Necrostatin-1 Attenuates Ischemia Injury Induced Cell Death in Rat Tubular Cell Line NRK-52E through Decreased Drp1 Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
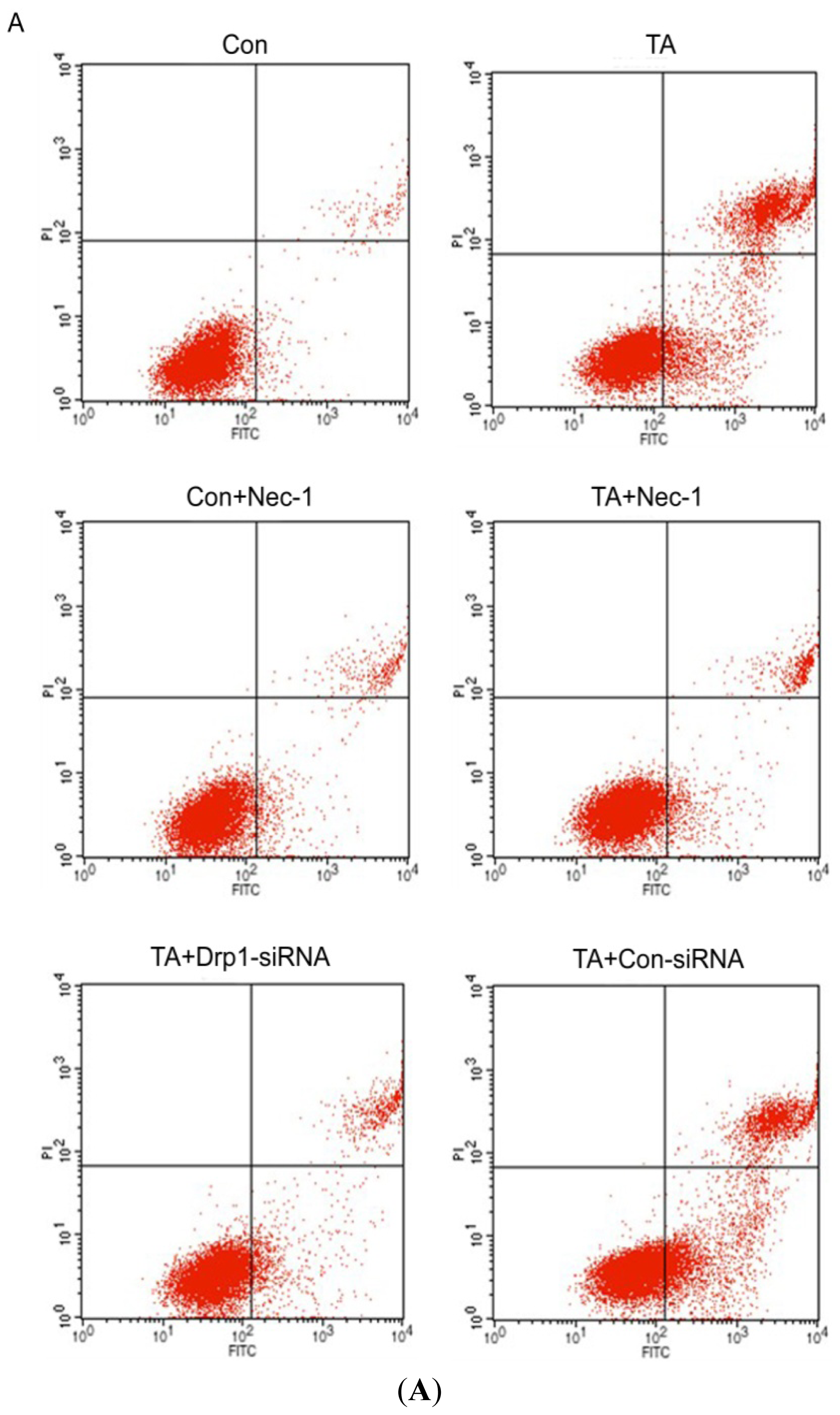
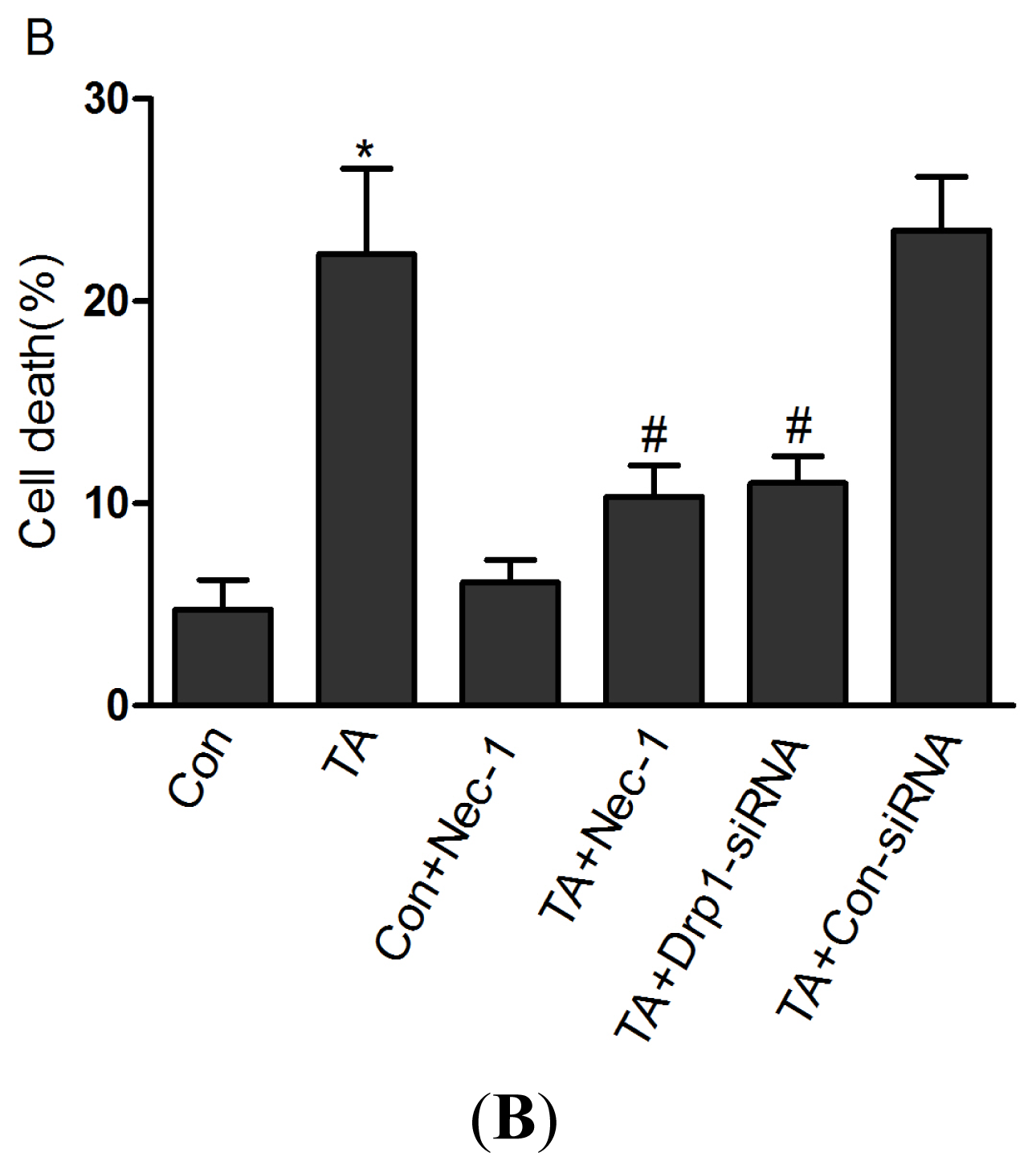
2.1.1. Nec-1 Inhibits Cell Death Induced by Simulated Ischemia Injury in Rat Tubular Cell Line NRK-52E with TNF-α Stimulation and ATP Depletion
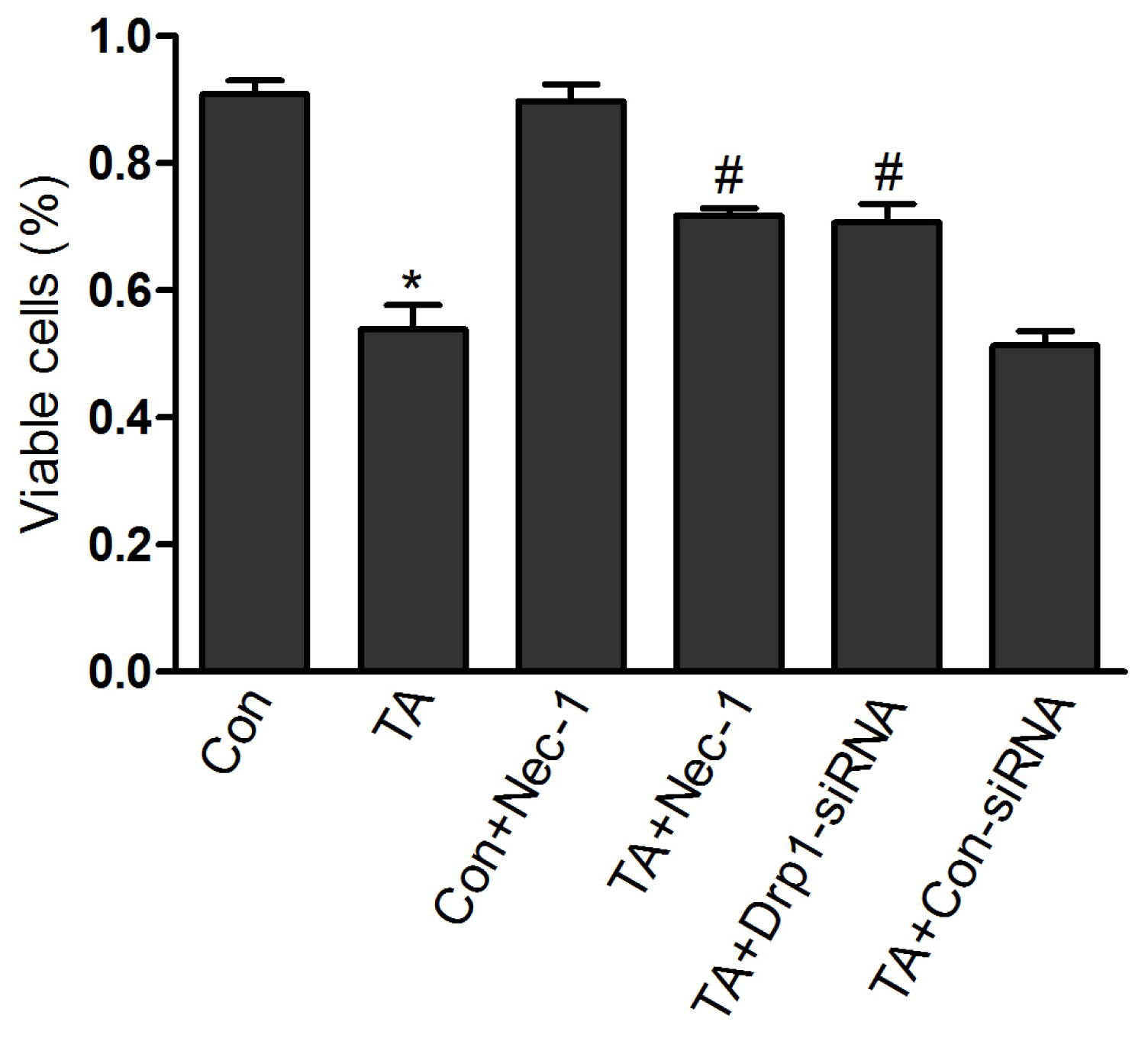
2.1.2. Nec-1 Increased Cell Viability in Rat Tubular Cell Line NRK-52E after TNF-α Stimulation and ATP Depletion
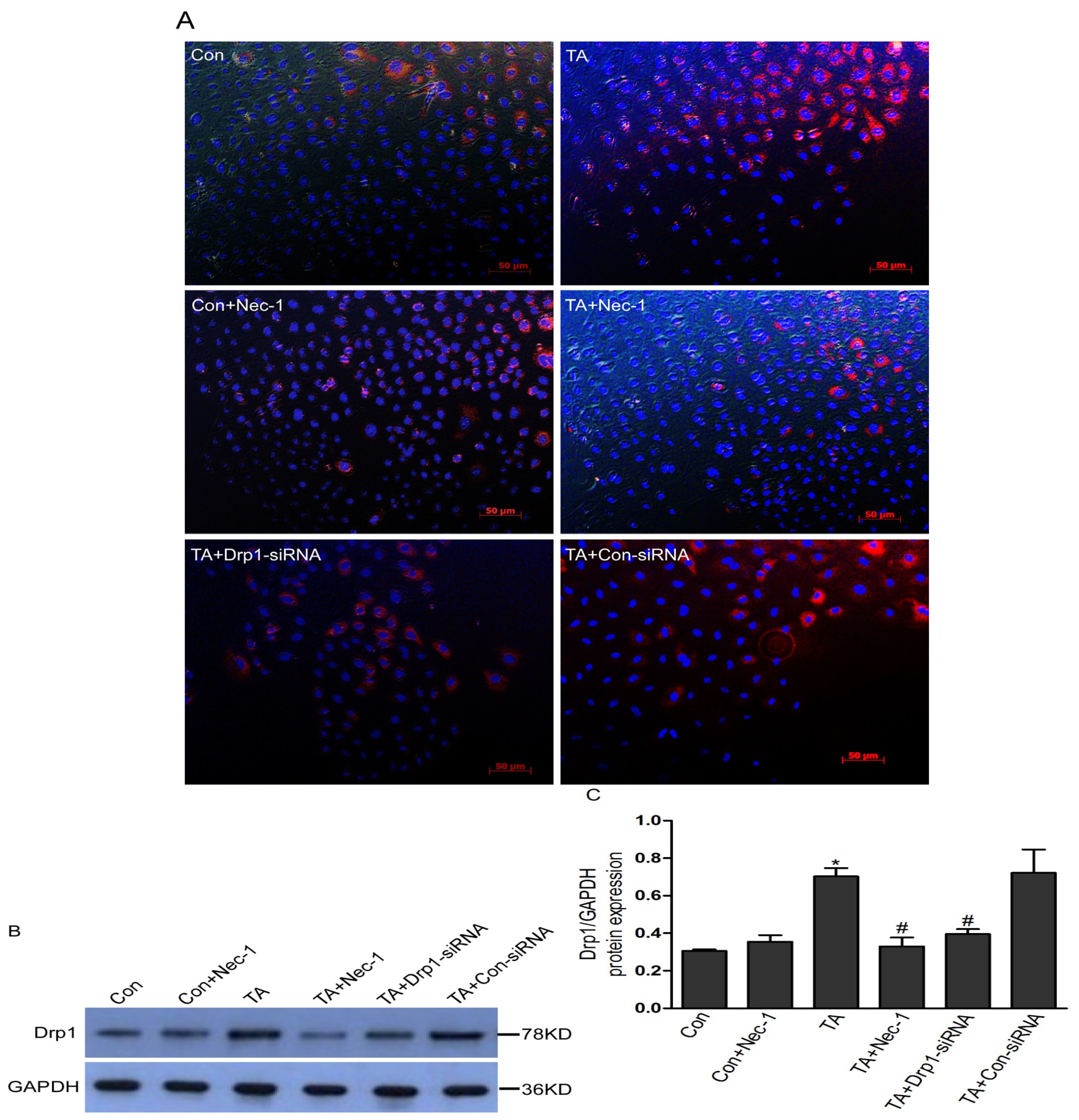
2.1.3. Nec-1 Inhibits Increased Drp1 Protein Expression in Rat Tubular Cell Line NRK-52E after TNF-α Stimulation and ATP Depletion
2.1.4. Drp1 Mediates Ischemia Injury-Induced Cell Death in Rat Tubular Cell Line NRK-52E with TNF-α Stimulation and ATP Depletion
2.2. Discussion
3. Experimental Section
3.1. Reagents
3.2. Cell Cultures
3.3. ATP Depletion
3.4. Cell Treatment
3.5. Annexin V and Propidium Iodide Stains
3.6. Hoechst 33258 Stains
3.7. Cell Viability Assay
3.8. Immunofluorescence
3.9. Western Blotting
