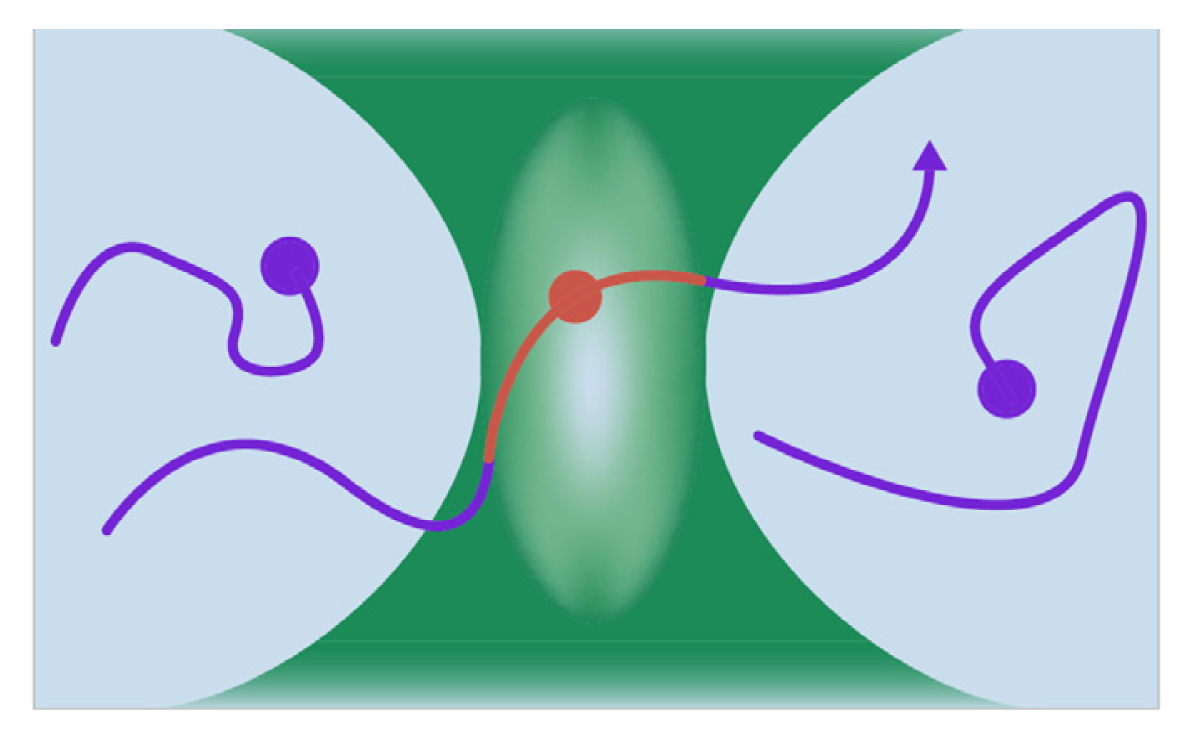
7.2. Fluorescence Correlation Spectroscopy
8. Model Membrane Systems to Study the Nonclassical Protein Release
8.1. Vesicles and Liposomes
8.2. Black Lipid Membranes
8.3. Hybrid Bilayer Membranes
8.4. Solid Supported Membranes
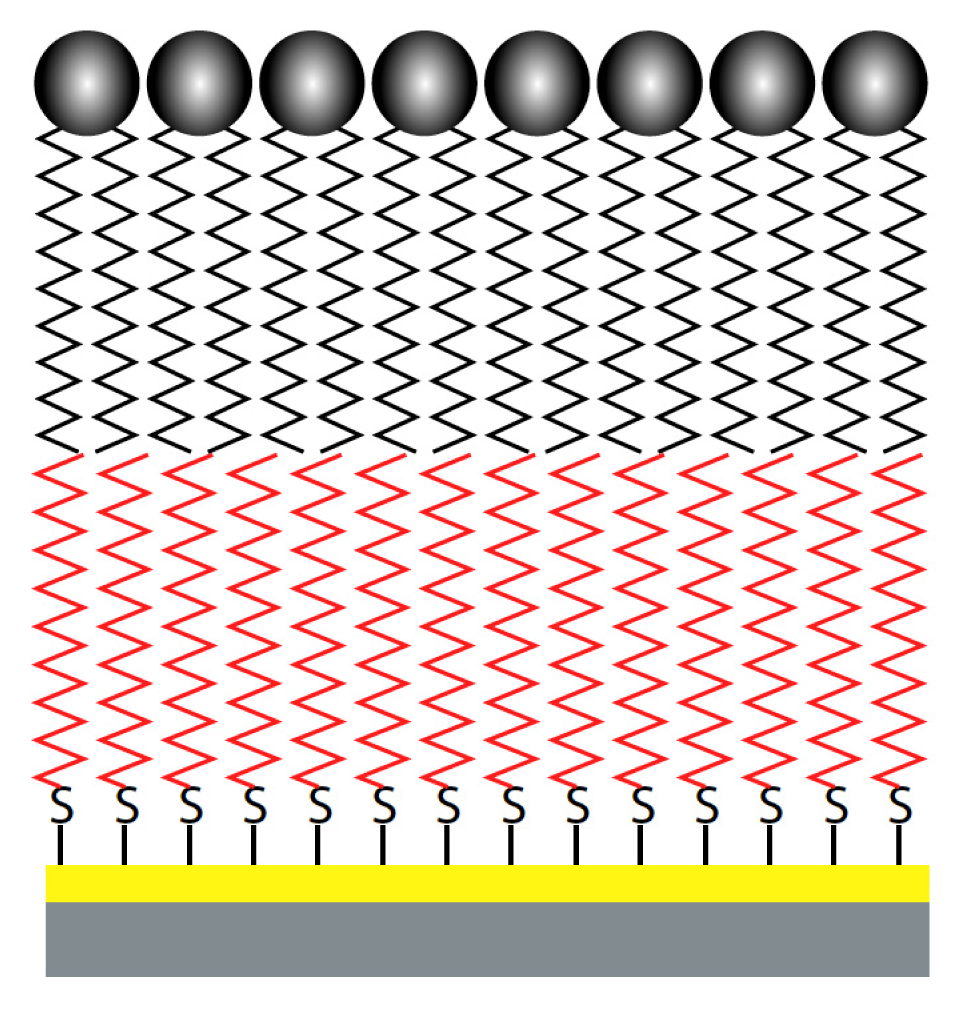
8.5. Cushioned Membranes
9. Conclusions
Acknowledgements
Abbreviations
| Anx 2 | annexin 2 |
| BLM | black lipid membrane |
| ER | endoplasmic reticulum |
| FCS | fluorescence correlation spectroscopy |
| FGF1 | fibroblast growth factor 1 |
| HBM | hybrid bilayer membrane |
| IL1 | interleukin 1 |
| ITC | isothermal titration calorimetry |
| MRC | multiprotein release complex |
| PC | phosphatidylcholine |
| PI | phosphatidylinositol |
| PG | phosphatidylglycerol |
| PL | phospholipid |
| PS | phosphatidylserine |
| SAM | self-assembled monolayers |
| SFG | sum frequency generation |
| SFS | sum frequency generation vibrational spectroscopy |
| SphK1 | sphingosine kinase 1 |
| Syt1 | synaptotagmin |
Conflict of Interest
References
- Blobel, G. Protein targeting (Nobel lecture). Chembiochem 2000, 1, 86–102. [Google Scholar]
- Prudovsky, I.; Tarantini, F.; Landriscina, M.; Neivandt, D.; Soldi, R.; Kirov, A.; Small, D.; Kathir, K.M.; Rajalingam, D.; Kumar, T.K. Secretion without Golgi. J. Cell Biochem 2008, 103, 1327–1343. [Google Scholar]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol 2009, 10, 148–155. [Google Scholar]
- Jackson, A.; Friedman, S.; Zhan, X.; Engleka, K.A.; Forough, R.; Maciag, T. Heat shock induces the release of fibroblast growth factor 1 from NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1992, 89, 10691–10695. [Google Scholar]
- Mignatti, P.; Morimoto, T.; Rifkin, D.B. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J. Cell Physiol 1992, 151, 81–93. [Google Scholar]
- Florkiewicz, R.Z.; Majack, R.A.; Buechler, R.D.; Florkiewicz, E. Quantitative export of FGF-2 occurs through an alternative, energy-dependent, non-ER/Golgi pathway. J. Cell Physiol 1995, 162, 388–399. [Google Scholar]
- Rubartelli, A.; Cozzolino, F.; Talio, M.; Sitia, R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J 1990, 9, 1503–1510. [Google Scholar]
- Andrei, C.; Dazzi, C.; Lotti, L.; Torrisi, M.R.; Chimini, G.; Rubartelli, A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell 1999, 10, 1463–1475. [Google Scholar]
- Tarantini, F.; Micucci, I.; Bellum, S.; Landriscina, M.; Garfinkel, S.; Prudovsky, I.; Maciag, T. The precursor but not the mature form of IL1alpha blocks the release of FGF1 in response to heat shock. J. Biol. Chem 2001, 276, 5147–5151. [Google Scholar]
- Mandinova, A.; Soldi, R.; Graziani, I.; Bagala, C.; Bellum, S.; Landriscina, M.; Tarantini, F.; Prudovsky, I.; Maciag, T. S100A13 mediates the copper-dependent stress-induced release of IL-1α from both human U937 and murine NIH 3T3 cells. J. Cell Sci 2003, 116, 2687–2696. [Google Scholar]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem 2012, 287, 6941–6948. [Google Scholar]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 2002, 3, 995–1001. [Google Scholar]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003, 22, 5551–5560. [Google Scholar]
- Shalak, V.; Kaminska, M.; Mitnacht-Kraus, R.; Vandenabeele, P.; Clauss, M.; Mirande, M. The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J. Biol. Chem 2001, 276, 23769–23776. [Google Scholar]
- Lue, H.; Kleemann, R.; Calandra, T.; Roger, T.; Bernhagen, J. Macrophage migration inhibitory factor (MIF): Mechanisms of action and role in disease. Microbes Infect 2002, 4, 449–460. [Google Scholar]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem 2002, 277, 6667–6675. [Google Scholar]
- Zemskov, E.A.; Mikhailenko, I.; Hsia, R.C.; Zaritskaya, L.; Belkin, A.M. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS One 2011, 6, e19414. [Google Scholar]
- Chapman, L.P.; Epton, M.J.; Buckingham, J.C.; Morris, J.F.; Christian, H.C. Evidence for a role of the adenosine 5′-triphosphate-binding cassette transporter A1 in the externalization of annexin I from pituitary folliculo-stellate cells. Endocrinology 2003, 144, 1062–1073. [Google Scholar]
- Peterson, E.A.; Sutherland, M.R.; Nesheim, M.E.; Pryzdial, E.L. Thrombin induces endothelial cell-surface exposure of the plasminogen receptor annexin 2. J. Cell Sci 2003, 116, 2399–2408. [Google Scholar]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J. Biol. Chem 2004, 279, 43411–43418. [Google Scholar]
- Sango, K.; Tokashiki, A.; Ajiki, K.; Horie, M.; Kawano, H.; Watabe, K.; Horie, H.; Kadoya, T. Synthesis, localization and externalization of galectin-1 in mature dorsal root ganglion neurons and Schwann cells. Eur. J. Neurosci 2004, 19, 55–64. [Google Scholar]
- Mehul, B.; Hughes, R.C. Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J. Cell Sci 1997, 110, 1169–1178. [Google Scholar]
- Keryer-Bibens, C.; Pioche-Durieu, C.; Villemant, C.; Souquere, S.; Nishi, N.; Hirashima, M.; Middeldorp, J.; Busson, P. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer 2006, 6, 283. [Google Scholar]
- Landriscina, M.; Soldi, R.; Bagala, C.; Micucci, I.; Bellum, S.; Tarantini, F.; Prudovsky, I.; Maciag, T. S100A13 participates in the release of fibroblast growth factor 1 in response to heat shock in vitro. J. Biol. Chem 2001, 276, 22544–22552. [Google Scholar]
- Davey, G.E.; Murmann, P.; Heizmann, C.W. Intracellular Ca2+ and Zn2+ levels regulate the alternative cell density-dependent secretion of S100B in human glioblastoma cells. J. Biol. Chem 2001, 276, 30819–30826. [Google Scholar]
- Flatmark, K.; Maelandsmo, G.M.; Mikalsen, S.O.; Nustad, K.; Varaas, T.; Rasmussen, H.; Meling, G.I.; Fodstad, O.; Paus, E. Immunofluorometric assay for the metastasis-related protein S100A4: Release of S100A4 from normal blood cells prohibits the use of S100A4 as a tumor marker in plasma and serum. Tumour Biol 2004, 25, 31–40. [Google Scholar]
- Chlebova, K.; Bryja, V.; Dvorak, P.; Kozubik, A.; Wilcox, W.R.; Krejci, P. High molecular weight FGF2: The biology of a nuclear growth factor. Cell. Mol. Life Sci 2009, 66, 225–235. [Google Scholar]
- Johnson, H.M.; Subramaniam, P.S.; Olsnes, S.; Jans, D.A. Trafficking and signaling pathways of nuclear localizing protein ligands and their receptors. Bioessays 2004, 26, 993–1004. [Google Scholar]
- Davidson, J.; Milton, A.S.; Rotondo, D. A study of the pyrogenic actions of interleukin-1 α and interleukin-1 β: Interactions with a steroidal and a non-steroidal anti-inflammatory agent. Br. J. Pharmacol 1990, 100, 542–546. [Google Scholar]
- Tomaszewski, M.; Charchar, F.J.; Lynch, M.D.; Padmanabhan, S.; Wang, W.Y.; Miller, W.H.; Grzeszczak, W.; Maric, C.; Zukowska-Szczechowska, E.; Dominiczak, A.F. Fibroblast growth factor 1 gene and hypertension: From the quantitative trait locus to positional analysis. Circulation 2007, 116, 1915–1924. [Google Scholar]
- Coulier, F.; Pontarotti, P.; Roubin, R.; Hartung, H.; Goldfarb, M.; Birnbaum, D. Of worms and men: An evolutionary perspective on the fibroblast growth factor (FGF) and FGF receptor families. J. Mol. Evol 1997, 44, 43–56. [Google Scholar]
- Itoh, N.; Ornitz, D.M. Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn 2008, 237, 18–27. [Google Scholar]
