Validation of Reference Genes for the Determination of Platelet Transcript Level in Healthy Individuals and in Patients with the History of Myocardial Infarction


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
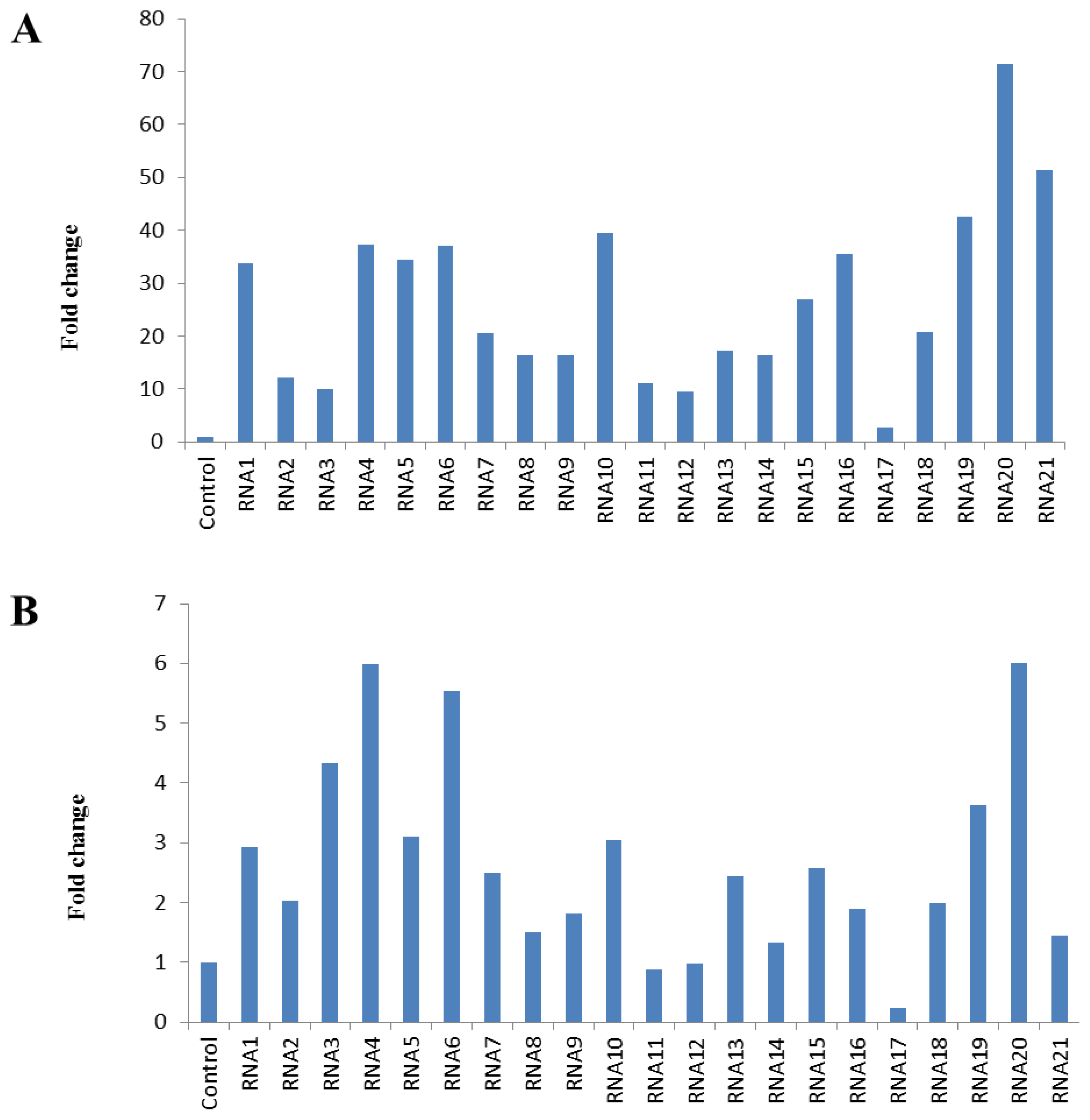
2.1. Evaluation of RNA Integrity, Contamination and Inhibition
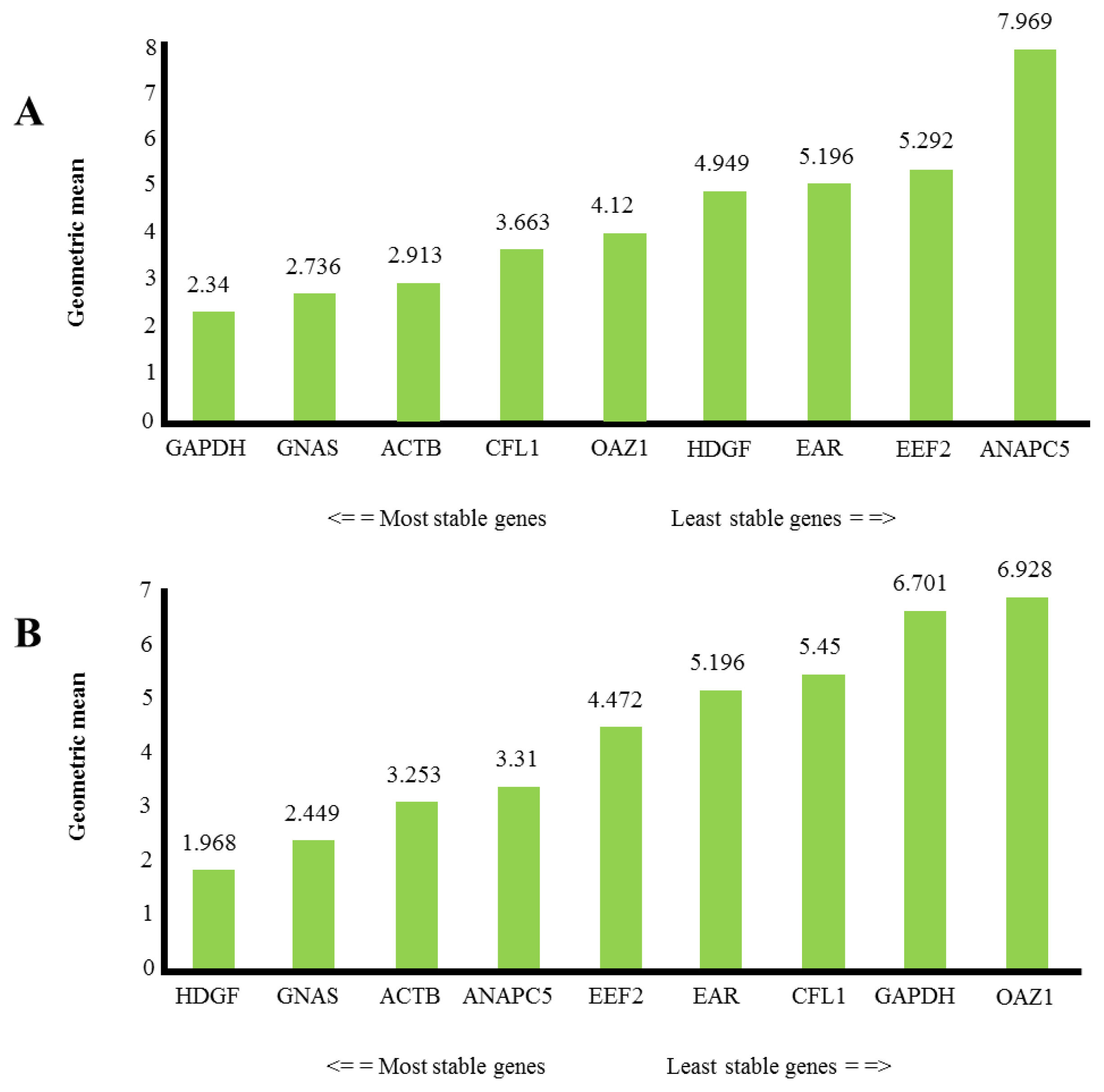
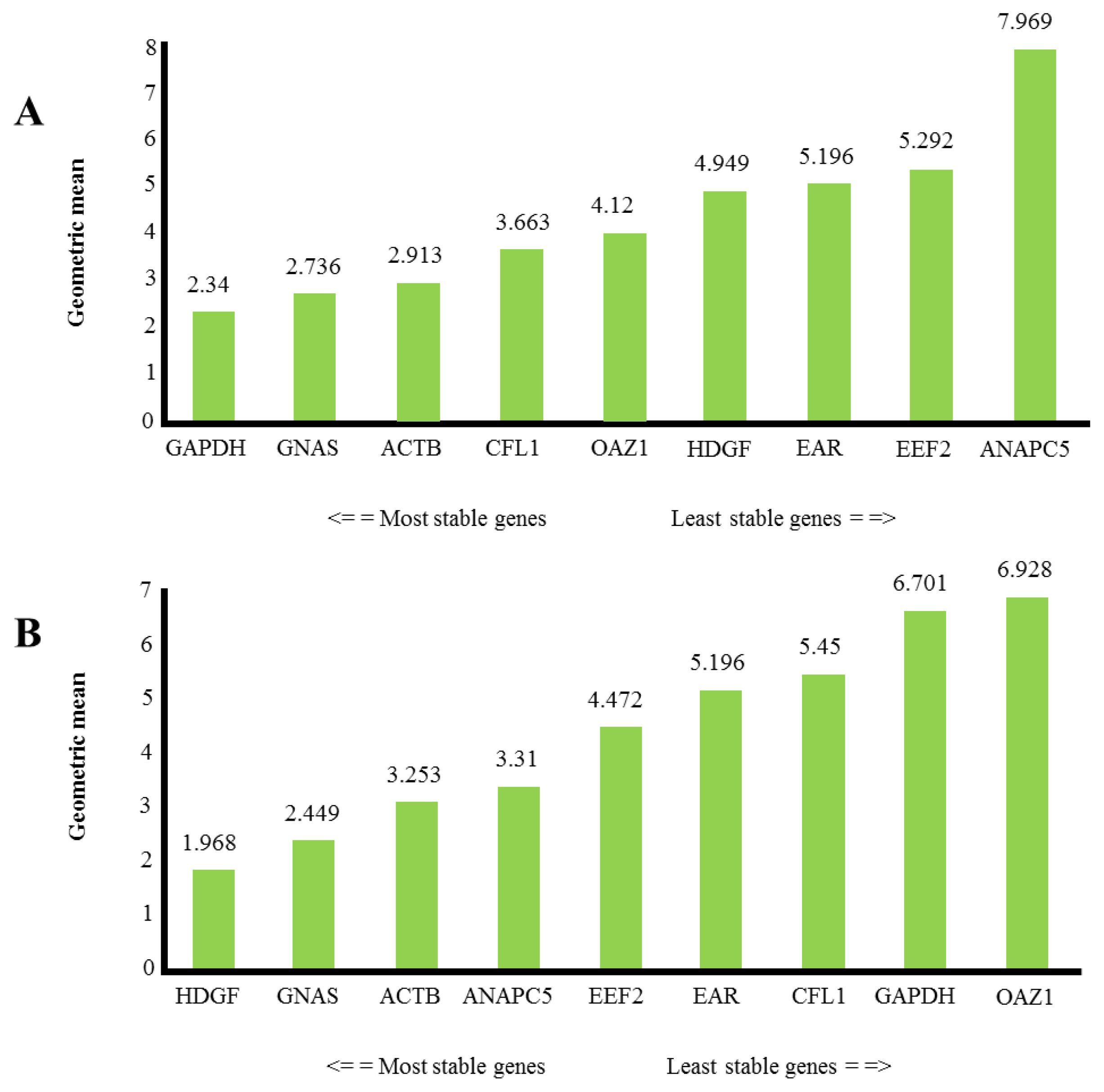
2.2. Expression Level of Putative Reference Genes
3. Discussion
4. Experimental Section
4.1. Subjects
4.2. Sample Collection and Preparation
4.3. Optimizing Primer Concentrations and PCR Efficiency
4.4. Reverse Transcription
4.5. qPCR
4.6. Data Analysis
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol 2000, 25, 169–193. [Google Scholar]
- Dheda, K.; Huggett, J.F.; Bustin, S.A.; Johnson, M.A.; Rook, G.; Zumla, A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004, 37. [Google Scholar]
- De Jonge, H.J.; Fehrmann, R.S.; de Bont, E.S.; Hofstra, R.M.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.; van der Zee, A.G.; te Meerman, G.J.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS One 2007, 2, e898. [Google Scholar]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc 2006, 1, 1559–1582. [Google Scholar]
- Brogren, H.; Karlsson, L.; Andersson, M.; Wang, L.; Erlinge, D.; Jern, S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood 2004, 104, 3943–3948. [Google Scholar]
- Censarek, P.; Steger, G.; Paolini, C.; Hohlfeld, T.; Grosser, T.; Zimmermann, N.; Fleckenstein, D.; Schror, K.; Weber, A.A. Alternative splicing of platelet cyclooxygenase-2 mRNA in patients after coronary artery bypass grafting. Thromb. Haemost 2007, 98, 1309–1315. [Google Scholar]
