Synthesis, Characterisation, and Evaluation of a Cross-Linked Disulphide Amide-Anhydride-Containing Polymer Based on Cysteine for Colonic Drug Delivery

Abstract
:1. Introduction
2. Results and Discussion
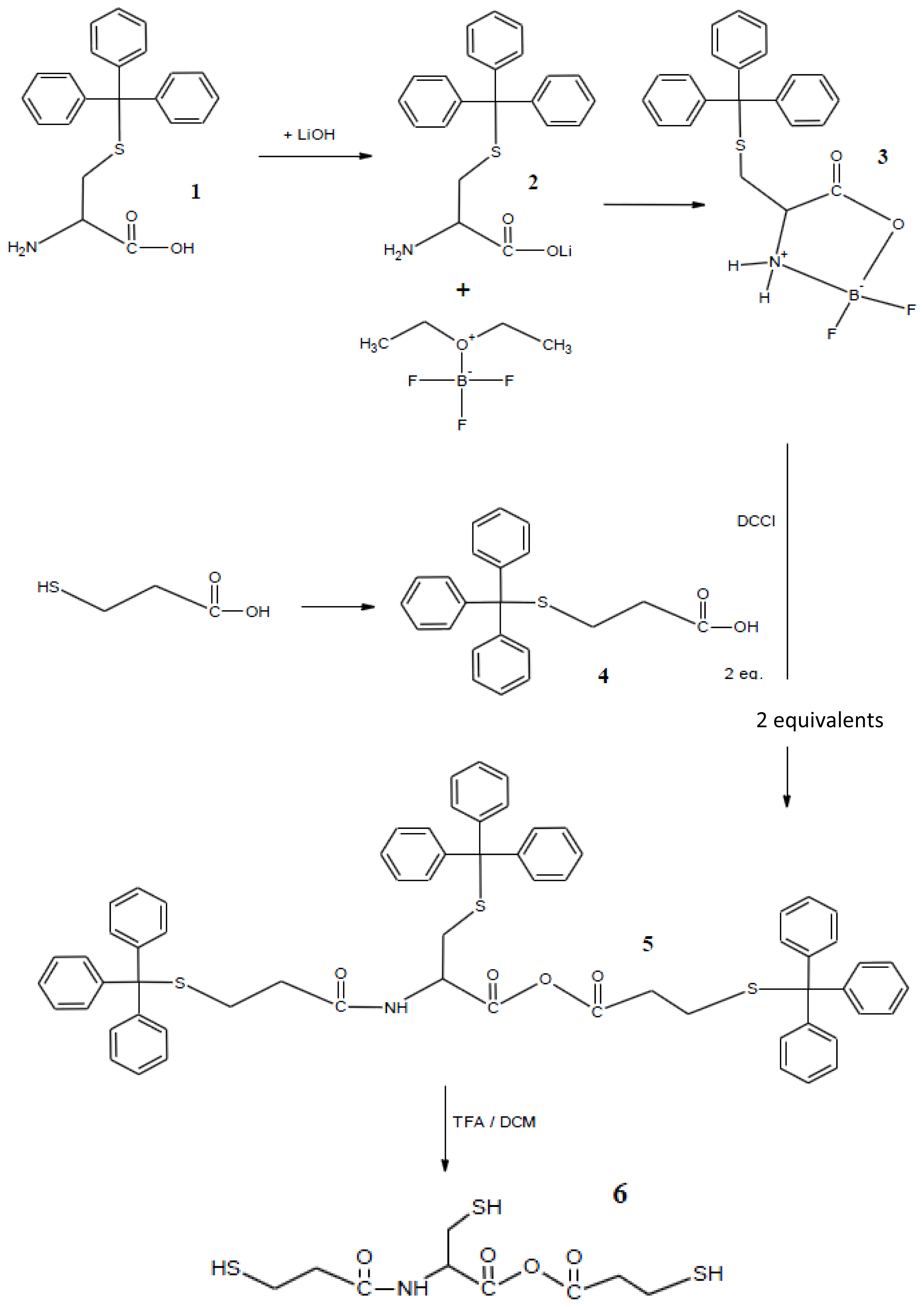
2.1. Synthesis
2.2. Physical Characterisation of Disulphide Cross-Linked Polymers
- Polymer P10: FTIR (KBr disc) = 3432 cm−1 (–NH stretch), 2925 cm−1 (–CH2–), 1704 cm−1 (–CO–O–CO–), 1654, 1593 cm−1 (–NHCO–), 1180 cm−1 (C–O stretch).
