Connecting Chromatin Modifying Factors to DNA Damage Response

Abstract
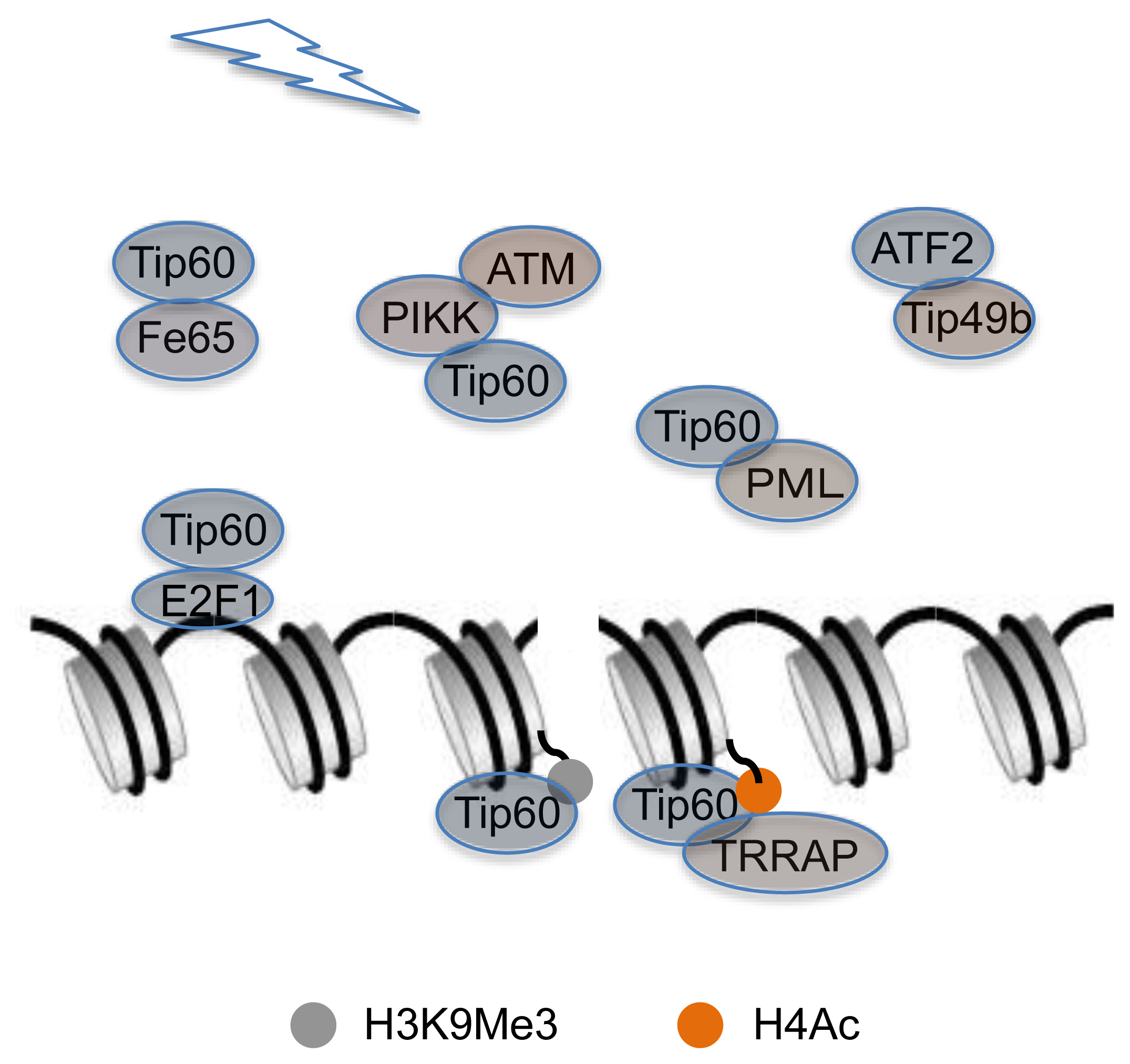
: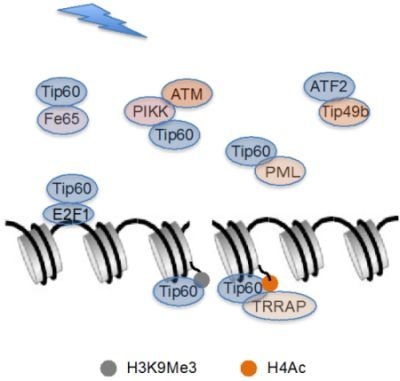
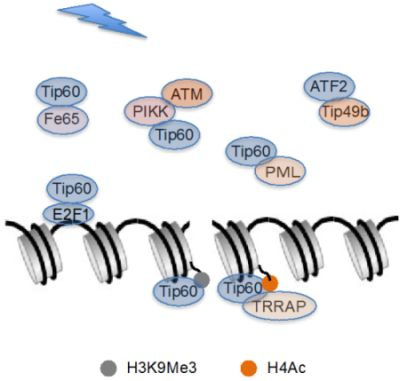{kind=link}
{kind=link}
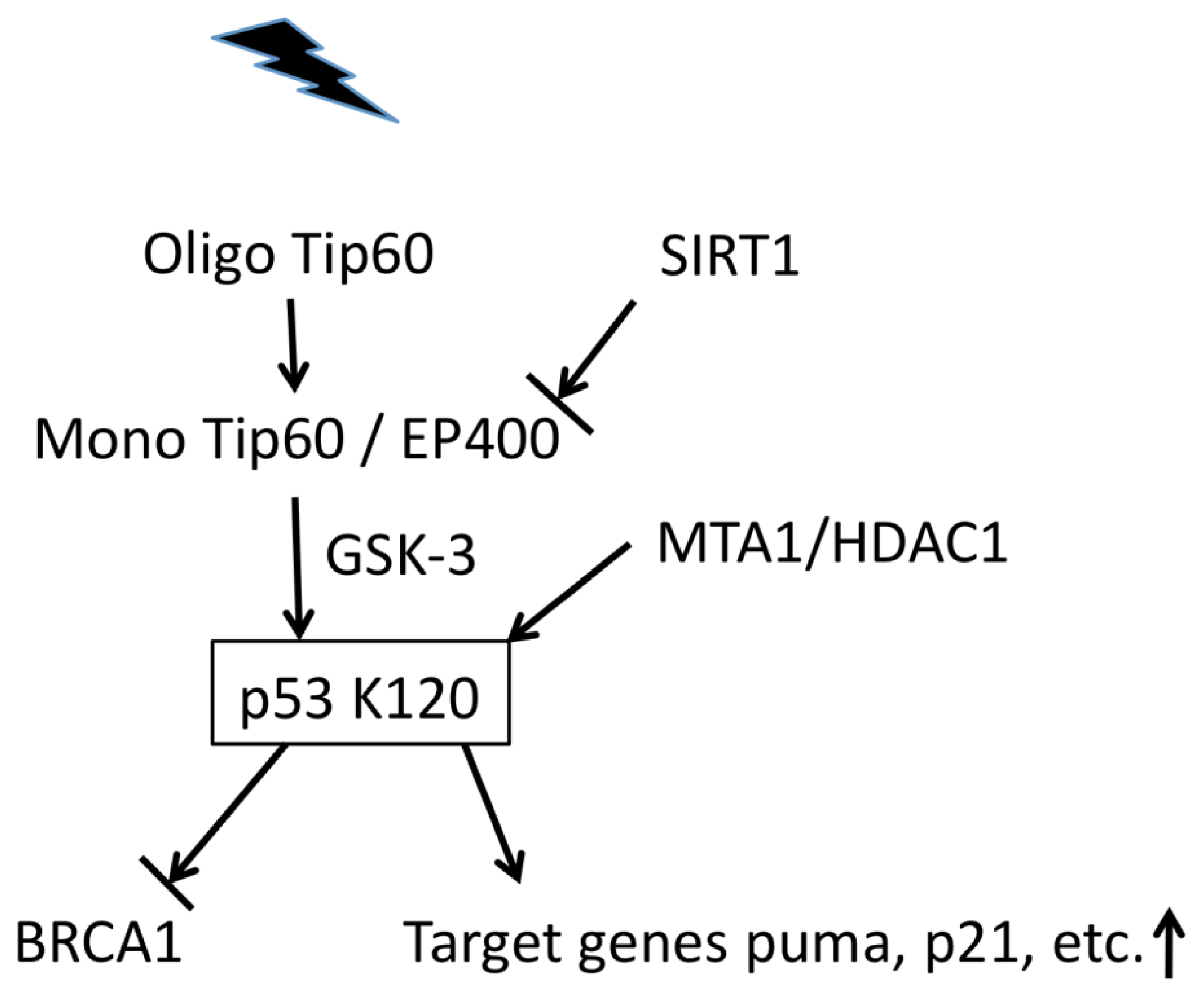
{kind=link}
{kind=link}
1. Introduction
2. Signal Transduction Pathways in DNA Damage Response Communicate with Chromatin-Remodeling Factors
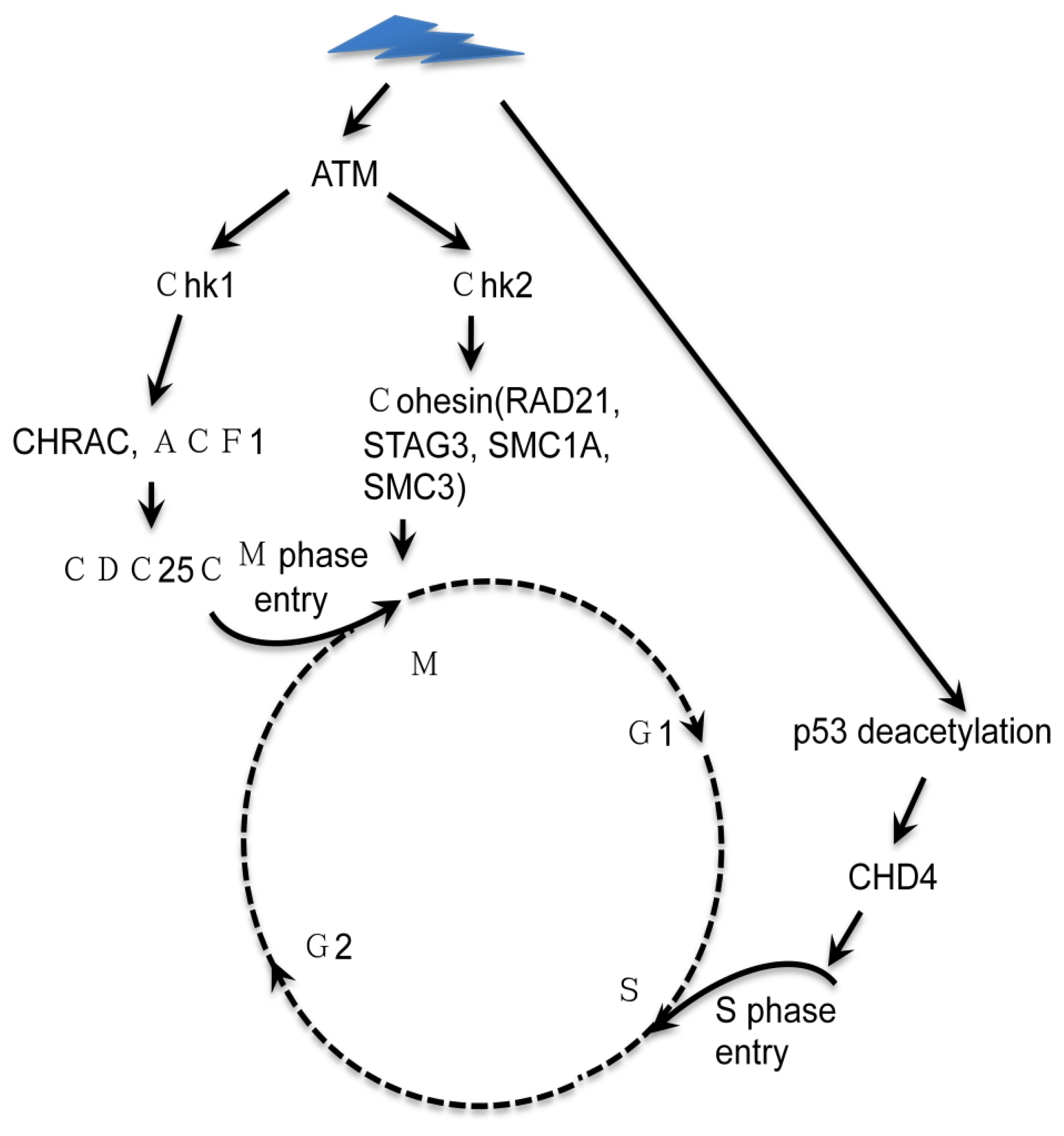
3. Interplay of Chromatin Remodeling and DDR during Cell Cycle Progression
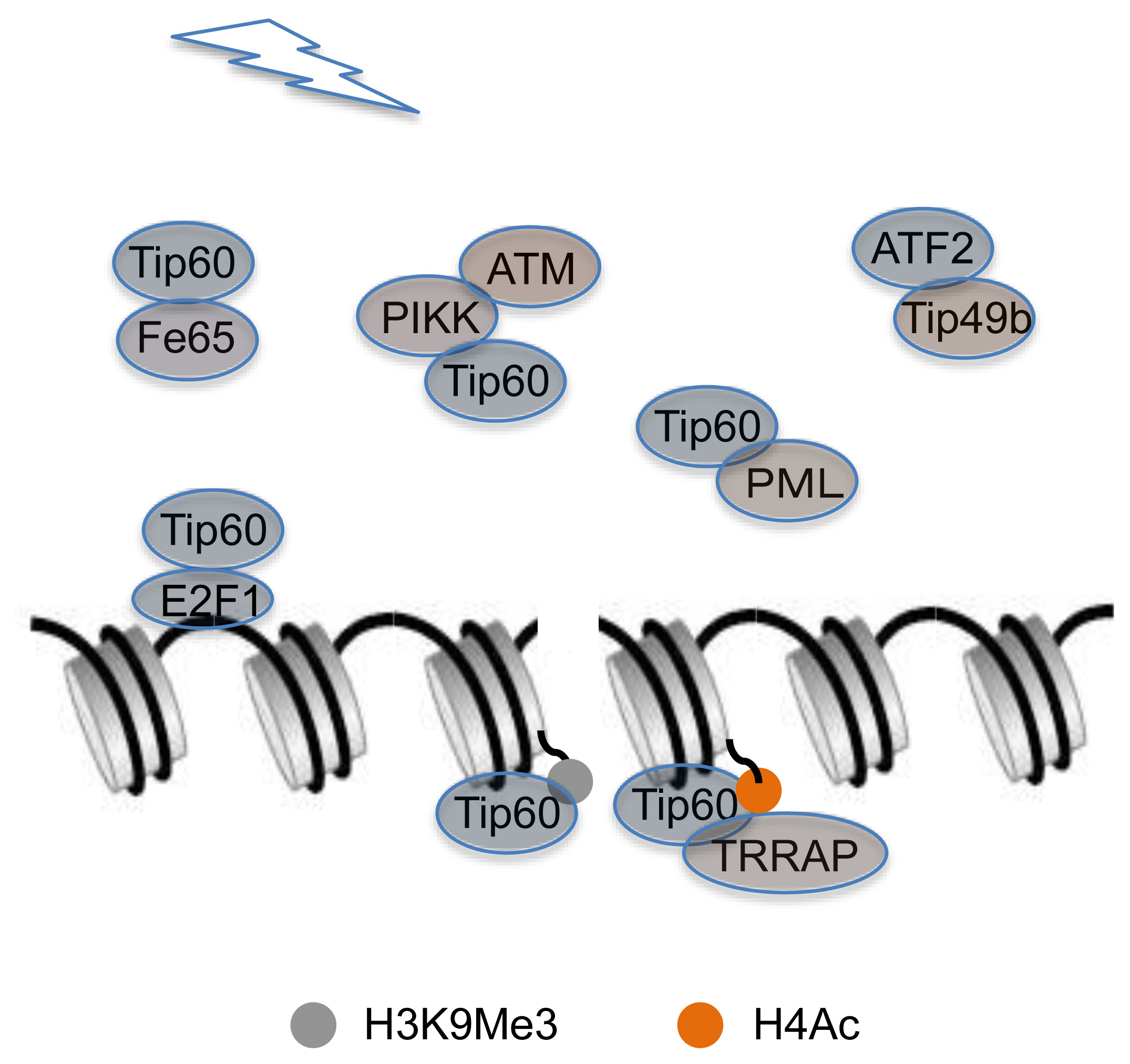
4. Interaction between DDR-Related Proteins and Chromatin-Remodeling Factor
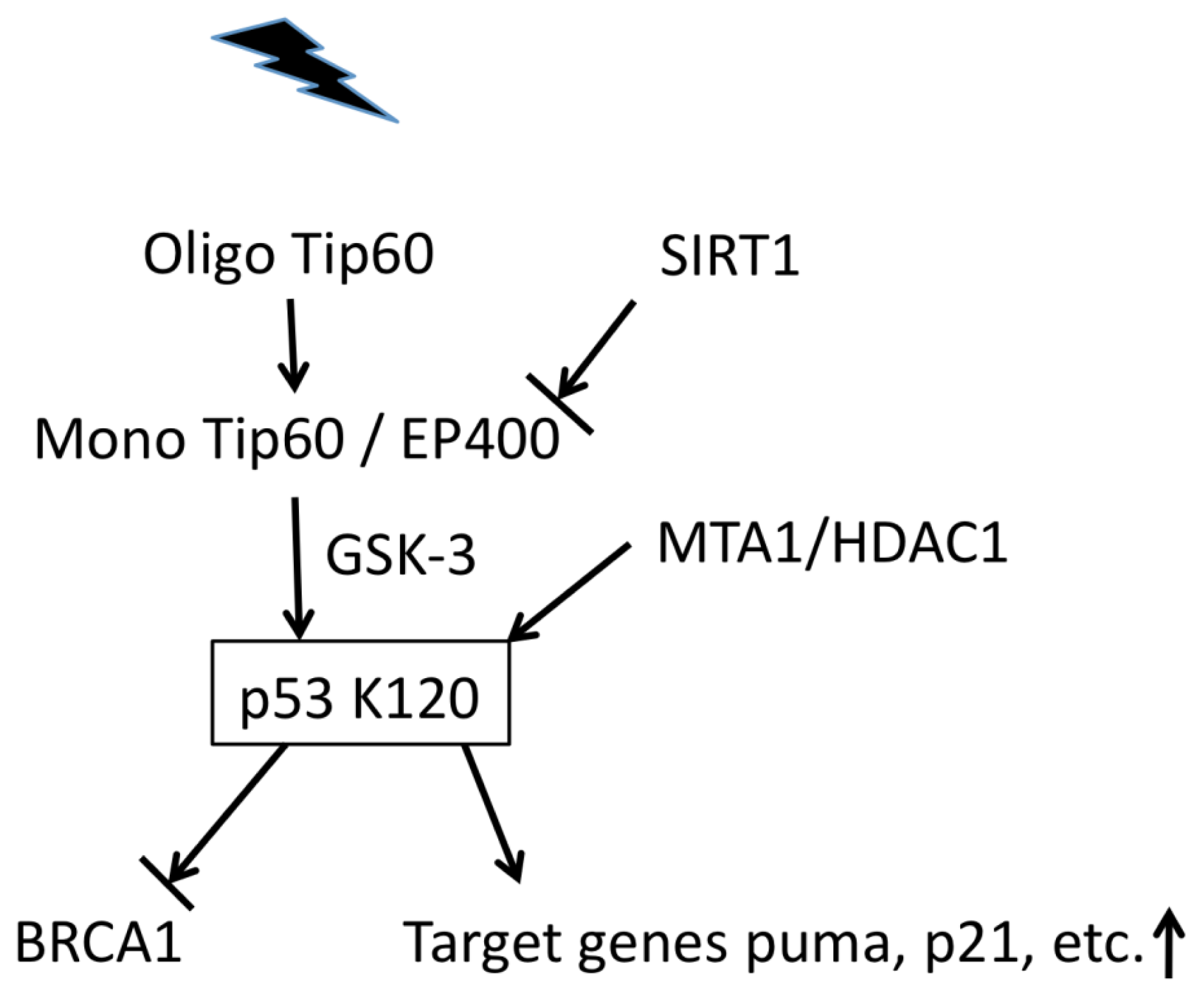
5. Modification of Chromatin-Remodeling Factors Is Linked to the DNA Damage Response
6. Direct Effects of Chromatin-Remodeling Factors on the Recruitment of DNA Damage Response Proteins
7. Conclusion and Perspective
Acknowledgements
Conflict of Interest
References
- Sherman, M.H.; Bassing, C.H.; Teitell, M.A. Regulation of cell differentiation by the DNA damage response. Trends Cell Biol 2011, 21, 312–319. [Google Scholar]
- Downs, J.A.; Nussenzweig, M.C.; Nussenzweig, A. Chromatin dynamics and the preservation of genetic information. Nature 2007, 447, 951–958. [Google Scholar]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar]
- Van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar]
- Cohn, M.A.; D’Andrea, A.D. Chromatin recruitment of DNA repair proteins: Lessons from the fanconi anemia and double-strand break repair pathways. Mol. Cell 2008, 32, 306–312. [Google Scholar]
- Morrison, A.J.; Shen, X. Chromatin remodelling beyond transcription: The INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol 2009, 10, 373–384. [Google Scholar]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar]