- Rox, J.M.; Bugert, P.; Muller, J.; Schorr, A.; Hanfland, P.; Madlener, K.; Kluter, H.; Potzsch, B. Gene expression analysis in platelets from a single donor: Evaluation of a PCR-based amplification technique. Clin. Chem 2004, 50, 2271–2278. [Google Scholar]
- Amisten, S.; Braun, O.O.; Bengtsson, A.; Erlinge, D. Gene expression profiling for the identification of G-protein coupled receptors in human platelets. Thromb. Res 2008, 122, 47–57. [Google Scholar]
- Amisten, S. A rapid and efficient platelet purification protocol for platelet gene expression studies. Methods Mol. Biol 2012, 788, 155–172. [Google Scholar]
- Toyama, K.; Karasawa, M.; Yamane, A.; Irisawa, H.; Yokohama, A.; Saitoh, T.; Handa, H.; Matsushima, T.; Sawamura, M.; Miyawaki, S.; et al. JAK2-V617F mutation analysis of granulocytes and platelets from patients with chronic myeloproliferative disorders: Advantage of studying platelets. Br. J. Haematol 2007, 139, 64–69. [Google Scholar]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004, 64, 5245–5250. [Google Scholar]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol 2006, 7, 33. [Google Scholar]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett 2004, 26, 509–515. [Google Scholar]
- Zhu, X.; Li, X.; Chen, W.; Chen, J.; Lu, W.; Chen, L.; Fu, D. Evaluation of new reference genes in papaya for accurate transcript normalization under different experimental conditions. PLoS One 2012, 7, e44405. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 2009, 55, 611–622. [Google Scholar]