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Coca, S.G.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis 2009, 53, 961–973. [Google Scholar]
- Havasi, A.; Borkan, S. Apoptosis and acute kidney injury. Kidney Int 2011, 80, 29–40. [Google Scholar]
- Padanilam, B.J. Cell death induced by acute renal injury: A perspective on the contributions of apoptosis and necrosis. Am. J. Physiol. Renal Physiol 2003, 284, F608–F627. [Google Scholar]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol 2012, 180, 517–525. [Google Scholar]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 2012, 82, 1271–1283. [Google Scholar]
- Vandenabeele, P.; Galluzzi, L.; vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell. Biol 2010, 11, 700–714. [Google Scholar]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 2005, 1, 112–119. [Google Scholar]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol 2008, 4, 313–321. [Google Scholar]
- Oerlemans, M.I.; Liu, J.; Arslan, F.; den Ouden, K.; van Middelaar, B.J.; Doevendans, P.A.; Sluijter, J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol 2012, 107, 270. [Google Scholar]
- Qinli, Z.; Meiqing, L.; Xia, J.; Li, X.; Weili, G.; Xiuliang, J.; Junwei, J.; Hailan, Y.; Ce, Z.; Qiao, N. Necrostatin-1 inhibits the degeneration of neural cells induced by aluminum exposure. Restor. Neurol. Neurosci 2013, 31, 543–555. [Google Scholar]
- Tristao, V.R.; Goncalves, P.F.; Dalboni, M.A.; Batista, M.C.; Durao Mde, S., Jr.; Monte, J.C. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren. Fail 2012, 34, 373–377. [Google Scholar]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 2012, 81, 751–761. [Google Scholar]
- Ouyang, Z.; Zhu, S.; Jin, J.; Li, J.; Qiu, Y.; Huang, M.; Huang, Z. Necroptosis contributes to the cyclosporin A-induced cytotoxicity in NRK-52E cells. Pharmazie 2012, 67, 725–732. [Google Scholar]
- Wang, Y.Q.; Wang, L.; Zhang, M.Y.; Wang, T.; Bao, H.J.; Liu, W.L.; Dai, D.K.; Zhang, L.; Chang, P.; Dong, W.W.; et al. Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem. Res 2012, 37, 1849–1858. [Google Scholar]
- Han, W.; Xie, J.; Fang, Y.; Wang, Z.; Pan, H. Nec-1 enhances shikonin-induced apoptosis in leukemia cells by inhibition of RIP-1 and ERK1/2. Int. J. Mol. Sci 2012, 13, 7212–7225. [Google Scholar]