- Wu, J.I.; Lessard, J.; Crabtree, G.R. Understanding the words of chromatin regulation. Cell 2009, 136, 200–206. [Google Scholar]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem 2009, 78, 273–304. [Google Scholar]
- Mills, A.A. Throwing the cancer switch: Reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol 2006, 7, 739–750. [Google Scholar]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol 2003, 4, 712–720. [Google Scholar]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol 2010, 11, 196–207. [Google Scholar]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar]
- Bao, Y.; Shen, X. Chromatin remodeling in DNA double-strand break repair. Curr. Opin. Genet. Dev 2007, 17, 126–131. [Google Scholar]
- Kranz, D.; Dohmesen, C.; Dobbelstein, M. BRCA1 and Tip60 determine the cellular response to ultraviolet irradiation through distinct pathways. J. Cell Biol 2008, 182, 197–213. [Google Scholar]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J.; et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol 2010, 8, e1000533. [Google Scholar]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 2011, 42, 584–596. [Google Scholar]
- Tyteca, S.; Vandromme, M.; Legube, G.; Chevillard-Briet, M.; Trouche, D. Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. EMBO J 2006, 25, 1680–1689. [Google Scholar]
- Mattera, L.; Escaffit, F.; Pillaire, M.J.; Selves, J.; Tyteca, S.; Hoffmann, J.S.; Gourraud, P.A.; Chevillard-Briet, M.; Cazaux, C.; Trouche, D. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 2009, 28, 1506–1517. [Google Scholar]
