Covalent Coupling of Nanoparticles with Low-Density Functional Ligands to Surfaces via Click Chemistry

Abstract
:1. Introduction
2. Results and Discussion
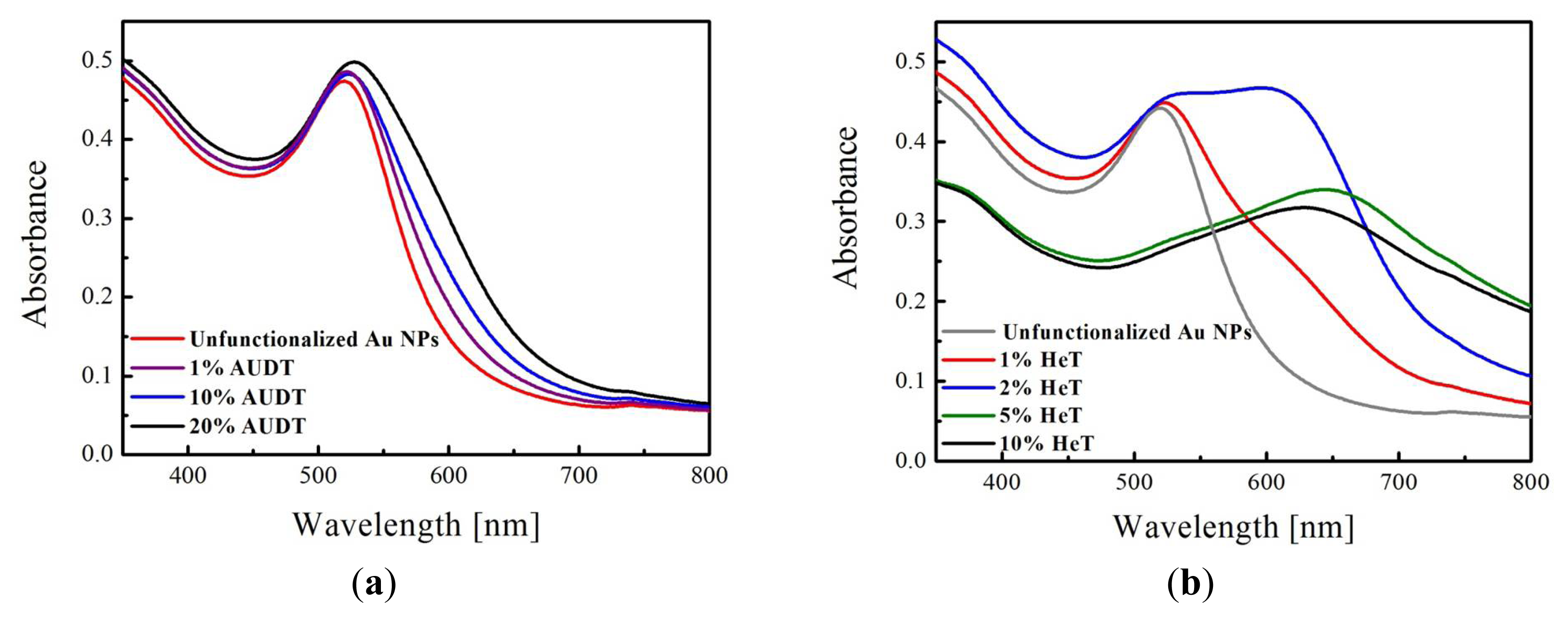
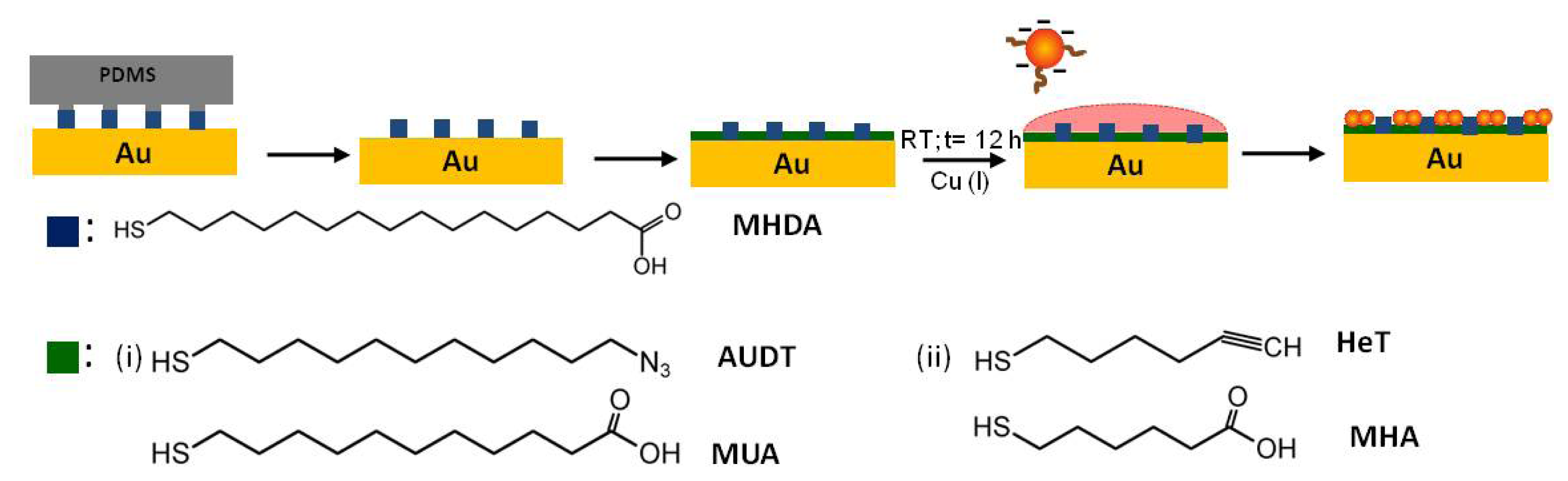
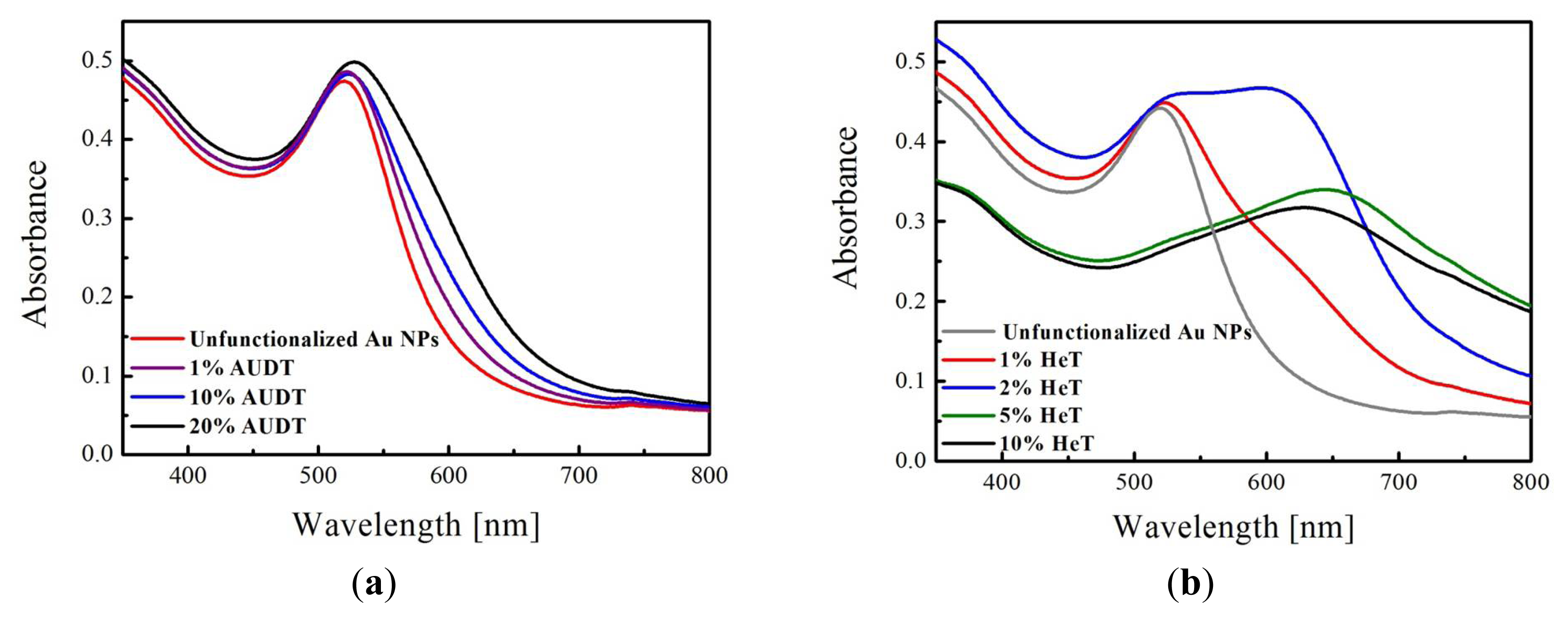
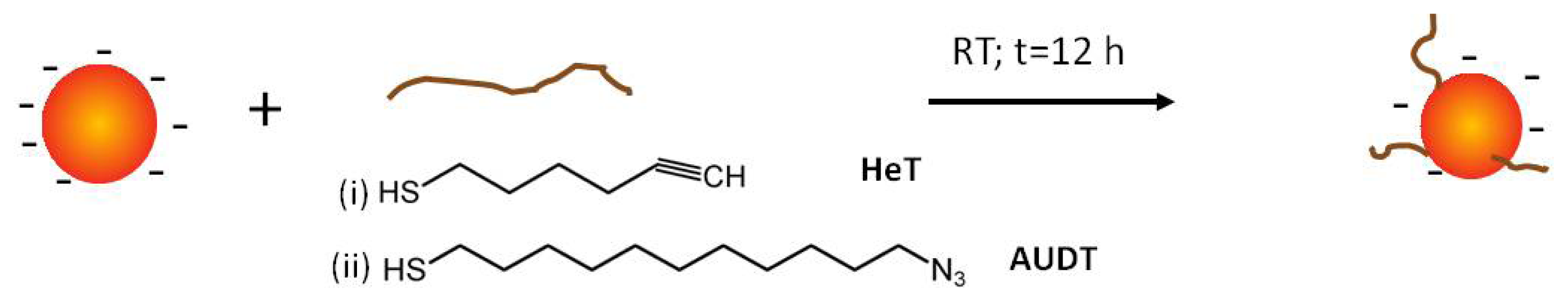
2.1. Functionalization of Au NPs
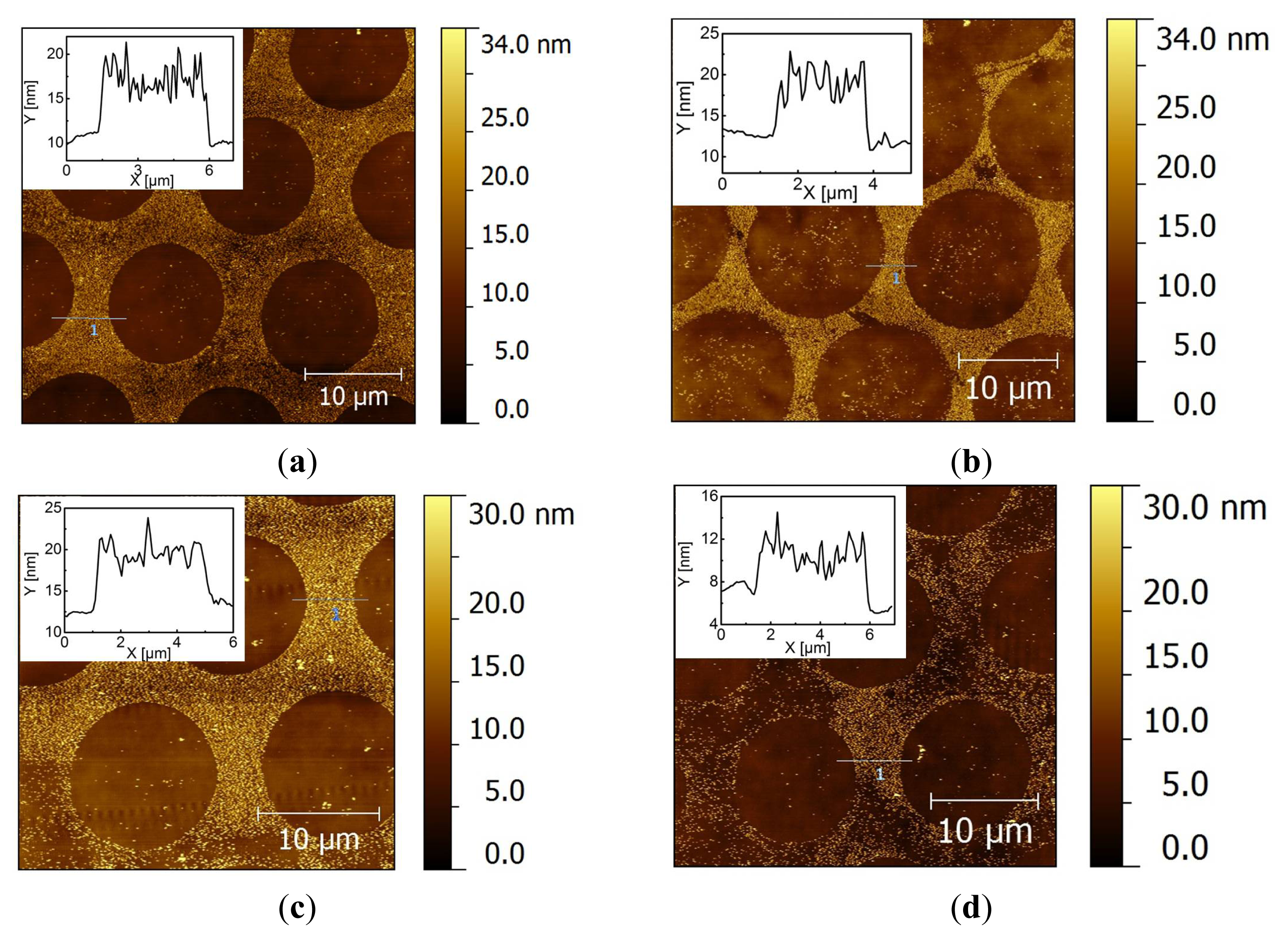
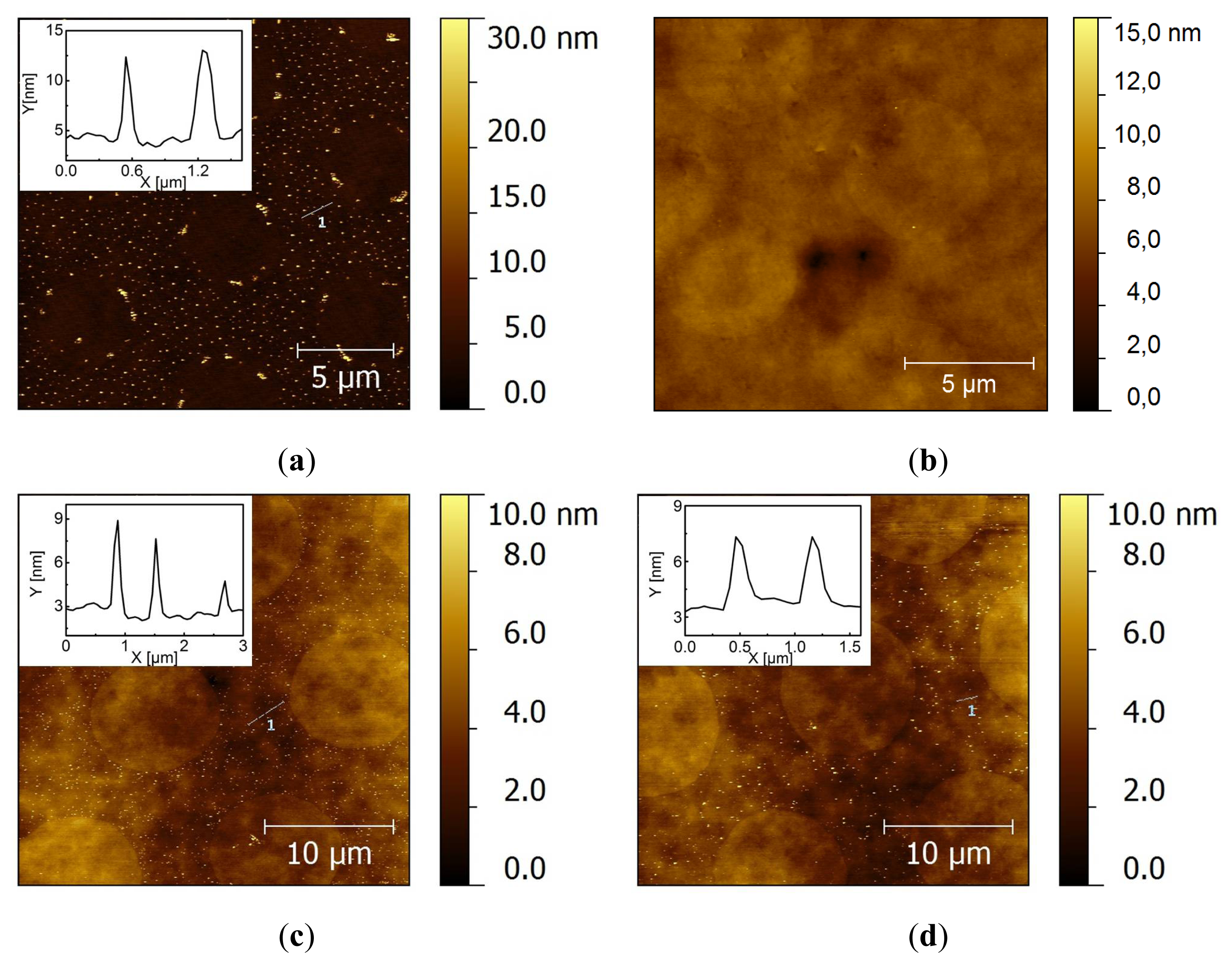
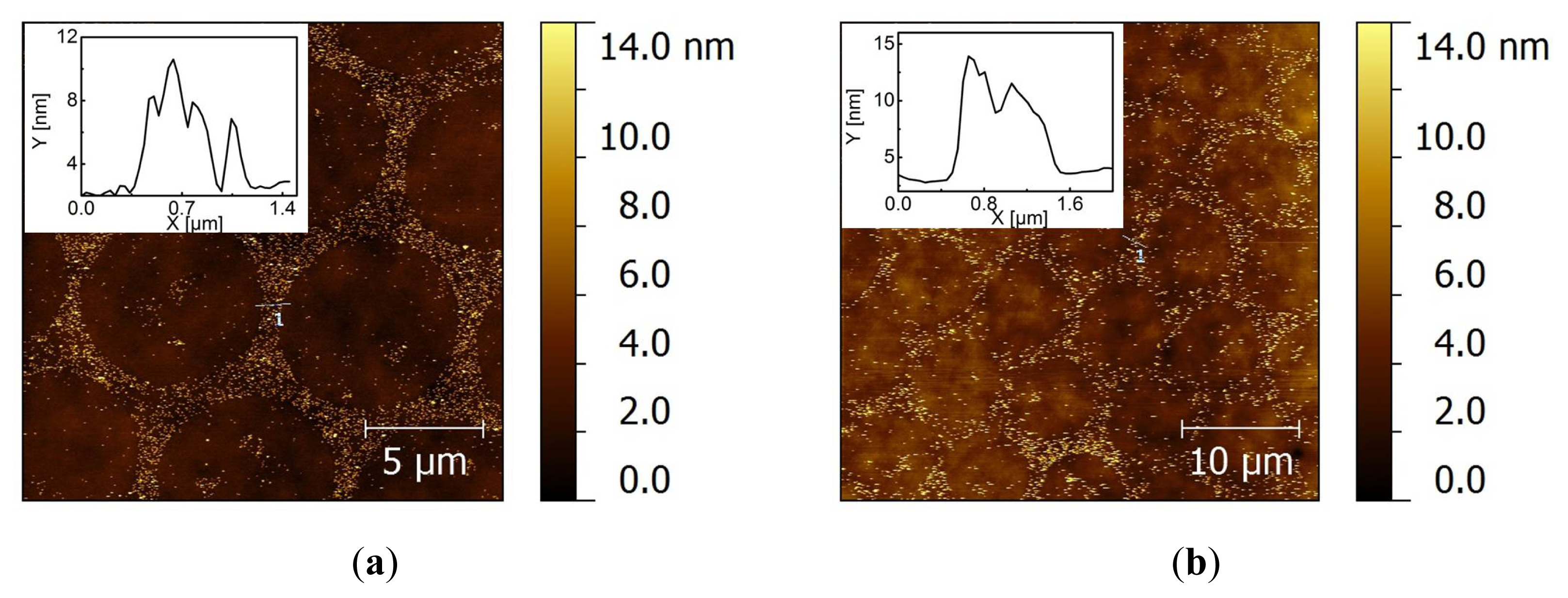
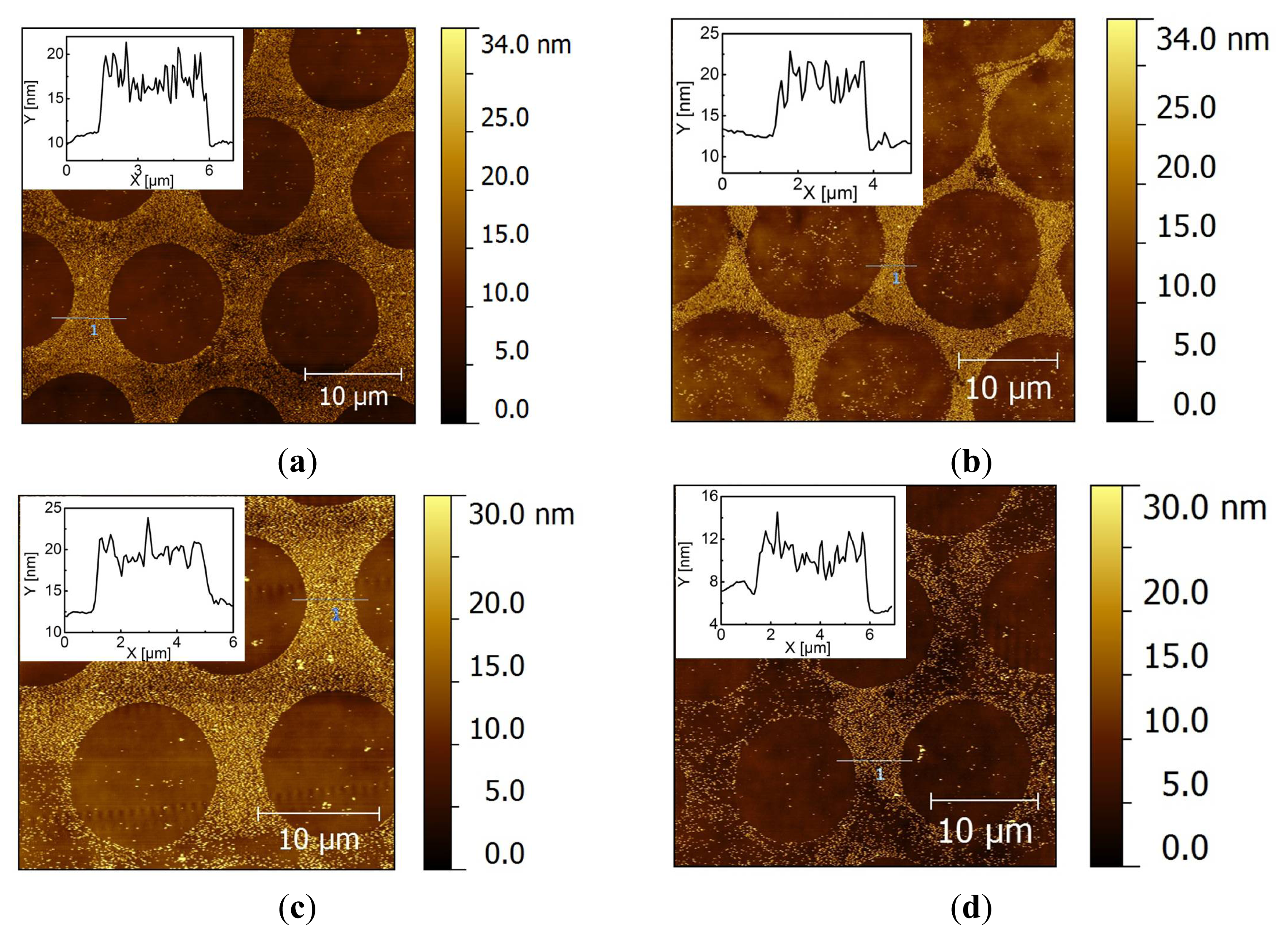
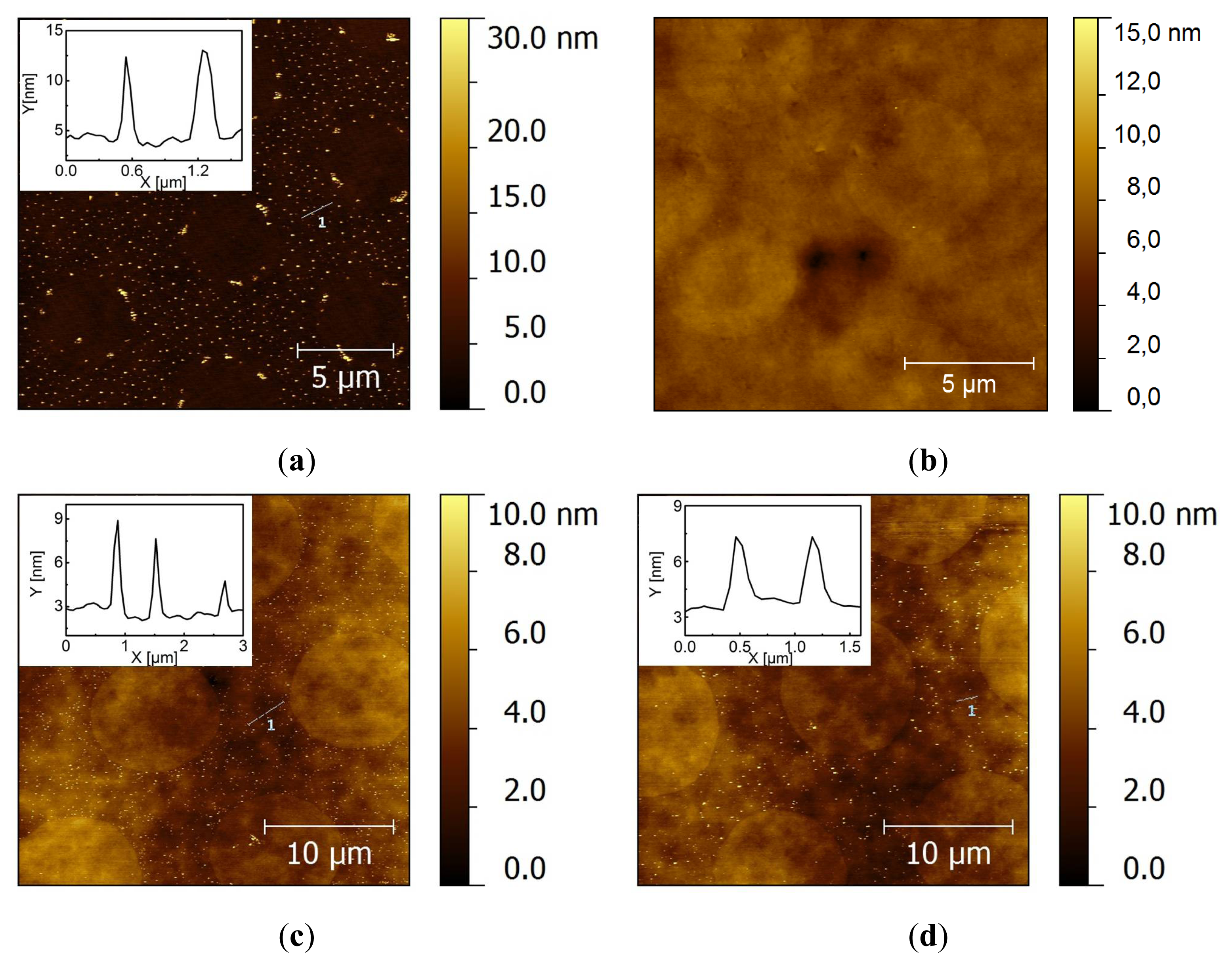
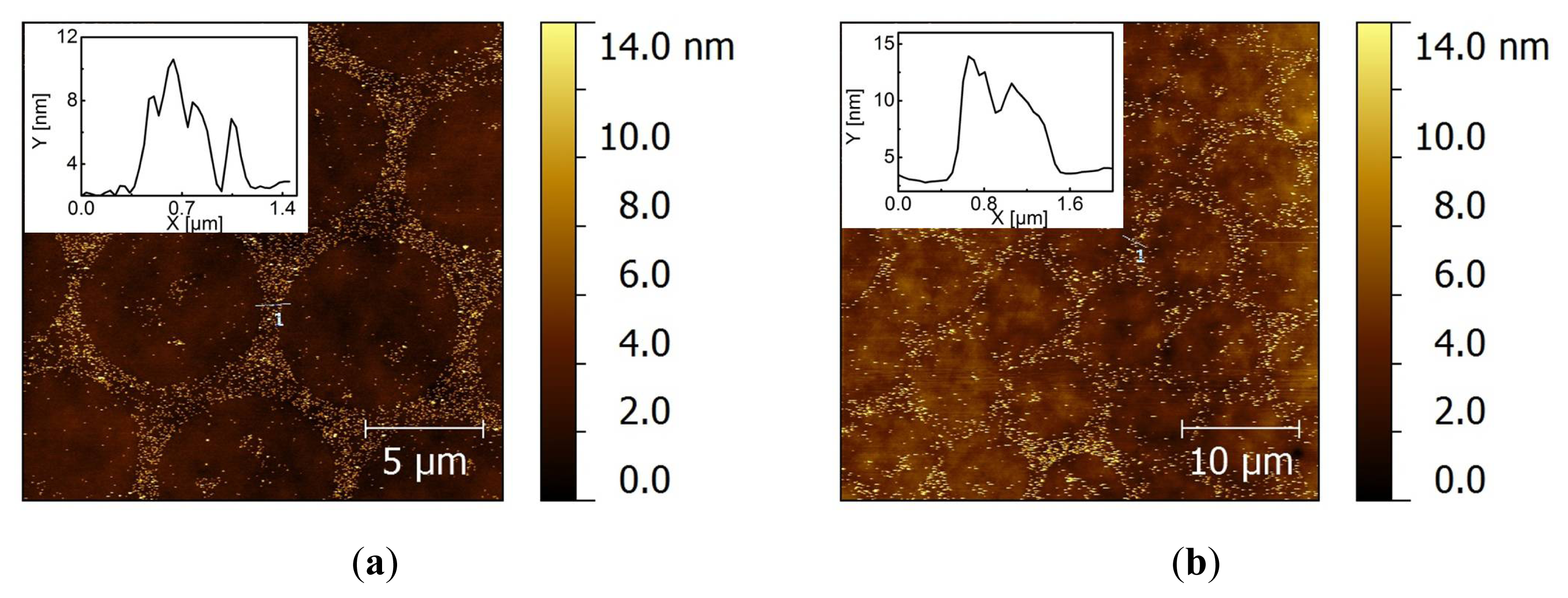
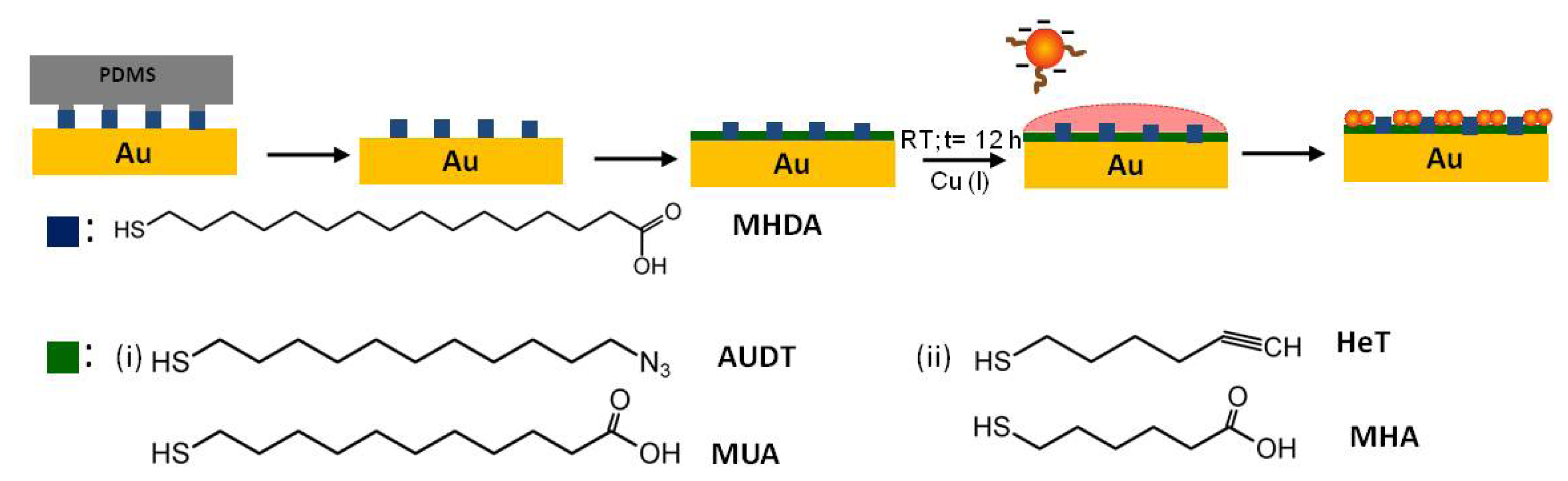
2.2. Covalent Bonding of Functionalized Au NPs onto Surfaces
3. Experimental Section
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Klein, D. An approach to electrical studies of single nanocrystals. Appl. Phys. Lett 1996, 68, 2574–2577. [Google Scholar]
- Sato, T.; Ahmed, H.; Brown, D.; Johnson, B.F.G. Single electron transistor using a molecularly linked gold colloidal particle chain. J. Appl. Phys 1997, 82, 696–702. [Google Scholar]
- Crooks, R.M.; Zhao, M.; Sun, L.; Chechik, V.; Yeung, L.K. Dendrimer-encapsulated metal nanoparticles: Synthesis, characterization, and applications to catalysis. Acc. Chem. Res 2000, 34, 181–190. [Google Scholar]