- Linkermann, A.; Heller, J.O.; Prokai, A.; Weinberg, J.M.; de Zen, F.; Himmerkus, N.; Szabo, A.J.; Brasen, J.H.; Kunzendorf, U.; Krautwald, S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J. Am. Soc. Nephrol 2013, 24, 1545–1557. [Google Scholar]
- Ueda, N.; Kaushal, G.P.; Shah, S.V. Apoptotic mechanisms in acute renal failure. Am. J. Med 2000, 108, 403–415. [Google Scholar]
- Obitsu, S.; Sakata, K.; Teshima, R.; Kondo, K. Eleostearic acid induces RIP1-mediated atypical apoptosis in a kinase-independent manner via ERK phosphorylation, ROS generation and mitochondrial dysfunction. Cell Death Dis 2013, 4, e674. [Google Scholar]
- Pasupuleti, N.; Leon, L.; Carraway, K.L., 3rd; Gorin, F. 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase- independent necroptosis mediated by apoptosis-inducing factor. J. Pharmacol. Exp. Ther 2013, 344, 600–615. [Google Scholar]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; de Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia- reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar]
- Chavez-Valdez, R.; Martin, L.J.; Flock, D.L.; Northington, F.J. Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience 2012, 219, 192–203. [Google Scholar]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol 1998, 143, 351–358. [Google Scholar]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar]
- Tanaka, A.; Youle, R.J. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol. Cell 2008, 29, 409–410. [Google Scholar]
- Thomas, K.J.; Jacobson, M.R. Defects in mitochondrial fission protein dynamin-related protein 1 are linked to apoptotic resistance and autophagy in a lung cancer model. PLoS One 2012, 7, e45319. [Google Scholar]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 2007, 14, 1086–1094. [Google Scholar]
- Mohib, K.; Wang, S.; Guan, Q.; Mellor, A.L.; Sun, H.; Du, C.; Jevnikar, A.M. Indoleamine 2,3-dioxygenase expression promotes renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol 2008, 295, F226–F234. [Google Scholar]
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol 1998, 274, F315–F327. [Google Scholar]