- Li, D.Q.; Ohshiro, K.; Reddy, S.D.; Pakala, S.B.; Lee, M.H.; Zhang, Y.; Rayala, S.K.; Kumar, R. E3 ubiquitin ligase COP1 regulates the stability and functions of MTA1. Proc. Natl. Acad. Sci. USA 2009, 106, 17493–17498. [Google Scholar]
- Li, D.Q.; Divijendra Natha Reddy, S.; Pakala, S.B.; Wu, X.; Zhang, Y.; Rayala, S.K.; Kumar, R. MTA1 coregulator regulates p53 stability and function. J. Biol. Chem 2009, 284, 34545–34552. [Google Scholar]
- Li, D.Q.; Ohshiro, K.; Khan, M.N.; Kumar, R. Requirement of MTA1 in ATR-mediated DNA damage checkpoint function. J. Biol. Chem 2010, 285, 19802–19812. [Google Scholar]
- Li, D.Q.; Pakala, S.B.; Reddy, S.D.; Ohshiro, K.; Peng, S.H.; Lian, Y.; Fu, S.W.; Kumar, R. Revelation of p53-independent function of MTA1 in DNA damage response via modulation of the p21 WAF1-proliferating cell nuclear antigen pathway. J. Biol. Chem 2010, 285, 10044–10052. [Google Scholar]
- Calonge, T.M.; Eshaghi, M.; Liu, J.; Ronai, Z.; O’Connell, M.J. Transformation/transcription domain-associated protein (TRRAP)-mediated regulation of Wee1. Genetics 2010, 185, 81–93. [Google Scholar]
- Pogribny, I.; Koturbash, I.; Tryndyak, V.; Hudson, D.; Stevenson, S.M.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol. Cancer Res 2005, 3, 553–561. [Google Scholar]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar]
- Cortazar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470, 419–423. [Google Scholar]
- Li, D.Q.; Kumar, R. Mi-2/NuRD complex making inroads into DNA-damage response pathway. Cell Cycle 2010, 9, 2071–2079. [Google Scholar]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Danielsen, J.M.; Menard, P.; Sand, J.C.; Stucki, M.; et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol 2010, 190, 731–740. [Google Scholar] [Green Version]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 2010, 29, 3130–3139. [Google Scholar]