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targ 2009, 9, 639–651. [Google Scholar]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol 2010, 188, 527–536. [Google Scholar]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res 2006, 66, 4795–4801. [Google Scholar]
- Mambula, S.S.; Calderwood, S.K. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol 2006, 177, 7849–7857. [Google Scholar]
- Schäfer, T.; Zentgraf, H.; Zehe, C.; Brügger, B.; Bernhagen, J.; Nickel, W. Unconventional secretion of fibroblast growth factor 2 is mediated by direct translocation across the plasma membrane of mammalian cells. J. Biol. Chem 2004, 279, 6244–6251. [Google Scholar]
- Graziani, I.; Doyle, A.; Sterling, S.; Kirov, A.; Tarantini, F.; Landriscina, M.; Kumar, T.K.; Neivandt, D.; Prudovsky, I. Protein folding does not prevent the nonclassical export of FGF1 and S100A13. Biochem. Biophys. Res. Commun 2009, 381, 350–354. [Google Scholar]
- Zehe, C.; Engling, A.; Wegehingel, S.; Schafer, T.; Nickel, W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc. Natl. Acad. Sci. USA 2006, 103, 15479–15484. [Google Scholar]
- Kirov, A.; Al-Hashimi, H.; Solomon, P.; Mazur, C.; Thorpe, P.E.; Sims, P.J.; Tarantini, F.; Kumar, T.K.; Prudovsky, I. Phosphatidylserine externalization and membrane blebbing are involved in the nonclassical export of FGF1. J. Cell Biochem 2012, 113, 956–966. [Google Scholar]
- Friesel, R.; Maciag, T. Fibroblast growth factor prototype release and fibroblast growth factor receptor signaling. Thromb. Haemost 1999, 82, 748–754. [Google Scholar]
- Zakrzewska, M.; Marcinkowska, E.; Wiedlocha, A. FGF-1: From biology through engineering to potential medical applications. Crit. Rev. Clin. Lab. Sci 2008, 45, 91–135. [Google Scholar]
- Kirov, A.; Duarte, M.; Guay, J.; Karolak, M.; Yan, C.; Oxburgh, L.; Prudovsky, I. Transgenic expression of nonclassically secreted FGF suppresses kidney repair. PLoS One 2012, 7, e36485. [Google Scholar]
- Mouta Carreira, C.; Landriscina, M.; Bellum, S.; Prudovsky, I.; Maciag, T. The comparative release of FGF1 by hypoxia and temperature stress. Growth Factors 2001, 18, 277–285. [Google Scholar]
- Ananyeva, N.M.; Tijurmin, A.V.; Berliner, J.A.; Chisolm, G.M.; Liau, G.; Winkles, J.A.; Haudenschild, C.C. Oxidized LDL mediates the release of fibroblast growth factor-1. Arterioscler. Thromb. Vasc. Biol 1997, 17, 445–453. [Google Scholar]
- Small, D.; Kovalenko, D.; Soldi, R.; Mandinova, A.; Kolev, V.; Trifonova, R.; Bagala, C.; Kacer, D.; Battelli, C.; Liaw, L.; et al. Notch activation suppresses fibroblast growth factor-dependent cellular transformation. J. Biol. Chem. 2003, 278, 16405–16413. [Google Scholar]
- Kacer, D.; McIntire, C.; Kirov, A.; Kany, E.; Roth, J.; Liaw, L.; Small, D.; Friesel, R.; Basilico, C.; Tarantini, F.; et al. Regulation of non-classical FGF1 release and FGF-dependent cell transformation by CBF1-mediated notch signaling. J. Cell Physiol. 2011, 226, 3064–3075. [Google Scholar]
- Duarte, M.; Kolev, V.; Soldi, R.; Kirov, A.; Graziani, I.; Oliveira, S.M.; Kacer, D.; Friesel, R.; Maciag, T.; Prudovsky, I. Thrombin induces rapid PAR1-mediated non-classical FGF1 release. Biochem. Biophys. Res. Commun 2006, 350, 604–609. [Google Scholar]
- Duarte, M.; Kolev, V.; Kacer, D.; Mouta-Bellum, C.; Soldi, R.; Graziani, I.; Kirov, A.; Friesel, R.; Liaw, L.; Small, D.; et al. Novel cross-talk between three cardiovascular regulators: Thrombin cleavage fragment of Jagged1 induces fibroblast growth factor 1 expression and release. Mol. Biol. Cell 2008, 19, 4863–4874. [Google Scholar]
- Ruf, W.; Mueller, B.M. Thrombin generation and the pathogenesis of cancer. Semin. Thromb. Hemost 2006, 32, 61–68. [Google Scholar]
- LaVallee, T.M.; Tarantini, F.; Gamble, S.; Carreira, C.M.; Jackson, A.; Maciag, T. Synaptotagmin-1 is required for fibroblast growth factor-1 release. J. Biol. Chem 1998, 273, 22217–22223. [Google Scholar]
- Soldi, R.; Mandinova, A.; Venkataraman, K.; Hla, T.; Vadas, M.; Pitson, S.; Duarte, M.; Graziani, I.; Kolev, V.; Kacer, D.; et al. Sphingosine kinase 1 is a critical component of the copper-dependent FGF1 export pathway. Exp. Cell Res. 2007, 313, 3308–3318. [Google Scholar]
- Tarantini, F.; Gamble, S.; Jackson, A.; Maciag, T. The cysteine residue responsible for the release of fibroblast growth factor-1 residues in a domain independent of the domain for phosphatidylserine binding. J. Biol. Chem 1995, 270, 29039–29042. [Google Scholar]
- Landriscina, M.; Bagala, C.; Mandinova, A.; Soldi, R.; Micucci, I.; Bellum, S.; Prudovsky, I.; Maciag, T. Copper induces the assembly of a multiprotein aggregate implicated in the release of fibroblast growth factor 1 in response to stress. J. Biol. Chem 2001, 276, 25549–25557. [Google Scholar]
- Cao, R.; Yan, B.; Yang, H.; Zu, X.; Wen, G.; Zhong, J. Effect of human S100A13 gene silencing on FGF-1 transportation in human endothelial cells. J. Formos. Med. Assoc 2010, 109, 632–640. [Google Scholar]
- Rescher, U.; Gerke, V. S100A10/p11: Family, friends and functions. Pflugers Arch 2008, 455, 575–582. [Google Scholar]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech 2003, 60, 540–551. [Google Scholar]
- Luk, E.; Jensen, L.T.; Culotta, V.C. The many highways for intracellular trafficking of metals. J. Biol. Inorg. Chem 2003, 8, 803–809. [Google Scholar]
- Kim, D.Y.; Bovet, L.; Maeshima, M.; Martinoia, E.; Lee, Y. The ABC transporter AtPDR8 is a cadmium extrusion pump conferring heavy metal resistance. Plant J 2007, 50, 207–218. [Google Scholar]
- Lopez, E.; Arce, C.; Oset-Gasque, M.J.; Canadas, S.; Gonzalez, M.P. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic. Biol. Med 2006, 40, 940–951. [Google Scholar]
- Pufahl, R.A.; Singer, C.P.; Peariso, K.L.; Lin, S.J.; Schmidt, P.J.; Fahrni, C.J.; Culotta, V.C.; Penner-Hahn, J.E.; O’Halloran, T.V. Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science 1997, 278, 853–856. [Google Scholar]
- Miras, R.; Morin, I.; Jacquin, O.; Cuillel, M.; Guillain, F.; Mintz, E. Interplay between glutathione, Atx1 and copper. 1. Copper(I) glutathionate induced dimerization of Atx1. J. Biol. Inorg. Chem 2008, 13, 195–205. [Google Scholar]
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr 2008, 88, 826S–829S. [Google Scholar]
- Shin, L.J.; Lo, J.C.; Yeh, K.C. Copper chaperone antioxidant protein1 is essential for copper homeostasis. Plant Physiol 2012, 159, 1099–1110. [Google Scholar]
- Banci, L.; Bertini, I.; Cefaro, C.; Ciofi-Baffoni, S.; Gallo, A. Functional role of two interhelical disulfide bonds in human Cox17 protein from a structural perspective. J. Biol. Chem 2011, 286, 34382–34390. [Google Scholar]
- Sivaraja, V.; Kumar, T.K.; Yu, C. Resonance assignments for mouse S100A13. J. Biomol. NMR 2005, 32, 257. [Google Scholar]
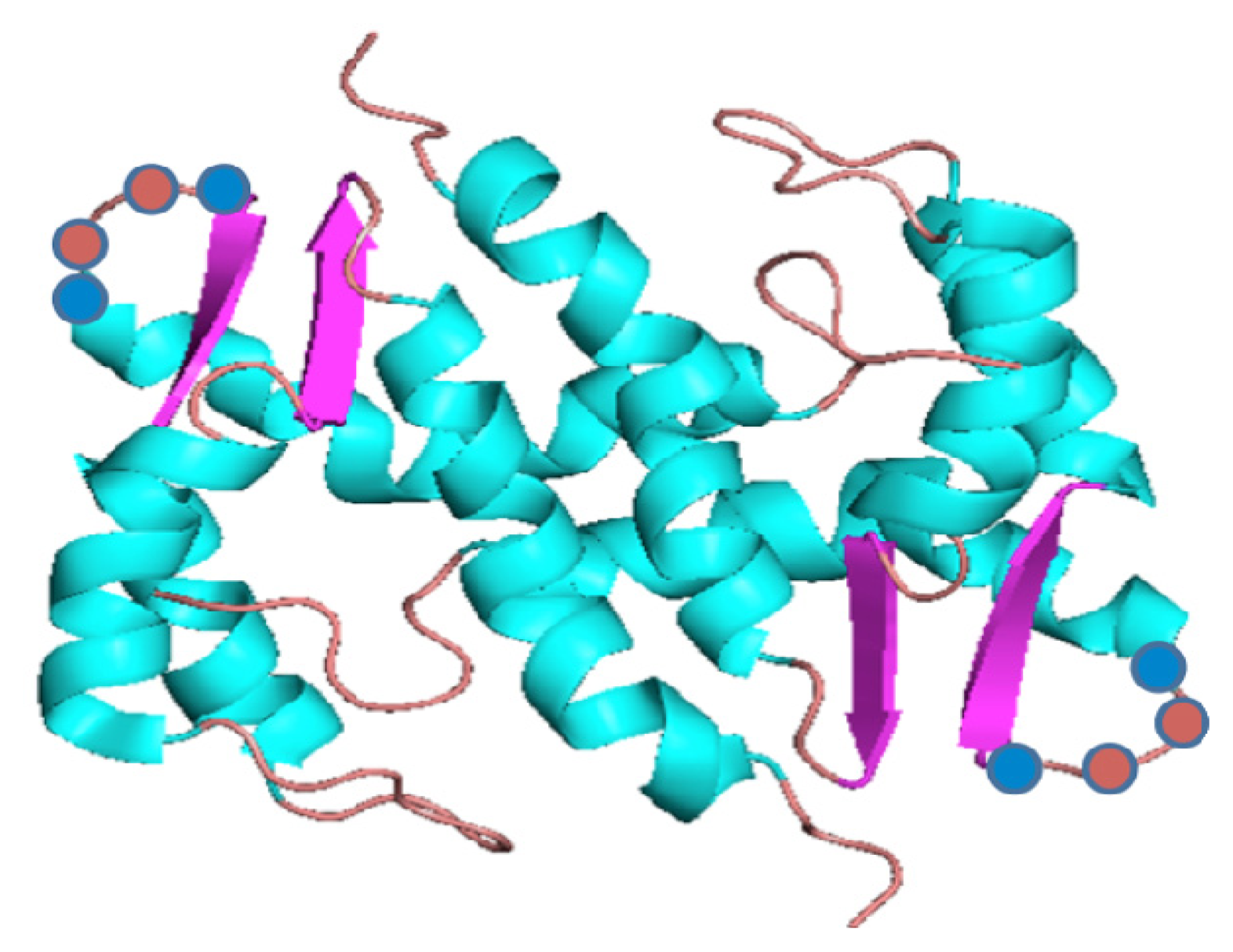
- Sivaraja, V.; Kumar, T.K.; Prudovsky, I.; Yu, C. Three-dimensional solution structure of a unique S100 protein. Biochem. Biophys. Res. Commun 2005, 335, 1140–1148. [Google Scholar]
- Sivaraja, V.; Kumar, T.K.; Rajalingam, D.; Graziani, I.; Prudovsky, I.; Yu, C. Copper binding affinity of S100A13, a key component of the FGF-1 nonclassical copper-dependent release complex. Biophys. J 2006, 91, 1832–1843. [Google Scholar]
- Yao, J.; Kwon, S.E.; Gaffaney, J.D.; Dunning, F.M.; Chapman, E.R. Uncoupling the roles of synaptotagmin I during endo- and exocytosis of synaptic vesicles. Nat. Neurosci 2012, 15, 243–249. [Google Scholar]