- Kim, B.-S.; Sung, J.-H.; Ju, W.-K.; Lee, M.-W.; Choi, C.-H.; Park, S.-G.; Lee, S.-G.; Lee, E.-H.; O, B.-H. Antireflective silicon subwavelength structure formed by self-aggregated gold nano particle as a catalyst. Microelectron. Eng 2011, 88, 2597–2600. [Google Scholar]
- Daniel, M.-C.; Astruc, D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev 2004, 35, 293–346. [Google Scholar]
- Hutchings, G.J.; Brust, M.; Schmidbaur, H. Gold—An introductory perspective. Chem. Soc. Rev 2008, 37, 1759–1765. [Google Scholar]
- Morpurgo, A.F.; Marcus, C.M.; Robinson, D.B. Controlled fabrication of metallic electrodes with atomic separation. Appl. Phys. Lett 1999, 74, 2084–2086. [Google Scholar]
- Reed, M.A.; Zhou, C.; Muller, C.J.; Burgin, T.P.; Tour, J.M. Conductance of a molecular junction. Science 1997, 278, 252–254. [Google Scholar]
- Davidovic, D.; Tinkham, M. Coulomb blockade and discrete energy levels in Au nanoparticles. Appl. Phys. Lett 1998, 73, 3959–3961. [Google Scholar]
- Negishi, R.; Hasegawa, T.; Terabe, K.; Aono, M.; Ebihara, T.; Tanaka, H.; Ogawa, T. Fabrication of nanoscale gaps using a combination of self-assembled molecular and electron beam lithographic techniques. Appl. Phys. Lett 2006, 88, 223111–223113. [Google Scholar]