- Couteau, F.; Zetka, M. DNA damage during meiosis induces chromatin remodeling and synaptonemal complex disassembly. Dev. Cell 2011, 20, 353–363. [Google Scholar]
- Sanchez-Molina, S.; Mortusewicz, O.; Bieber, B.; Auer, S.; Eckey, M.; Leonhardt, H.; Friedl, A.A.; Becker, P.B. Role for hACF1 in the G2/M damage checkpoint. Nucl. Acids Res 2011, 39, 8445–8456. [Google Scholar]
- Xu, H.; Tomaszewski, J.M.; McKay, M.J. Can corruption of chromosome cohesion create a conduit to cancer? Nat. Rev. Cancer 2011, 11, 199–210. [Google Scholar]
- Unal, E.; Arbel-Eden, A.; Sattler, U.; Shroff, R.; Lichten, M.; Haber, J.E.; Koshland, D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 2004, 16, 991–1002. [Google Scholar]
- Strom, L.; Lindroos, H.B.; Shirahige, K.; Sjogren, C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 2004, 16, 1003–1015. [Google Scholar]
- Dorsett, D.; Strom, L. The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr. Biol 2012, 22, R240–R250. [Google Scholar]
- Unal, E.; Heidinger-Pauli, J.M.; Koshland, D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 2007, 317, 245–248. [Google Scholar]
- Strom, L.; Karlsson, C.; Lindroos, H.B.; Wedahl, S.; Katou, Y.; Shirahige, K.; Sjogren, C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 2007, 317, 242–245. [Google Scholar]
- Watrin, E.; Peters, J.M. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J 2009, 28, 2625–2635. [Google Scholar] [Green Version]
- Lans, H.; Marteijn, J.A.; Schumacher, B.; Hoeijmakers, J.H.; Jansen, G.; Vermeulen, W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet 2010, 6, e1000941. [Google Scholar]