- Jahn, R.; Fasshauer, D. Molecular machines governing exocytosis of synaptic vesicles. Nature 2012, 490, 201–207. [Google Scholar]
- Rizo, J.; Sudhof, T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices-guilty as charged? Annu. Rev. Cell Dev. Biol 2012, 28, 279–308. [Google Scholar]
- Bagala, C.; Kolev, V.; Mandinova, A.; Soldi, R.; Mouta, C.; Graziani, I.; Prudovsky, I.; Maciag, T. The alternative translation of synaptotagmin 1 mediates the non-classical release of FGF1. Biochem. Biophys. Res. Commun 2003, 310, 1041–1047. [Google Scholar]
- Rajalingam, D.; Kumar, T.K.; Yu, C. The C2A domain of synaptotagmin exhibits a high binding affinity for copper: Implications in the formation of the multiprotein FGF release complex. Biochemistry 2005, 44, 14431–14442. [Google Scholar]
- Rajalingam, D.; Kumar, T.K.; Soldi, R.; Graziani, I.; Prudovsky, I.; Yu, C. Molecular mechanism of inhibition of nonclassical FGF-1 export. Biochemistry 2005, 44, 15472–15479. [Google Scholar]
- Rajalingam, D.; Graziani, I.; Prudovsky, I.; Yu, C.; Kumar, T.K. Relevance of partially structured states in the non-classical secretion of acidic fibroblast growth factor. Biochemistry 2007, 46, 9225–9238. [Google Scholar]
- Mohan, S.K.; Rani, S.G.; Yu, C. The heterohexameric complex structure, a component in the non-classical pathway for fibroblast growth factor 1 (FGF1) secretion. J. Biol. Chem 2010, 285, 15464–15475. [Google Scholar]
- Prudovsky, I.; Bagala, C.; Tarantini, F.; Mandinova, A.; Soldi, R.; Bellum, S.; Maciag, T. The intracellular translocation of the components of the fibroblast growth factor 1 release complex precedes their assembly prior to export. J. Cell Biol 2002, 158, 201–208. [Google Scholar]
- Pitson, S.M.; Xia, P.; Leclercq, T.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Wattenberg, B.W.; Vadas, M.A. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J. Exp. Med 2005, 201, 49–54. [Google Scholar]
- Pomorski, T.; Hrafnsdottir, S.; Devaux, P.; van Meer, G. Lipid distribution and transport across cellular membranes. Semin. Cell Dev. Biol 2001, 12, 139–148. [Google Scholar]
- Bevers, E.M.; Comfurius, P.; Dekkers, D.W.; Zwaal, R.F. Lipid translocation across the plasma membrane of mammalian cells. Biochim. Biophys. Acta 1999, 1439, 317–330. [Google Scholar]
- Sims, P.J.; Wiedmer, T. Unraveling the mysteries of phospholipid scrambling. Thromb. Haemost 2001, 86, 266–275. [Google Scholar]
- Marqueze, B.; Berton, F.; Seagar, M. Synaptotagmins in membrane traffic: Which vesicles do the tagmins tag? Biochimie 2000, 82, 409–420. [Google Scholar]
- Heizmann, C.W.; Fritz, G.; Schafer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci 2002, 7, d1356–d1368. [Google Scholar]
- Stahelin, R.V.; Hwang, J.H.; Kim, J.H.; Park, Z.Y.; Johnson, K.R.; Obeid, L.M.; Cho, W. The mechanism of membrane targeting of human sphingosine kinase 1. J. Biol. Chem 2005, 280, 43030–43038. [Google Scholar]
- Jost, M.; Weber, K.; Gerke, V. Annexin II contains two types of Ca(2+)-binding sites. Biochem. J 1994, 298, 553–559. [Google Scholar]
- Steringer, J.P.; Bleicken, S.; Andreas, H.; Zacherl, S.; Laussmann, M.; Temmerman, K.; Contreras, F.X.; Bharat, T.A.; Lechner, J.; Muller, H.M.; et al. PI(4,5)P2 Dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem. 2012, 287, 27659–27669. [Google Scholar]
- Mach, H.; Middaugh, C.R. Interaction of partially structured states of acidic fibroblast growth factor with phospholipid membranes. Biochemistry 1995, 34, 9913–9920. [Google Scholar]
- Graziani, I.; Bagala, C.; Duarte, M.; Soldi, R.; Kolev, V.; Tarantini, F.; Kumar, T.K.; Doyle, A.; Neivandt, D.; Yu, C.; et al. Release of FGF1 and p40 synaptotagmin 1 correlates with their membrane destabilizing ability. Biochem. Biophys. Res. Commun. 2006, 349, 192–199. [Google Scholar]
- Engling, A.; Backhaus, R.; Stegmayer, C.; Zehe, C.; Seelenmeyer, C.; Kehlenbach, A.; Schwappach, B.; Wegehingel, S.; Nickel, W. Biosynthetic FGF-2 is targeted to non-lipid raft microdomains following translocation to the extracellular surface of CHO cells. J. Cell Sci 2002, 115, 3619–3631. [Google Scholar]
- Ebert, A.D.; Laussmann, M.; Wegehingel, S.; Kaderali, L.; Erfle, H.; Reichert, J.; Lechner, J.; Beer, H.D.; Pepperkok, R.; Nickel, W. Tec-kinase-mediated phosphorylation of fibroblast growth factor 2 is essential for unconventional secretion. Traffic 2010, 11, 813–826. [Google Scholar]
- Mach, H.; Ryan, J.A.; Burke, C.J.; Volkin, D.B.; Middaugh, C.R. Partially structured self-associating states of acidic fibroblast growth factor. Biochemistry 1993, 32, 7703–7711. [Google Scholar]
- Kueltzo, L.A.; Middaugh, C.R. Nonclassical transport proteins and peptides: An alternative to classical macromolecule delivery systems. J. Pharm. Sci 2003, 92, 1754–1772. [Google Scholar]
- Burke, C.J.; Volkin, D.B.; Mach, H.; Middaugh, C.R. Effect of polyanions on the unfolding of acidic fibroblast growth factor. Biochemistry 1993, 32, 6419–6426. [Google Scholar]
- Fan, H.; Li, H.; Zhang, M.; Middaugh, C.R. Effects of solutes on empirical phase diagrams of human fibroblast growth factor 1. J. Pharm. Sci 2007, 96, 1490–1503. [Google Scholar]
- Sanz, J.M.; Gimenez-Gallego, G. A partly folded state of acidic fibroblast growth factor at low pH. Eur. J. Biochem 1997, 246, 328–335. [Google Scholar]
- Wiedlocha, A.; Madshus, I.H.; Mach, H.; Middaugh, C.R.; Olsnes, S. Tight folding of acidic fibroblast growth factor prevents its translocation to the cytosol with diphtheria toxin as vector. EMBO J 1992, 11, 4835–4842. [Google Scholar]
- Wesche, J.; Wiedlocha, A.; Falnes, P.O.; Choe, S.; Olsnes, S. Externally added aFGF mutants do not require extensive unfolding for transport to the cytosol and the nucleus in NIH/3T3 cells. Biochemistry 2000, 39, 15091–15100. [Google Scholar]
- Backhaus, R.; Zehe, C.; Wegehingel, S.; Kehlenbach, A.; Schwappach, B.; Nickel, W. Unconventional protein secretion: Membrane translocation of FGF-2 does not require protein unfolding. J. Cell Sci 2004, 117, 1727–1736. [Google Scholar]
- Kathir, K.M.; Gao, L.; Rajalingam, D.; Daily, A.E.; Brixey, S.; Liu, H.; Davis, D.; Adams, P.; Prudovsky, I.; Kumar, T.K. NMR characterization of copper and lipid interactions of the C2B domain of synaptotagmin I-relevance to the non-classical secretion of the human acidic fibroblast growth factor (hFGF-1). Biochim. Biophys. Acta 2010, 1798, 297–302. [Google Scholar]
- Carreira, C.M.; LaVallee, T.M.; Tarantini, F.; Jackson, A.; Lathrop, J.T.; Hampton, B.; Burgess, W.H.; Maciag, T. S100A13 is involved in the regulation of fibroblast growth factor-1 and p40 synaptotagmin-1 release in vitro. J. Biol. Chem 1998, 273, 22224–22231. [Google Scholar]
- Kathir, K.M.; Ibrahim, K.; Rajalingam, D.; Prudovsky, I.; Yu, C.; Kumar, T.K. S100A13-lipid interactions-role in the non-classical release of the acidic fibroblast growth factor. Biochim. Biophys. Acta 2007, 1768, 3080–3089. [Google Scholar]
- Santamaria-Kisiel, L.; Rintala-Dempsey, A.C.; Shaw, G.S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J 2006, 396, 201–214. [Google Scholar]
- Hajjar, K.A.; Jacovina, A.T.; Chacko, J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J. Biol. Chem 1994, 269, 21191–21197. [Google Scholar]
- Flood, E.C.; Hajjar, K.A. The annexin A2 system and vascular homeostasis. Vascul. Pharmacol 2011, 54, 59–67. [Google Scholar]