- Balzani, V.; Credi, A.; Venturi, M. The bottom-up approach to molecular-level devices and machines. Chem. Eur. J 2002, 8, 5524–5532. [Google Scholar]
- Lu, W.; Lieber, C.M. Nanoelectronics from the bottom up. Nat. Mater 2007, 6, 841–850. [Google Scholar]
- Mizuta, H.; Oda, S. Bottom-up approach to silicon nanoelectronics. Microelectron. J 2008, 39, 171–176. [Google Scholar]
- Cederquist, K.B.; Keating, C.D. Curvature effects in DNA:Au nanoparticle conjugates. ACS Nano 2009, 3, 256–260. [Google Scholar]
- Tao, H.; Yi, C.; Tzang, C.-H.; Zhu, J.; Yang, M. DNA-directed self-assembly of gold nanoparticles into binary and ternary nanostructures. Nanotechnology 2007, 18, 015102. [Google Scholar]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar]
- Bernard, L.; Kamdzhilov, Y.; Calame, M.; van der Molen, S.J.; Liao, J.; Schönenberger, C. Spectroscopy of molecular junction networks obtained by place exchange in 2d nanoparticle arrays. J. Phys. Chem. C 2007, 111, 18445–18450. [Google Scholar]
- Bastus, N.G.; Comenge, J.; Puntes, V. Kinetically controlled seeded growth synthesis of citrate-stabilized gold nanoparticles of up to 200 nm: Size focusing versus ostwald ripening. Langmuir 2011, 27, 11098–11105. [Google Scholar]