- Sinha, S.; Malonia, S.K.; Mittal, S.P.; Singh, K.; Kadreppa, S.; Kamat, R.; Mukhopadhyaya, R.; Pal, J.K.; Chattopadhyay, S. Coordinated regulation of p53 apoptotic targets BAX and PUMA by SMAR1 through an identical MAR element. EMBO J 2010, 29, 830–842. [Google Scholar]
- Van den Broeck, A.; Nissou, D.; Brambilla, E.; Eymin, B.; Gazzeri, S. Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis 2012, 33, 320–325. [Google Scholar]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol 2009, 11, 1376–1382. [Google Scholar]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem 2006, 281, 15741–15746. [Google Scholar]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar]
- Wu, Q.; Hu, H.; Lan, J.; Emenari, C.; Wang, Z.; Chang, K.S.; Huang, H.; Yao, X. PML3 orchestrates the nuclear dynamics and function of TIP60. J. Biol. Chem 2009, 284, 8747–8759. [Google Scholar]
- Cho, S.G.; Bhoumik, A.; Broday, L.; Ivanov, V.; Rosenstein, B.; Ronai, Z. TIP49b, a regulator of activating transcription factor 2 response to stress and DNA damage. Mol. Cell. Biol 2001, 21, 8398–8413. [Google Scholar]
- Stante, M.; Minopoli, G.; Passaro, F.; Raia, M.; Vecchio, L.D.; Russo, T. Fe65 is required for Tip60-directed histone H4 acetylation at DNA strand breaks. Proc. Natl. Acad. Sci. USA 2009, 106, 5093–5098. [Google Scholar]
- Minopoli, G.; Stante, M.; Napolitano, F.; Telese, F.; Aloia, L.; de Felice, M.; di Lauro, R.; Pacelli, R.; Brunetti, A.; Zambrano, N.; et al. Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. J. Biol. Chem 2007, 282, 831–835. [Google Scholar]
- Sy, S.M.; Huen, M.S.; Chen, J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J. Biol. Chem 2009, 284, 21127–21131. [Google Scholar]