- Lambert, A.G.; Davies, P.B.; Neivandt, D.J. Implementing the theory of sum frequency generation vibrational spectroscopy: A tutorial review. Appl. Spectrosc. Rev 2005, 40, 103–145. [Google Scholar]
- Shen, Y.R. Surface properties probed by second-harmonic and sum-frequency generation. Nature 1989, 337, 519–525. [Google Scholar]
- Adamson, A.W.; Gast, A.P. Physical Chemistry of Surfaces, 6th ed; John Wiley & Sons: New York, NY, USA, 1999. [Google Scholar]
- Walker, R.A.; Conboy, J.C.; Richmond, G.L. Molecular structure and ordering of phospholipids at a liquid-liquid interface. Langmuir 1997, 13, 3070–3073. [Google Scholar]
- Zhu, X.D.; Suhr, H.; Shen, Y.R. Surface vibrational spectroscopy by infrared-visible sum frequency generation. Phys. Rev. B 1987, 35, 3047–3050. [Google Scholar]
- Harris, A.L.; Chidsey, C.E.D.; Levinos, N.J.; Loiacono, D.N. Monolayer vibrational spectroscopy by infrared-visible sum generation at metal and semiconductor surfaces. Chem. Phys. Lett 1987, 141, 350–356. [Google Scholar]
- Hunt, J.H.; Guyot-Sionnest, P.; Shen, Y.R. Observation of C–H stretching vibrations of monolayers of molecules optical sum frequency generation. Chem. Phys. Lett 1987, 133, 189–192. [Google Scholar]
- Watry, M.R.; Richmond, G.L. Comparison of the adsorption of linear alkanesulfonate and linear alkylbenzenesulfonate surfactants at liquid interfaces. J. Am. Chem. Soc 2000, 122, 875–883. [Google Scholar]
- Conboy, J.C.; Messmer, M.C.; Richmond, G.L. Investigation of surfactant conformation and order at the liquid-liquid interface by total internal reflection sum-frequency vibrational spectroscopy. J. Phys. Chem 1996, 100, 7617–7622. [Google Scholar]
- Conboy, J.C.; Messmer, M.C.; Richmond, G.L. Dependence of alkyl chain conformation of simple ionic surfactants on head group functionality as studied by vibrational sum-frequency spectroscopy. J. Phys. Chem. B 1997, 101, 6724–6733. [Google Scholar]
- Walker, R.A.; Gragson, D.E.; Richmond, G.L. Induced changes in solvent structure by phospholipid monolayer formation at a liquid–liquid interface. Colloids Surf. A 1999, 154, 175–185. [Google Scholar]
- Smiley, B.L.; Richmond, G.L. Assembly of long chain phosphatidylcholines at a liquid-liquid interface. Biopolymers 2000, 57, 117–125. [Google Scholar]
- Watry, M.R.; Richmond, G.L. Effects of halothane on phosphatidylcholine, -ethanolamine, -glycerol, and -serine monolayer order at a liquid/liquid interface. Langmuir 2002, 18, 8881–8887. [Google Scholar]
- Watry, M.R.; Tarbuck, T.L.; Richmond, G.L. Vibrational sum-frequency studies of a series of phospholipid monolayers and the associated water structure at the vapor/water interface. J. Phys. Chem. B 2003, 107, 512–518. [Google Scholar]
- Pohle, W.; Saβ, M.; Selle, C.; Wolfrum, K.; Löbau, J. Probing phospholipid chain fluidity by vibrational spectroscopy including sum-frequency generation. Vib. Spectrosc 1999, 19, 321–327. [Google Scholar]
- Lobau, J.; Sass, M.; Pohle, W.; Selle, C.; Koch, M.H.J.; Wolfrum, K. Chain fluidity and phase behaviour of phospholipids as revealed by FTIR and sum-frequency spectroscopy. J. Mol. Struct 1999, 481, 407–411. [Google Scholar]
- Roke, S.; Schins, J.; Muller, M.; Bonn, M. Vibrational spectroscopic investigation of the phase diagram of a biomimetic lipid monolayer. Phys. Rev. Lett 2003, 90, 128101–128104. [Google Scholar]
- Smits, M.; Sovago, M.; Wurpel, G.W.H.; Kim, D.; Muller, M.; Bonn, M. Polarization-resolved broad-bandwidth sum-frequency generation spectroscopy of monolayer relaxation. J. Phys. Chem. C 2007, 111, 8878–8883. [Google Scholar]
- Sovago, M.; Wurpel, G.W.H.; Smits, M.; Müller, M.; Bonn, M. Calcium-induced phospholipid ordering depends on surface pressure. J. Am. Chem. Soc 2007, 129, 11079–11084. [Google Scholar]
- Campen, R.K.; Ngo, T.T.M.; Sovago, M.; Ruysschaert, J.-M.; Bonn, M. Molecular restructuring of water and lipids upon the interaction of DNA with lipid monolayers. J. Am. Chem. Soc 2010, 132, 8037–8047. [Google Scholar]
- RzeŸnicka, I.I.; Sovago, M.; Backus, E.H.G.; Bonn, M.; Yamada, T.; Kobayashi, T.; Kawai, M. Duramycin-induced destabilization of a phosphatidylethanolamine monolayer at the air-water interface observed by vibrational sum-frequency generation spectroscopy. Langmuir 2010, 26, 16055–16062. [Google Scholar]
- Wurpel, G.W.H.; Sovago, M.; Bonn, M. Sensitive probing of DNA binding to a cationic lipid monolayer. J. Am. Chem. Soc 2007, 129, 8420–8421. [Google Scholar]
- Smits, M.; Ghosh, A.; Bredenbeck, J.; Yamamoto, S.; Müller, M.; Bonn, M. Ultrafast energy flow in model biological membranes. New J. Phys 2007, 9, 390. [Google Scholar]
- Sovago, M.; Vartiainen, E.; Bonn, M. Observation of buried water molecules in phospholipid membranes by surface sum-frequency generation spectroscopy. J. Chem. Phys 2009, 131, 161107. [Google Scholar]
- Bonn, M.; Bakker, H.J.; Ghosh, A.; Yamamoto, S.; Sovago, M.; Campen, R.K. Structural inhomogeneity of interfacial water at lipid monolayers revealed by surface-specific vibrational pump-probe spectroscopy. J. Am. Chem. Soc 2010, 132, 14971–14978. [Google Scholar]
- Ohe, C.; Sasaki, T.; Noi, M.; Goto, Y.; Itoh, K. Sum frequency generation spectroscopic study of the condensation effect of cholesterol on a lipid monolayer. Anal. Bioanal. Chem 2007, 388, 73–79. [Google Scholar]
- Bonn, M.; Roke, S.; Berg, O.; Juurlink, L.B.F.; Stamouli, A.; Müller, M. A molecular view of cholesterol-induced condensation in a lipid monolayer. J. Phys. Chem. B 2004, 108, 19083–19085. [Google Scholar]
- Ohe, C.; Ida, Y.; Matsumoto, S.; Sasaki, T.; Goto, Y.; Noi, M.; Tsurumaru, T.; Itoh, K. Investigations of polymyxin B-phospholipid interactions by vibrational sum frequency generation spectroscopy. J. Phys. Chem. B 2004, 108, 18081–18087. [Google Scholar]
- Ma, G.; Liu, J.; Fu, L.; Yan, E.C.Y. Probing water and biomolecules at the air-water interface with a broad bandwidth vibrational sum frequency generation spectrometer from 3800 to 900 cm−1. Appl. Spectrosc 2009, 63, 528–537. [Google Scholar]
- Fu, L.; Ma, G.; Yan, E.C.Y. In situ misfolding of human islet amyloid polypeptide at interfaces probed by vibrational sum frequency generation. J. Am. Chem. Soc 2010, 132, 5405–5412. [Google Scholar]
- Viswanath, P.; Aroti, A.; Motschmann, H.; Leontidis, E. Vibrational sum frequency generation spectroscopic investigation of the interaction of thiocyanate ions with zwitterionic phospholipid monolayers at the air-water interface. J. Phys. Chem. B 2009, 113, 14816–14823. [Google Scholar]
- Hill, K.; Pénzes, C.B.; Schnöller, D.; Horváti, K.; Bősze, S.; Hudecz, F.; Keszthelyi, T.; Kiss, É. Characterisation of the membrane affinity of an isoniazide peptide conjugate by tensiometry, atomic force microscopy and sum-frequency vibrational spectroscopy, using a phospholipid Langmuir monolayer model. Phys. Chem. Chem. Phys. 2010, 12, 11498–11506. [Google Scholar]
- Liljeblad, J.F.D.; Bulone, V.; Tyrode, E.; Rutland, M.W.; Johnson, C.M. Phospholipid monolayers probed by vibrational sum frequency spectroscopy: Instability of unsaturated phospholipids. Biophys. J 2010, 98, L50–L52. [Google Scholar]
- Liljeblad, J.F.D.; Bulone, V.; Rutland, M.W.; Johnson, C.M. Supported phospholipid monolayers. The molecular structure investigated by vibrational sum frequency spectroscopy. J. Phys. Chem. C 2011, 115, 10617–10629. [Google Scholar]