- Ji, X.; Song, X.; Li, J.; Bai, Y.; Yang, W.; Peng, X. Size control of gold nanocrystals in citrate reduction: The third role of citrate. J. Am. Chem. Soc 2007, 129, 13939–13948. [Google Scholar]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Disc. Faraday Soc 1951, 11, 55–75. [Google Scholar]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiolderivatized gold nanoparticles in a two-phase liquid-liquid system. Chem. Commun. 1994, 801–802. [Google Scholar]
- Jana, N.R.; Peng, X. Single-phase and gram-scale routes toward nearly monodisperse Au and other noble metal nanocrystals. J. Am. Chem. Soc 2003, 125, 14280–14281. [Google Scholar]
- Goulet, P.J.G.; Lennox, R.B. New insights into brust-schiffrin metal nanoparticle synthesis. J. Am. Chem. Soc 2010, 132, 9582–9584. [Google Scholar]
- Maneeprakorn, W.; Malik, M.A.; O’Brien, P. Developing chemical strategies for the assembly of nanoparticles into mesoscopic objects. J. Am. Chem. Soc 2010, 132, 1780–1781. [Google Scholar]
- Lin, S.-Y.; Tsai, Y.-T.; Chen, C.-C.; Lin, C.-M.; Chen, C.-H. Two-step functionalization of neutral and positively charged thiols onto citrate-stabilized au nanoparticles. J. Phys. Chem. B 2004, 108, 2134–2139. [Google Scholar]
- Aslan, K.; Perez-Luna, V.H. Surface modification of colloidal gold by chemisorption of alkanethiols in the presence of a nonionic surfactant. Langmuir 2002, 18, 6059–6065. [Google Scholar]