- Hayakawa, T.; Zhang, F.; Hayakawa, N.; Ohtani, Y.; Shinmyozu, K.; Nakayama, J.; Andreassen, P.R. MRG15 binds directly to PALB2 and stimulates homology-directed repair of chromosomal breaks. J. Cell Sci 2010, 123, 1124–1130. [Google Scholar]
- Bleuyard, J.Y.; Buisson, R.; Masson, J.Y.; Esashi, F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO Rep 2012, 13, 135–141. [Google Scholar]
- Chen, M.; Pereira-Smith, O.M.; Tominaga, K. Loss of the chromatin regulator MRG15 limits neural stem/progenitor cell proliferation via increased expression of the p21 Cdk inhibitor. Stem Cell Res 2011, 7, 75–88. [Google Scholar]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R.J.; Kaneda, Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009, 460, 287–291. [Google Scholar]
- Marango, J.; Shimoyama, M.; Nishio, H.; Meyer, J.A.; Min, D.J.; Sirulnik, A.; Martinez-Martinez, Y.; Chesi, M.; Bergsagel, P.L.; Zhou, M.M.; et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 2008, 111, 3145–3154. [Google Scholar]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar]
- Tian, C.; Xing, G.; Xie, P.; Lu, K.; Nie, J.; Wang, J.; Li, L.; Gao, M.; Zhang, L.; He, F. KRAB-type zinc-finger protein Apak specifically regulates p53-dependent apoptosis. Nat. Cell Biol 2009, 11, 580–591. [Google Scholar]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol 2010, 17, 1144–1151. [Google Scholar]
- Chung, Y.L.; Tsai, T.Y. Promyelocytic leukemia nuclear bodies link the DNA damage repair pathway with hepatitis B virus replication: implications for hepatitis B virus exacerbation during chemotherapy and radiotherapy. Mol. Cancer Res 2009, 7, 1672–1685. [Google Scholar]
- Mao, Z.; Hine, C.; Tian, X.; van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar]