- Ma, G.; Allen, H.C. Real-time investigation of lung surfactant respreading with surface vibrational spectroscopy. Langmuir 2006, 22, 11267–11274. [Google Scholar]
- Ma, G.; Allen, H.C. New insights into lung surfactant monolayers using vibrational sum frequency generation spectroscopy. Photochem. Photobiol 2006, 82, 1517–1529. [Google Scholar]
- Ma, G.; Allen, H.C. Condensing effect of palmitic acid on DPPC in mixed Langmuir monolayers. Langmuir 2007, 23, 589–597. [Google Scholar]
- Ma, G.; Allen, H.C. DPPC Langmuir monolayer at the air-water interface: Probing the tail and head groups by vibrational sum frequency generation spectroscopy. Langmuir 2006, 22, 5341–5349. [Google Scholar]
- Chen, X.; Hua, W.; Huang, Z.; Allen, H.C. Interfacial water structure associated with phospholipid membranes studied by phase-sensitive vibrational sum frequency generation spectroscopy. J. Am. Chem. Soc 2010, 132, 11336–11342. [Google Scholar]
- Casillas-Ituarte, N.N.; Chen, X.; Castada, H.; Allen, H.C. Na+ and Ca2+ effect on the hydration and orientation of the phosphate group of DPPC at air-water and air-hydrated silica interfaces. J. Phys. Chem. B 2010, 114, 9485–9495. [Google Scholar]
- Chen, X.; Allen, H.C. Interactions of dimethylsulfoxide with a dipalmitoylphosphatidylcholine monolayer studied by vibrational sum frequency generation. J. Phys. Chem. A 2009, 113, 12655–12662. [Google Scholar]
- Harper, K.L.; Allen, H.C. Competition between DPPC and SDS at the air-aqueous interface. Langmuir 2007, 23, 8925–8931. [Google Scholar]
- Can, S.Z.; Chang, C.F.; Walker, R.A. Spontaneous formation of DPPC monolayers at aqueous/vapor interfaces and the impact of charged surfactants. Biochim. Biophys. Acta 2008, 1778, 2368–2377. [Google Scholar]
- Nickolov, Z.S.; Britt, D.W.; Miller, J.D. Sum-frequency spectroscopy analysis of two-component Langmuir monolayers and the associated interfacial water structure. J. Phys. Chem. B 2006, 110, 15506–15513. [Google Scholar]
- Mondal, J.A.; Nihonyanagi, S.; Yamaguchi, S.; Tahara, T. Structure and orientation of water at charged lipid monolayer/water interfaces probed by heterodyne-detected vibrational sum frequency generation spectroscopy. J. Am. Chem. Soc 2010, 132, 10656–10657. [Google Scholar]
- Sung, W.; Seok, S.; Kim, D.; Tian, C.S.; Shen, Y.R. Sum-frequency spectroscopic study of Langmuir monolayers of lipids having oppositely charged headgroups. Langmuir 2010, 26, 18266–18272. [Google Scholar]
- Liu, J.; Conboy, J.C. Structure of a gel phase lipid bilayer prepared by the Langmuir-Blodgett/Langmuir-Schaefer method characterized by sum-frequency vibrational spectroscopy. Langmuir 2005, 21, 9091–9097. [Google Scholar]
- Liu, J.; Conboy, J.C. Phase transitions of a single lipid bilayer measured by sum-frequency vibrational spectroscopy. J. Am. Chem. Soc 2004, 126, 8894–8895. [Google Scholar]
- Liu, J.; Conboy, J.C. Asymmetric distribution of lipids in a phase segregated phospholipid bilayer observed by sum-frequency vibrational spectroscopy. J. Phys. Chem. C 2007, 111, 8988–8999. [Google Scholar]
- Liu, J.; Conboy, J.C. Phase behavior of planar supported lipid membranes composed of cholesterol and 1,2-distearoyl-sn-glycerol-3-phosphocholine examined by sum-frequency vibrational spectroscopy. Vib. Spectrosc 2009, 50, 106–115. [Google Scholar]
- Nguyen, T.T.; Rembert, K.; Conboy, J.C. Label-free detection of drug-membrane association using ultraviolet-visible sum-frequency generation. J. Am. Chem. Soc 2009, 131, 1401–1403. [Google Scholar]
- Liu, J.; Conboy, J.C. Direct measurement of transbilayer movement of phospholipids by sum-frequency vibrational spectroscopy. J. Am. Chem. Soc 2004, 126, 8376–8377. [Google Scholar]
- Liu, J.; Conboy, J.C. 1,2-diacyl-phosphatidylcholine flip-flop measured directly by sum-frequency vibrational spectroscopy. Biophys. J 2005, 89, 2522–2532. [Google Scholar]
- Anglin, T.C.; Conboy, J.C. Lateral pressure dependence of the phospholipid transmembrane diffusion rate in planar-supported lipid bilayers. Biophys. J 2008, 95, 186–193. [Google Scholar]
- Anglin, T.C.; Conboy, J.C. Kinetics and thermodynamics of flip-flop in binary phospholipid membranes measured by sum-frequency vibrational spectroscopy. Biochemistry 2009, 48, 10220–10234. [Google Scholar]
- Anglin, T.C.; Cooper, M.P.; Li, H.; Chandler, K.; Conboy, J.C. Free energy and entropy of activation for phospholipid flip-flop in planar supported lipid bilayers. J. Phys. Chem. B 2010, 114, 1903–1914. [Google Scholar]
- Anglin, T.C.; Brown, K.L.; Conboy, J.C. Phospholipid flip-flop modulated by transmembrane peptides WALP and melittin. J. Struct. Biol 2009, 168, 37–52. [Google Scholar]
- Brown, K.L.; Conboy, J.C. Electrostatic induction of lipid asymmetry. J. Am. Chem. Soc 2011, 133, 8794–8797. [Google Scholar]
- Tong, Y.; Li, N.; Liu, H.; Ge, A.; Osawa, M.; Ye, S. Mechanistic studies by sum-frequency generation spectroscopy: Hydrolysis of a supported phospholipid bilayer by phospholipase A2. Angew. Chem. Int. Ed. Engl 2010, 49, 2319–2323. [Google Scholar]
- Kim, J.; Kim, G.; Cremer, P.S. Investigations of water structure at the solid/liquid interface in the presence of supported lipid bilayers by vibrational sum frequency spectroscopy. Langmuir 2001, 17, 7255–7260. [Google Scholar]
- Petralli-Mallow, T.; Briggman, K.A.; Richter, L.J.; Stephenson, J.C.; Plant, A.L. Nonlinear optics as a detection scheme for biomimetic sensors: SFG spectroscopy of hybrid bilayer membrane formation. Proc. SPIE 1999, 25, 1–7. [Google Scholar]
- Anderson, N.A.; Richter, L.J.; Stephenson, J.C.; Briggman, K.A. Characterization and control of lipid layer fluidity in hybrid bilayer membranes. J. Am. Chem. Soc 2007, 129, 2094–2100. [Google Scholar]
- Levy, D.; Briggman, K.A. Cholesterol/phospholipid interactions in hybrid bilayer membranes. Langmuir 2007, 23, 7155–7161. [Google Scholar]
- Kett, P.J.; Casford, M.T.; Davies, P.B. Sum frequency generation (SFG) vibrational spectroscopy of planar phosphatidylethanolamine hybrid bilayer membranes under water. Langmuir 2010, 26, 9710–9719. [Google Scholar]
- Kett, P.J.; Casford, M.T.; Davies, P.B. Structure of mixed phosphatidylethanolamine and cholesterol monolayers in a supported hybrid bilayer membrane studied by sum frequency generation vibrational spectroscopy. J. Phys. Chem. B 2011, 115, 6465–6473. [Google Scholar]
- Lis, D.; Guthmuller, J.; Champagne, B.; Humbert, C.; Busson, B.; Tadjeddine, A.; Peremans, A.; Cecchet, F. Selective detection of the antigenic polar heads of model lipid membranes supported on metals from their vibrational nonlinear optical response. Chem. Phys. Lett 2010, 489, 12–15. [Google Scholar]
- Doyle, A.W.; Fick, J.; Himmelhaus, M.; Eck, W.; Graziani, I.; Prudovsky, I.; Grunze, M.; Maciag, T.; Neivandt, D. Protein deformation of lipid hybrid bilayer membranes studied by Sum Frequency Generation Vibrational Spectroscopy (SFS). Langmuir 2004, 20, 8961–8965. [Google Scholar]
- Chen, X.; Tang, H.; Even, M.A.; Wang, J.; Tew, G.N.; Chen, Z. Observing a molecular knife at work. J. Am. Chem. Soc 2006, 128, 2711–2714. [Google Scholar]
- Chen, X.; Chen, Z. SFG studies on interactions between antimicrobial peptides and supported lipid bilayers. Biochim. Biophys. Acta 2006, 1758, 1257–1273. [Google Scholar]