- Bartczak, D.; Kanaras, A.G. Diacetylene-containing ligand as a new capping agent for the preparation of water-soluble colloidal nanoparticles of remarkable stability. Langmuir 2010, 26, 7072–7077. [Google Scholar]
- Locatelli, E.; Ori, G.; Fournelle, M.; Lemor, R.; Montorsi, M.; Comes Franchini, M. Click chemistry for the assembly of gold nanorods and silver nanoparticles. Chem. Eur. J 2011, 17, 9052–9056. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed 2001, 40, 2004–2021. [Google Scholar]
- Nandivada, H.; Jiang, X.; Lahann, J. Click chemistry: Versatility and control in the hands of materials scientists. Adv. Mater 2007, 19, 2197–2208. [Google Scholar]
- Binder, W.H. “Click”-chemistry in polymer and material science: The update. Macromol. Rapid Commun 2008, 29, 951–951. [Google Scholar]
- Feng, D.; Zhang, Y.; Shi, W.; Li, X.; Ma, H. A simple and sensitive method for visual detection of phosgene based on the aggregation of gold nanoparticles. Chem. Commun 2010, 46, 9203–9205. [Google Scholar]
- Janczewski, D.; Tomczak, N.; Liu, S.; Han, M.-Y.; Vancso, G.J. Covalent assembly of functional inorganic nanoparticles by “Click” Chemistry in water. Chem. Commun 2010, 46, 3253–3255. [Google Scholar]