- Axtner, J.; Sommer, S. Validation of internal reference genes for quantitative real-time PCR in a non-model organism, the yellow-necked mouse, Apodemus flavicollis. BMC Res. Notes 2009, 2, 264. [Google Scholar]
- Seol, D.; Choe, H.; Zheng, H.; Jang, K.; Ramakrishnan, P.S.; Lim, T.H.; Martin, J.A. Selection of reference genes for normalization of quantitative real-time PCR in organ culture of the rat and rabbit intervertebral disc. BMC Res. Notes 2011, 4, 162. [Google Scholar]
- Yan, J.; Yuan, F.; Long, G.; Qin, L.; Deng, Z. Selection of reference genes for quantitative real-time RT-PCR analysis in citrus. Mol. Biol. Rep 2012, 39, 1831–1838. [Google Scholar]
- Zampieri, M.; Ciccarone, F.; Guastafierro, T.; Bacalini, M.G.; Calabrese, R.; Moreno-Villanueva, M.; Reale, A.; Chevanne, M.; Burkle, A.; Caiafa, P. Validation of suitable internal control genes for expression studies in aging. Mech. Ageing Dev 2010, 131, 89–95. [Google Scholar]
- Karlovich, C.; Duchateau-Nguyen, G.; Johnson, A.; McLoughlin, P.; Navarro, M.; Fleurbaey, C.; Steiner, L.; Tessier, M.; Nguyen, T.; Wilhelm-Seiler, M.; et al. A longitudinal study of gene expression in healthy individuals. BMC Med. Genomics 2009, 2, 33. [Google Scholar]
- Nolan, T.; Hands, R.E.; Ogunkolade, W.; Bustin, S.A. SPUD: A quantitative PCR assay for the detection of inhibitors in nucleic acid preparations. Anal. Biochem 2006, 351, 308–310. [Google Scholar]
- Birschmann, I.; Mietner, S.; Dittrich, M.; Pfrang, J.; Dandekar, T.; Walter, U. Use of functional highly purified human platelets for the identification of new proteins of the IPP signaling pathway. Thromb. Res 2008, 122, 59–68. [Google Scholar]