- Avery, C.W.; Palermo, E.F.; McLaughlin, A.; Kuroda, K.; Chen, Z. Investigations of the interactions between synthetic antimicrobial polymers and substrate-supported lipid bilayers using sum frequency generation vibrational spectroscopy. Anal. Chem 2011, 83, 1342–1349. [Google Scholar]
- Yang, P.; Ramamoorthy, A.; Chen, Z. Membrane orientation of MSI-78 measured by sum frequency generation vibrational spectroscopy. Langmuir 2011, 27, 7760–7767. [Google Scholar]
- Chen, X.; Wang, J.; Kristalyn, C.B.; Chen, Z. Real-time structural investigation of a lipid bilayer during its interaction with melittin using sum frequency generation vibrational spectroscopy. Biophys. J 2007, 93, 866–875. [Google Scholar]
- Nguyen, K.T.; Le Clair, S.V.; Ye, S.; Chen, Z. Molecular interactions between magainin 2 and model membranes in situ. J. Phys. Chem. B 2009, 113, 12358–12363. [Google Scholar]
- Wang, T.; Li, D.; Lu, X.; Khmaladze, A.; Han, X.; Ye, S.; Yang, P.; Xue, G.; He, N.; Chen, Z. Single lipid bilayers constructed on polymer cushion studied by sum frequency generation vibrational spectroscopy. J. Phys. Chem. C 2011, 115, 7613–7620. [Google Scholar]
- Chen, X.; Boughton, A.P.; Tesmer, J.J.G.; Chen, Z. In situ investigation of heterotrimeric G protein βγ subunit binding and orientation on membrane bilayers. J. Am. Chem. Soc 2007, 129, 12658–12659. [Google Scholar]
- Chen, X.; Wang, J.; Boughton, A.P.; Kristalyn, C.B.; Chen, Z. Multiple orientation of melittin inside a single lipid bilayer determined by combined vibrational spectroscopic studies. J. Am. Chem. Soc 2007, 129, 1420–1427. [Google Scholar]
- Thennarasu, S.; Huang, R.; Lee, D.-K.; Yang, P.; Maloy, L.; Chen, Z.; Ramamoorthy, A. Limiting an antimicrobial peptide to the lipid-water interface enhances its bacterial membrane selectivity: A case study of MSI-367. Biochemistry 2010, 49, 10595–10605. [Google Scholar]
- Ye, S.; Nguyen, K.T.; Boughton, A.P.; Mello, C.M.; Chen, Z. Orientation difference of chemically immobilized and physically adsorbed biological molecules on polymers detected at the solid/liquid interfaces in situ. Langmuir 2010, 26, 6471–6477. [Google Scholar]
- Ye, S.; Nguyen, K.T.; Chen, Z. Interactions of alamethicin with model cell membranes investigated using sum frequency generation vibrational spectroscopy in real time in situ. J. Phys. Chem. B 2010, 114, 3334–3340. [Google Scholar]
- Nguyen, K.T.; King, J.T.; Chen, Z. Orientation determination of interfacial β-sheet structures in situ. J. Phys. Chem. B 2010, 114, 8291–8300. [Google Scholar]
- Nguyen, K.T.; Soong, R.; Lm, S.-C.; Waskell, L.; Ramamoorthy, A.; Chen, Z. Probing the spontaneous membrane insertion of a tail-anchored membrane protein by sum frequency generation spectroscopy. J. Am. Chem. Soc 2010, 132, 15112–15115. [Google Scholar]
- Pavinatto, F.J.; Pacholatti, C.P.; Montanha, E.A.; Caseli, L.; Silva, H.S.; Miranda, P.B.; Viitala, T.; Oliveira, O.N., Jr. Cholesterol mediates chitosan activity on phospholipid monolayers and Langmuir-Blodgett films. Langmuir 2009, 25, 10051–10061. [Google Scholar]
- Pavinatto, F.J.; Caseli, L.; Pavinatto, A.; dos Santos, D.S., Jr; Nobre, T.M.; Zaniquelli, M.E.D.; Silva, H.S.; Miranda, P.B.; de Oliveira, O.N., Jr. Probing chitosan and phospholipid interactions using Langmuir and Langmuir-Blodgett films as cell membrane models. Langmuir 2007, 23, 7666–7671. [Google Scholar]
- Nobre, T.M.; de Sousa e Silva, H.; Furriel, R.P.M.; Leone, F.A.; Miranda, P.B.; Zaniquelli, M.E.D. Molecular view of the interaction between iota-carrageenan and a phospholipid film and its role in enzyme immobilization. J. Phys. Chem. B 2009, 113, 7491–7497. [Google Scholar]
- Inoue, K.; Fujii, M.; Sakai, M. Development of a non-scanning vibrational sum-frequency generation detected infrared super-resolution microscope and its application to biological cells. Appl. Spectrosc 2010, 64, 275–281. [Google Scholar]
- Magde, D.; Elson, E.; Webb, W.W. Thermodynamic fluctuations in a reacting system-measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett 1972, 29, 705–708. [Google Scholar]
- Schwille, P.; Haustein, E. Fluorescence Correlation Spectroscopy: An Introduction to Its Concepts and Applications. In Biophysics Textbook; Max-Planck-Institute for Biophysical Chemistry: Göttingen, Germany, 2001. [Google Scholar]
- Haustein, E.; Schwille, P. Fluorescence correlation spectroscopy: Novel variations of an established technique. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 151–169. [Google Scholar]
- Hess, S.T.; Huang, S.; Heikal, A.A.; Webb, W.W. Biological and chemical applications of fluorescence correlation spectroscopy: A review. Biochemistry 2002, 41, 697–705. [Google Scholar]
- García-Sáez, A.J.; Schwille, P. Fluorescence correlation spectroscopy for the study of membrane dynamics and protein/lipid interactions. Methods 2008, 46, 116–122. [Google Scholar]
- Rochira, J.A.; Gudheti, M.V.; Gould, T.J.; Laughlin, R.R.; Nadeau, J.L.; Hess, S.T. Fluorescence intermittency limits brightness in CdSe/ZnS nanoparticles quantified by fluorescence correlation spectroscopy. J. Phys. Chem. C 2007, 111, 1695–1708. [Google Scholar]
- Schwille, P.; Korlach, J.; Webb, W.W. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry 1999, 36, 176–182. [Google Scholar]
- Burns, A.R.; Frankel, D.J.; Buranda, T. Local mobility in lipid domains of supported bilayers characterized by atomic force microscopy and fluorescence correlation spectroscopy. Biophys. J 2005, 89, 1081–1093. [Google Scholar]
- Zhang, L.; Granick, S. Lipid diffusion compared in outer and inner leaflets of planar supported bilayers. J. Chem. Phys 2005, 123, 211104. [Google Scholar]
- Krichevsky, O.; Bonnet, G. Fluorescence correlation spectroscopy: The technique and its applications. Rep. Prog. Phys 2002, 65, 251–297. [Google Scholar]
- Elliott, J.T.; Burden, D.L.; Woodward, J.T.; Sehgal, A.; Douglas, J.F. Phospholipid monolayers supported on spun cast polystyrene films. Langmuir 2003, 19, 2275–2283. [Google Scholar]
- Kahya, N.; Scherfeld, D.; Bacia, K.; Poolman, B.; Schwille, P. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem 2003, 278, 28109–28115. [Google Scholar]
- Kahya, N.; Schwille, P. Fluorescence correlation studies of lipid domains in model membranes. Mol. Membr. Biol 2006, 23, 29–39. [Google Scholar]
- Kahya, N.; Schwille, P. How phospholipid-cholesterol interactions modulate lipid lateral diffusion, as revealed by fluorescence correlation spectroscopy. J. Fluoresc 2006, 16, 671–678. [Google Scholar]
- Korlach, J.; Schwille, P.; Webb, W.W.; Feigenson, G.W. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 8461–8466. [Google Scholar]
- Chiantia, S.; Ries, J.; Schwille, P. Fluorescence correlation spectroscopy in membrane structure elucidation. Biochim. Biophys. Acta 2009, 1788, 225–233. [Google Scholar]
- Fahey, P.F.; Webb, W.W. Lateral diffusion in phospholipid bilayer membranes and multilamellar liquid crystals. Biochemistry 1978, 17, 3046–3053. [Google Scholar]
- Zhang, L.; Granick, S. Slaved diffusion in phospholipid bilayers. Proc. Natl. Acad. Sci. USA 2005, 102, 9118–9121. [Google Scholar]
- Campbell, A.S.; Yu, Y.; Granick, S.; Gewirth, A.A. PCB association with model phospholipid bilayers. Environ. Sci. Technol 2008, 42, 7496–7501. [Google Scholar]