- Jha, S.; Shibata, E.; Dutta, A. Human Rvb1/Tip49 is required for the histone acetyltransferase activity of Tip60/NuA4 and for the downregulation of phosphorylation on H2AX after DNA damage. Mol. Cell. Biol 2008, 28, 2690–2700. [Google Scholar]
- Garcia, S.N.; Kirtane, B.M.; Podlutsky, A.J.; Pereira-Smith, O.M.; Tominaga, K. Mrg15 null and heterozygous mouse embryonic fibroblasts exhibit DNA-repair defects post exposure to gamma ionizing radiation. FEBS Lett 2007, 581, 5275–5281. [Google Scholar]
- Cheng, Z.; Ke, Y.; Ding, X.; Wang, F.; Wang, H.; Wang, W.; Ahmed, K.; Liu, Z.; Xu, Y.; Aikhionbare, F.; et al. Functional characterization of TIP60 sumoylation in UV-irradiated DNA damage response. Oncogene 2008, 27, 931–941. [Google Scholar]
- Ikura, T.; Tashiro, S.; Kakino, A.; Shima, H.; Jacob, N.; Amunugama, R.; Yoder, K.; Izumi, S.; Kuraoka, I.; Tanaka, K.; et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol 2007, 27, 7028–7040. [Google Scholar]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–95. [Google Scholar]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grofte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 2012, 150, 697–709. [Google Scholar]
- Gospodinov, A.; Tsaneva, I.; Anachkova, B. RAD51 foci formation in response to DNA damage is modulated by TIP49. Int. J. Biochem. Cell Biol 2009, 41, 925–933. [Google Scholar]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol 2008, 18, 566–575. [Google Scholar]
- Papamichos-Chronakis, M.; Watanabe, S.; Rando, O.J.; Peterson, C.L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar]
- Hogan, C.J.; Aligianni, S.; Durand-Dubief, M.; Persson, J.; Will, W.R.; Webster, J.; Wheeler, L.; Mathews, C.K.; Elderkin, S.; Oxley, D.; et al. Fission yeast Iec1-ino80-mediated nucleosome eviction regulates nucleotide and phosphate metabolism. Mol. Cell. Biol 2010, 30, 657–674. [Google Scholar]