- Li, F.; Mao, H.P.; Ruchalski, K.L.; Wang, Y.H.; Choy, W.; Schwartz, J.H.; Borkan, S.C. Heat stress prevents mitochondrial injury in ATP-depleted renal epithelial cells. Am. J. Physiol. Cell Physiol 2002, 283, C917–C926. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, L.; Jiang, F.; Chen, Y.; Luo, J.; Liu, S.; Zhang, B.; Ye, Z.; Wang, W.; Liang, X.; Shi, W. Necrostatin-1 Attenuates Ischemia Injury Induced Cell Death in Rat Tubular Cell Line NRK-52E through Decreased Drp1 Expression. Int. J. Mol. Sci. 2013, 14, 24742-24754. https://doi.org/10.3390/ijms141224742
Zhang L, Jiang F, Chen Y, Luo J, Liu S, Zhang B, Ye Z, Wang W, Liang X, Shi W. Necrostatin-1 Attenuates Ischemia Injury Induced Cell Death in Rat Tubular Cell Line NRK-52E through Decreased Drp1 Expression. International Journal of Molecular Sciences. 2013; 14(12):24742-24754. https://doi.org/10.3390/ijms141224742
Chicago/Turabian StyleZhang, Li, Fen Jiang, Yuanhan Chen, Jialun Luo, Shuangxin Liu, Bin Zhang, Zhiming Ye, Wenjian Wang, Xinling Liang, and Wei Shi. 2013. "Necrostatin-1 Attenuates Ischemia Injury Induced Cell Death in Rat Tubular Cell Line NRK-52E through Decreased Drp1 Expression" International Journal of Molecular Sciences 14, no. 12: 24742-24754. https://doi.org/10.3390/ijms141224742