- Zhu, J.; He, F.; Song, S.; Wang, J.; Yu, J. How many human genes can be defined as housekeeping with current expression data? BMC Genomics 2008, 9, 172. [Google Scholar]
- Marullo, M.; Zuccato, C.; Mariotti, C.; Lahiri, N.; Tabrizi, S.J.; di Donato, S.; Cattaneo, E. Expressed Alu repeats as a novel, reliable tool for normalization of real-time quantitative RT-PCR data. Genome Biol 2010, 11, R9. [Google Scholar]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001, 29, e45. [Google Scholar]
- Pestana, E.A.; Belak, S.; Diallo, A.; Crowther, J.R.; Viljoen, G.J. Early, Rapid and Sensitive Veterinary Molecular Diagnostics—Real Time PCR Applications; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Pachot, A.; Blond, J.L.; Mougin, B.; Miossec, P. Peptidylpropyl isomerase B (PPIB): A suitable reference gene for mRNA quantification in peripheral whole blood. J. Biotechnol 2004, 114, 121–124. [Google Scholar]
- Mikeska, T.; Dobrovic, A. Validation of a primer optimisation matrix to improve the performance of reverse transcription—Quantitative real-time PCR assays. BMC Res. Notes 2009, 2, 112. [Google Scholar]
- Tichopad, A.; Kitchen, R.; Riedmaier, I.; Becker, C.; Stahlberg, A.; Kubista, M. Design and optimization of reverse-transcription quantitative PCR experiments. Clin. Chem 2009, 55, 1816–1823. [Google Scholar]
- Universal ProbeLibrary System. Available online: https://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp accessed on 5 February 2013.