- Tao, Y.; Xi, S.; Shan, J.; Maunakea, A.; Che, A.; Briones, V.; Lee, E.Y.; Geiman, T.; Huang, J.; Stephens, R.; et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc. Natl. Acad. Sci. USA 2011, 108, 5626–5631. [Google Scholar]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489, 83–90. [Google Scholar]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar]
- Tkach, J.M.; Yimit, A.; Lee, A.Y.; Riffle, M.; Costanzo, M.; Jaschob, D.; Hendry, J.A.; Ou, J.; Moffat, J.; Boone, C.; et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell. Biol 2012, 14, 966–976. [Google Scholar]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med 2011, 17, 330–339. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; de Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; d’Adda di Fagagna, F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lai, W.; Li, H.; Liu, S.; Tao, Y. Connecting Chromatin Modifying Factors to DNA Damage Response. Int. J. Mol. Sci. 2013, 14, 2355-2369. https://doi.org/10.3390/ijms14022355
Lai W, Li H, Liu S, Tao Y. Connecting Chromatin Modifying Factors to DNA Damage Response. International Journal of Molecular Sciences. 2013; 14(2):2355-2369. https://doi.org/10.3390/ijms14022355
Chicago/Turabian StyleLai, Weiwei, Hongde Li, Shuang Liu, and Yongguang Tao. 2013. "Connecting Chromatin Modifying Factors to DNA Damage Response" International Journal of Molecular Sciences 14, no. 2: 2355-2369. https://doi.org/10.3390/ijms14022355
APA StyleLai, W., Li, H., Liu, S., & Tao, Y. (2013). Connecting Chromatin Modifying Factors to DNA Damage Response. International Journal of Molecular Sciences, 14(2), 2355-2369. https://doi.org/10.3390/ijms14022355