- Dertinger, T.; Pacheco, V.; von der Hocht, I.; Hartmann, R.; Gregor, I.; Enderlein, J. Two-focus fluorescence correlation spectroscopy: A new tool for accurate and absolute diffusion measurements. Chemphyschem 2007, 8, 433–443. [Google Scholar]
- Guo, L.; Har, J.Y.; Sankaran, J.; Hong, Y.; Kannan, B.; Wohland, T. Molecular diffusion measurement in lipid bilayers over wide concentration ranges: A comparative study. Chemphyschem 2008, 9, 721–728. [Google Scholar]
- Renner, L.; Osaki, T.; Chiantia, S.; Schwille, P.; Pompe, T.; Werner, C. Supported lipid bilayers on spacious and pH-responsive polymer cushions with varied hydrophilicity. J. Phys. Chem. B 2008, 112, 6373–6378. [Google Scholar]
- Benda, A.; Beneš, M.; Mareček, V.; Lhotský, A.; Hermens, W.Th.; Hof, M. How to determine diffusion coefficients in planar phospholipid systems by confocal fluorescence correlation spectroscopy. Langmuir 2003, 19, 4120–4126. [Google Scholar]
- Dertinger, T.; von der Hocht, I.; Benda, A.; Hof, M.; Enderlein, J. Surface sticking and lateral diffusion of lipids in supported bilayers. Langmuir 2006, 22, 9339–9344. [Google Scholar]
- Miszta, A.; Machán, R.; Benda, A.; Ouellette, A.J.; Hermens, W.T.; Hof, M. Combination of ellipsometry, laser scanning microscopy and Z-scan fluorescence correlation spectroscopy elucidating interaction of cryptdin-4 with supported phospholipid bilayers. J. Pept. Sci 2008, 14, 503–509. [Google Scholar]
- Stefl, M.; Kułakowska, A.; Hof, M. Simultaneous characterization of lateral lipid and prothrombin diffusion coefficients by z-scan fluorescence correlation spectroscopy. Biophys. J 2009, 97, L01–L03. [Google Scholar]
- Beranova, L.; Cwiklik, L.; Jurkiewicz, P.; Hof, M.; Jungwirth, P. Oxidation changes physical properties of phospholipid bilayers: Fluorescence spectroscopy and molecular simulations. Langmuir 2010, 26, 6140–6144. [Google Scholar]
- Machán, R.; Miszta, A.; Hermens, W.; Hof, M. Real-time monitoring of melittin-induced pore and tubule formation from supported lipid bilayers and its physiological relevance. Chem. Phys. Lipids 2010, 163, 200–206. [Google Scholar]
- Macháň, R.; Hof, M.; Chernovets, T.; Zhmak, M.N.; Ovchinnikova, T.V.; Sýkora, J. Formation of arenicin-1 microdomains in bilayers and their specific lipid interaction revealed by Z-scan FCS. Anal. Bioanal. Chem 2011, 399, 3547–3554. [Google Scholar]
- Przybylo, M.; Sýkora, J.; Humpolíčková, J.; Benda, A.; Zan, A.; Hof, M. Lipid diffusion in giant unilamellar vesicles is more than 2 times faster than in supported phospholipid bilayers under identical conditions. Langmuir 2006, 22, 9096–9099. [Google Scholar]
- Kriegsmann, J.; Gregor, I.; von der Hocht, I.; Klare, J.; Engelhard, M.; Enderlein, J.; Fitter, J. Translational diffusion and interaction of a photoreceptor and its cognate transducer observed in giant unilamellar vesicles by using dual-focus FCS. Chembiochem 2009, 10, 1823–1829. [Google Scholar]
- Gielen, E.; vandeVen, M.; Margineanu, A.; Dedecker, P.; van der Auweraer, M.; Engelborghs, Y.; Hofkens, J.; Ameloot, M. On the use of Z-scan fluorescence correlation experiments on giant unilamellar vesicles. Chem. Phys. Lett 2009, 469, 110–114. [Google Scholar]
- Humpolíčková, J.; Gielen, E.; Benda, A.; Fagulova, V.; Vercammen, J.; VandeVen, M.; Hof, M.; Ameloot, M.; Engelborghs, Y. Probing diffusion laws within cellular membranes by Z-scan fluorescence correlation spectroscopy. Biophys. J 2006, 91, L23–L25. [Google Scholar]
- Ganguly, S.; Chattopadhyay, A. Cholesterol depletion mimics the effect of cytoskeletal destabilization on membrane dynamics of the serotonin1A receptor: A zFCS study. Biophys. J 2010, 99, 1397–1407. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 5th ed; Taylor & Francis Group: New York, NY, USA, 2008. [Google Scholar]
- Šegota, S.; Težak, Đ. Spontaneous formation of vesicles. Adv. Colloid Interfac. 2006, 121, 51–75. [Google Scholar]
- Kleinfeld, A.M.; Chu, P.; Romero, C. Transport of long-chain native fatty acids across lipid bilayer membranes indicates that transbilayer flip-flop is rate limiting. Biochemistry 1997, 36, 14146–14158. [Google Scholar]
- Wong, J.Y.; Majewski, J.; Seitz, M.; Park, C.K.; Israelachvili, J.N.; Smith, G.S. Polymer-cushioned bilayers. I. A structural study of various preparation methods using neutron reflectometry. Biophys. J 1999, 77, 1445–1457. [Google Scholar]
- Uchiyama-Kokubu, N.; Naito, M.; Nakajima, M.; Tsuruo, T. Transport of somatostatin and substance P by human P-glycoprotein. FEBS Lett 2004, 574, 55–61. [Google Scholar]
- De Beer, A.G.F.; de Aguiar, H.B.; Nijsen, J.F.W.; Roke, S. Detection of buried microstructures by nonlinear light scattering spectroscopy. Phys. Rev. Lett 2009, 102, 095502. [Google Scholar]
- De Beer, A.G.F.; Roke, S. Sum frequency generation scattering from the interface of an isotropic particle: Geometrical and chiral effects. Phys. Rev. B 2007, 75, 245438. [Google Scholar]
- Tien, H.T.; Ottova, A.L. The lipid bilayer concept and its experimental realization: From soap bubbles, kitchen sink, to bilayer lipid membranes. J. Membr. Sci 2001, 189, 83–117. [Google Scholar]
- Castellana, E.T.; Cremer, P.S. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf. Sci. Rep 2006, 61, 429–444. [Google Scholar]
- Römer, W.; Lam, Y.H.; Fischer, D.; Watts, A.; Fischer, W.B.; Göring, P.; Wehrspohn, R.B.; Gösele, U.; Steinem, C. Channel activity of a viral transmembrane peptide in micro-BLMs: Vpu1–32 from HIV-1. J. Am. Chem. Soc 2004, 126, 16267–16274. [Google Scholar]
- Duschl, C.; Liley, M.; Lang, H.; Ghandi, A.; Zakeeruddin, S.M.; Stahlberg, H.; Dubochet, J.; Nemetz, A.; Knoll, W.; Vogel, H. Sulphur-bearing lipids for the covalent attachment of supported lipid bilayer to gold surfaces: A detailed characterisation and analysis. Mat. Sci. Eng. C 1996, 4, 7–18. [Google Scholar]
- Plant, A.L. Supported hybrid bilayer membranes as rugged cell membrane mimics. Langmuir 1999, 15, 5128–5135. [Google Scholar]
- Dubois, L.H.; Nuzzo, R.G. Synthesis, structure, and properties of model organic surfaces. Annu. Rev. Phys. Chem 1992, 43, 437–463. [Google Scholar]
- Roberts, G.G. Langmuir-Blodgett films. Contemp. Phys 1984, 25, 109–128. [Google Scholar]
- Rao, N.M.; Silin, V.; Ridge, K.D.; Woodward, J.T.; Plant, A.L. Cell membrane hybrid bilayers containing the G-protein-coupled receptor CCR5. Anal. Biochem 2002, 307, 117–130. [Google Scholar]
- Anderson, N.A.; Richter, L.J.; Stephenson, J.C.; Briggman, K.A. Determination of lipid phase transition temperatures in hybrid bilayer membranes. Langmuir 2006, 22, 8333–8336. [Google Scholar]
- Zhang, L.; Vidu, R.; Waring, A.J.; Lehrer, R.I.; Longo, M.L.; Stroeve, P. Electrochemical and surface properties of solid-supported, mobile phospholipid bilayers on a polyion/alkylthiol layer pair used for detection of antimicrobial peptide insertion. Langmuir 2002, 18, 1318–1331. [Google Scholar]
- Tien, H.T.; Ottova, A.L. From self-assembled bilayer lipid membranes (BLMs) to supported BLMs on metal and gel substrates to practical applications. Coll. Surf. A 1999, 149, 217–233. [Google Scholar]
- Tien, H.T.; Wurster, S.H.; Ottova, A.L. Electrochemistry of supported bilayer lipid membranes: Background and techniques for biosensor development. Bioelectrochem. Bioenerg 1997, 42, 77–94. [Google Scholar]