- Li, H.; Yao, Y.; Han, C.; Zhan, J. Triazole-ester modified silver nanoparticles: Click synthesis and Cd2+ colorimetric sensing. Chem. Commun. 2009, 4812–4814. [Google Scholar]
- Xu, X.; Daniel, W.L.; Wei, W.; Mirkin, C.A. Colorimetric Cu2+ detection using DNA-modified gold-nanoparticle aggregates as probes and click chemistry. Small 2010, 6, 623–626. [Google Scholar]
- Zhang, M.-X.; Huang, B.-H.; Sun, X.-Y.; Pang, D.-W. Clickable gold nanoparticles as the building block of nanobioprobes. Langmuir 2010, 26, 10171–10176. [Google Scholar]
- Zhou, Y.; Wang, S.; Zhang, K.; Jiang, X. Visual detection of copper(ii) by azide- and alkyne-functionalized gold nanoparticles using click chemistry. Angew. Chem. Int. Ed 2008, 47, 7454–7456. [Google Scholar]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev 2005, 105, 1103–1170. [Google Scholar]
- Hostetler, M.J.; Wingate, J.E.; Zhong, C.-J.; Harris, J.E.; Vachet, R.W.; Clark, M.R.; Londono, J.D.; Green, S.J.; Stokes, J.J.; Wignall, G.D.; et al. Alkanethiolate gold cluster molecules with core diameters from 1.5 to 5.2 nm: Core and monolayer properties as a function of core size. Langmuir 1998, 14, 17–30. [Google Scholar]