- Lefever, S.; Vandesompele, J.; Speleman, F.; Pattyn, F. RTPrimerDB: The portal for real-time PCR primers and probes. Nucleic Acids Res 2009, 37, D942–D945. [Google Scholar]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 2007, 35, W71–W74. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zsóri, K.S.; Muszbek, L.; Csiki, Z.; Shemirani, A.H. Validation of Reference Genes for the Determination of Platelet Transcript Level in Healthy Individuals and in Patients with the History of Myocardial Infarction. Int. J. Mol. Sci. 2013, 14, 3456-3466. https://doi.org/10.3390/ijms14023456
Zsóri KS, Muszbek L, Csiki Z, Shemirani AH. Validation of Reference Genes for the Determination of Platelet Transcript Level in Healthy Individuals and in Patients with the History of Myocardial Infarction. International Journal of Molecular Sciences. 2013; 14(2):3456-3466. https://doi.org/10.3390/ijms14023456
Chicago/Turabian StyleZsóri, Katalin S., László Muszbek, Zoltán Csiki, and Amir H. Shemirani. 2013. "Validation of Reference Genes for the Determination of Platelet Transcript Level in Healthy Individuals and in Patients with the History of Myocardial Infarction" International Journal of Molecular Sciences 14, no. 2: 3456-3466. https://doi.org/10.3390/ijms14023456
APA StyleZsóri, K. S., Muszbek, L., Csiki, Z., & Shemirani, A. H. (2013). Validation of Reference Genes for the Determination of Platelet Transcript Level in Healthy Individuals and in Patients with the History of Myocardial Infarction. International Journal of Molecular Sciences, 14(2), 3456-3466. https://doi.org/10.3390/ijms14023456