- Ottova, A.; Tvarozek, V.; Racek, J.; Sabo, J.; Ziegler, W.; Hianik, T.; Tien, H.T. Self-assembled BLMs: Biomembrane models and biosensor applications. Supramol. Sci 1997, 4, 101–112. [Google Scholar]
- Ottova, A.; Tien, H.T. The 40th anniversary of bilayer lipid membrane research. Bioelectrochemistry 2002, 56, 171–173. [Google Scholar]
- Asaka, K.; Ottova, A.; Tien, H.T. Mediated electron transfer across supported bilayer lipid membrane (s-BLM). Thin Solid Films 1999, 354, 201–207. [Google Scholar]
- Asaka, K.; Tien, H.T.; Ottova, A. Voltammetric study of charge transfer across supported bilayer lipid membranes (s-BLMs). J. Biochem. Biophys. Methods 1999, 40, 27–37. [Google Scholar]
- Tamm, L.K.; McConnell, H.M. Supported phospholipid bilayers. Biophys. J 1985, 47, 105–113. [Google Scholar]
- Hinterdorfer, P.; Baber, G.; Tamm, L.K. Reconstitution of membrane fusion sites. J. Biol. Chem 1994, 269, 20360–20368. [Google Scholar]
- Salafsky, J.; Groves, J.T.; Boxer, S.G. Architecture and function of membrane proteins in planar supported bilayers: A study with photosynthetic reaction centers. Biochemistry 1996, 35, 14773–14781. [Google Scholar]
- Scomparin, C.; Lecuyer, S.; Ferreira, M.; Charitat, T.; Tinland, B. Diffusion in supported lipid bilayers: Influence of substrate and preparation technique on the internal dynamics. Eur. Phys. J. E 2009, 28, 211–220. [Google Scholar]
- Chi, L.F.; Johnston, R.R.; Ringsdorf, H.; Kimizuka, N.; Kunitake, T. Mobile supported monolayers of ionic amphiphiles: Variation of domain morphology via preadsorbed polyelectrolytes. Langmuir 1992, 8, 1360–1365. [Google Scholar]
- Sackmann, E. Supported membranes: Scientific and practical applications. Science 1996, 271, 43–48. [Google Scholar]
- Tanaka, M.; Sackmann, E. Polymer-supported membranes as models of the cell surface. Nature 2005, 437, 656–663. [Google Scholar]
- Baumgart, T.; Offenhäusser, A. Polysaccharide-supported planar bilayer lipid model membranes. Langmuir 2003, 19, 1730–1737. [Google Scholar]
- Purrucker, O.; Förtig, A.; Jordan, R.; Tanaka, M. Supported membranes with well-defined polymer tethers—Incorporation of cell receptors. ChemPhysChem 2004, 5, 327–335. [Google Scholar]
- Cremer, P.S.; Yang, T. Creating spatially addressed arrays of planar supported fluid phospholipid membranes. J. Am. Chem. Soc 1999, 121, 8130–8131. [Google Scholar]
- Albertorio, F.; Diaz, A.J.; Yang, T.; Chapa, V.A.; Kataoka, S.; Castellana, E.T.; Cremer, P.S. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir 2005, 21, 7476–7482. [Google Scholar]
- Albertorio, F.; Daniel, S.; Cremer, P.S. Supported lipopolymer membranes as nanoscale filters: Simultaneous protein recognition and size-selection assays. J. Am. Chem. Soc 2006, 128, 7168–7169. [Google Scholar]
- Holden, M.A.; Jung, S.-Y.; Yang, T.; Castellana, E.T.; Cremer, P.S. Creating fluid and air-stable solid supported lipid bilayers. J. Am. Chem. Soc 2004, 126, 6512–6513. [Google Scholar]
- Smith, E.A.; Coym, J.W.; Cowell, S.M.; Tokimoto, T.; Hruby, V.J.; Yamamura, H.I.; Wirth, M.J. Lipid bilayers on polyacrylamide brushes for inclusion of membrane proteins. Langmuir 2005, 21, 9644–9650. [Google Scholar]
- Atanasov, V.; Knorr, N.; Duran, R.S.; Ingebrandt, S.; Offenhäusser, A.; Knoll, W.; Köper, I. Membrane on a chip: A functional tethered lipid bilayer membrane on silicon oxide surfaces. Biophys. J 2005, 89, 1780–1788. [Google Scholar]
- Knoll, W.; Köper, I.; Naumann, R.; Sinner, E.-K. Tethered bimolecular lipid membranes—A novel model membrane platform. Electrochim. Acta 2008, 53, 6680–6689. [Google Scholar]
- Kügler, R.; Knoll, W. Polyelectrolyte-supported lipid membranes. Bioelectrochemistry 2002, 56, 175–178. [Google Scholar]
- Naumann, R.; Schiller, S.M.; Giess, F.; Grohe, B.; Hartman, K.B.; Kärcher, I.; Köper, I.; Lübben, J.; Vasilev, K.; Knoll, W. Tethered lipid bilayers on ultraflat gold surfaces. Langmuir 2003, 19, 5435–5443. [Google Scholar]
- Shen, W.W.; Boxer, S.G.; Knoll, W.; Frank, C.W. Polymer-supported lipid bilayers on benzophenone-modified substrates. Biomacromolecules 2001, 2, 70–79. [Google Scholar]
- Spinke, J.; Yang, J.; Wolf, H.; Liley, M.; Ringsdorf, H.; Knoll, W. Polymer-supported bilayer on a solid substrate. Biophys. J 1992, 63, 1667–1671. [Google Scholar]
- Munro, J.C.; Frank, C.W. In situ formation and characterization of poly(ethylene glycol)-supported lipid bilayers on gold surfaces. Langmuir 2004, 20, 10567–10575. [Google Scholar]
- Weng, K.C.; Stålgren, J.J.R.; Duval, D.J.; Risbud, S.H.; Frank, C.W. Fluid biomembranes supported on nanoporous aerogel/xerogel substrates. Langmuir 2004, 20, 7232–7239. [Google Scholar]
- Luo, G.; Liu, T.; Zhao, X.S.; Huang, Y.; Huang, C.; Cao, W. Investigation of polymer-cushioned phospholipid bilayers in the solid phase by atomic force microscopy. Langmuir 2001, 17, 4074–4080. [Google Scholar]
- Arya, A.; Krull, U.J.; Thompson, M.; Wong, H.E. Langmuir-Blodgett deposition of lipid films on hydrogel as a basis for biosensor development. Anal. Chim. Acta 1985, 173, 331–336. [Google Scholar]
- Kühner, M.; Tampé, R.; Sackmann, E. Lipid mono- and bilayer supported on polymer films: Composite polymer-lipid films on solid substrates. Biophys. J 1994, 67, 217–226. [Google Scholar]
- Wiegand, G.; Jaworek, T.; Wegner, G.; Sackmann, E. Heterogeneous surfaces of structured hairy-rod polymer films: Preparation and methods of functionalization. Langmuir 1997, 13, 3563–3569. [Google Scholar]
- Nissen, J.; Gritsch, S.; Wiegand, G.; Rädler, J.O. Wetting of phospholipid membranes on hydrophilic surfaces—Concepts towards self-healing membranes. Eur. Phys. J. B 1999, 10, 335–344. [Google Scholar]
- Györvary, E.; Wetzer, B.; Sleytr, U.B.; Sinner, A.; Offenhäusser, A.; Knoll, W. Lateral diffusion of lipids in silane-, dextran-, and S-layer-supported mono- and bilayers. Langmuir 1999, 15, 1337–1347. [Google Scholar]
- Baumgart, T.; Offenhäusser, A. Spreading of lipid monolayers on hydrophilic substrates at increased relative humidities. Langmuir 2002, 18, 5899–5908. [Google Scholar]
- Naumann, C.A.; Prucker, O.; Lehmann, T.; Rühe, J.; Knoll, W.; Frank, C.W. The polymer-supported phospholipid bilayer: Tethering as a new approach to substrate-membrane stabilization. Biomacromolecules 2002, 3, 27–35. [Google Scholar]
- Smith, H.; Jablin, M.; Vidyasagar, A.; Saiz, J.; Watkins, E.; Toomey, R.; Hurd, A.; Majewski, J. Model lipid membranes on a tunable polymer cushion. Phys. Rev. Lett 2009, 102, 228102–228105. [Google Scholar]



© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Prudovsky, I.; Kumar, T.K.S.; Sterling, S.; Neivandt, D. Protein-Phospholipid Interactions in Nonclassical Protein Secretion: Problem and Methods of Study. Int. J. Mol. Sci. 2013, 14, 3734-3772. https://doi.org/10.3390/ijms14023734
Prudovsky I, Kumar TKS, Sterling S, Neivandt D. Protein-Phospholipid Interactions in Nonclassical Protein Secretion: Problem and Methods of Study. International Journal of Molecular Sciences. 2013; 14(2):3734-3772. https://doi.org/10.3390/ijms14023734
Chicago/Turabian StylePrudovsky, Igor, Thallapuranam Krishnaswamy Suresh Kumar, Sarah Sterling, and David Neivandt. 2013. "Protein-Phospholipid Interactions in Nonclassical Protein Secretion: Problem and Methods of Study" International Journal of Molecular Sciences 14, no. 2: 3734-3772. https://doi.org/10.3390/ijms14023734