- Dubois, L.H.; Nuzzo, R.G. Synthesis, structure, and properties of model organic surfaces. Annu. Rev. Phys. Chem 1992, 43, 437–463. [Google Scholar]
- Wang, Y.; Zeiri, O.; Neyman, A.; Stellacci, F.; Weinstock, I.A. Nucleation and island growth of alkanethiolate ligand domains on gold nanoparticles. ACS Nano 2012, 6, 629–640. [Google Scholar]
- Zheng, H.; Lee, I.; Rubner, M.F.; Hammond, P.T. Two component particle arrays on patterned polyelectrolyte multilayer templates. Adv. Mater 2002, 14, 569–572. [Google Scholar]
- Robert, N.; Jinsang, K. Directed self-assembly of nanogold using a chemically modified nanopatterned surface. Nanotechnology 2012, 23, 045602–045610. [Google Scholar]
- Ma, L.-C.; Subramanian, R.; Huang, H.-W.; Ray, V.; Kim, C.-U.; Koh, S.J. Electrostatic funneling for precise nanoparticle placement: A route to wafer-scale integration. Nano Lett 2007, 7, 439–445. [Google Scholar]
- Gwyddion. Available online: http://www.gwyddion.net accessed on 2 February 2013.
- Weiss, E.A.; Kaufman, G.K.; Kriebel, J.K.; Li, Z.; Schalek, R.; Whitesides, G.M. Si/SiO2-templated formation of ultraflat metal surfaces on glass, polymer, and solder supports: Their use as substrates for self-assembled monolayers. Langmuir 2007, 23, 9686–9694. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (%) | 1 | 2 | 10 |
|---|---|---|---|
| Dh (AUDT) (nm) | 14 ± 3 | 14 ± 3 | 57 ± 4 |
| Dh (HeT) (nm) | 84 ± 3 | 81 ± 3 | precipitates |
Supplementary Files
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rianasari, I.; De Jong, M.P.; Huskens, J.; Van der Wiel, W.G. Covalent Coupling of Nanoparticles with Low-Density Functional Ligands to Surfaces via Click Chemistry. Int. J. Mol. Sci. 2013, 14, 3705-3717. https://doi.org/10.3390/ijms14023705
Rianasari I, De Jong MP, Huskens J, Van der Wiel WG. Covalent Coupling of Nanoparticles with Low-Density Functional Ligands to Surfaces via Click Chemistry. International Journal of Molecular Sciences. 2013; 14(2):3705-3717. https://doi.org/10.3390/ijms14023705
Chicago/Turabian StyleRianasari, Ina, Michel P. De Jong, Jurriaan Huskens, and Wilfred G. Van der Wiel. 2013. "Covalent Coupling of Nanoparticles with Low-Density Functional Ligands to Surfaces via Click Chemistry" International Journal of Molecular Sciences 14, no. 2: 3705-3717. https://doi.org/10.3390/ijms14023705
APA StyleRianasari, I., De Jong, M. P., Huskens, J., & Van der Wiel, W. G. (2013). Covalent Coupling of Nanoparticles with Low-Density Functional Ligands to Surfaces via Click Chemistry. International Journal of Molecular Sciences, 14(2), 3705-3717. https://doi.org/10.3390/ijms14023705