- Polymer P11: FTIR (KBr disc) = 3408 cm−1 (–NH stretch), 2938 cm−1 (–CH2–), 1708 cm−1 (–CO–O–CO–), 1654, 1593 cm−1 (–NHCO–), 1189 cm−1 (C–O stretch).
- Polymer 151: FTIR (KBr disc) = 3405 cm−1 (–NH stretch), 2917 cm−1 (–CH2–), 1687 cm−1 (–CO–O–CO–), 1654 cm−1 (–NHCO–), 1176 cm−1 (C–O stretch).
- Polymer 15: FTIR (KBr disc) = 3307 cm−1 (–NH stretch), 2913 cm−1 (–CH2–), 1704 cm−1 (–CO–O–CO–), 1597 cm−1 (–NHCO–), 1185 cm−1 (C–O stretch).
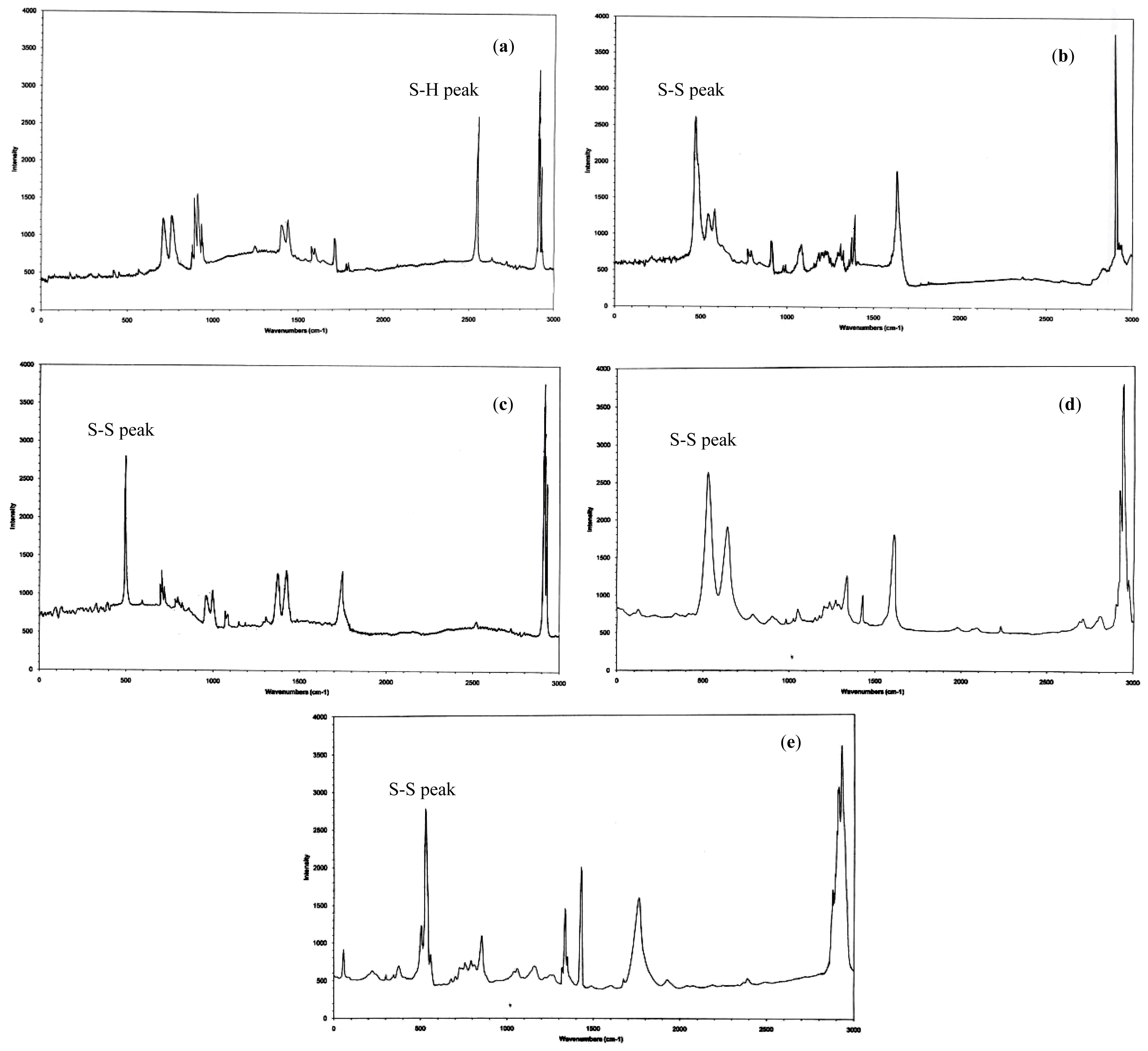
2.3. Raman Spectroscopy
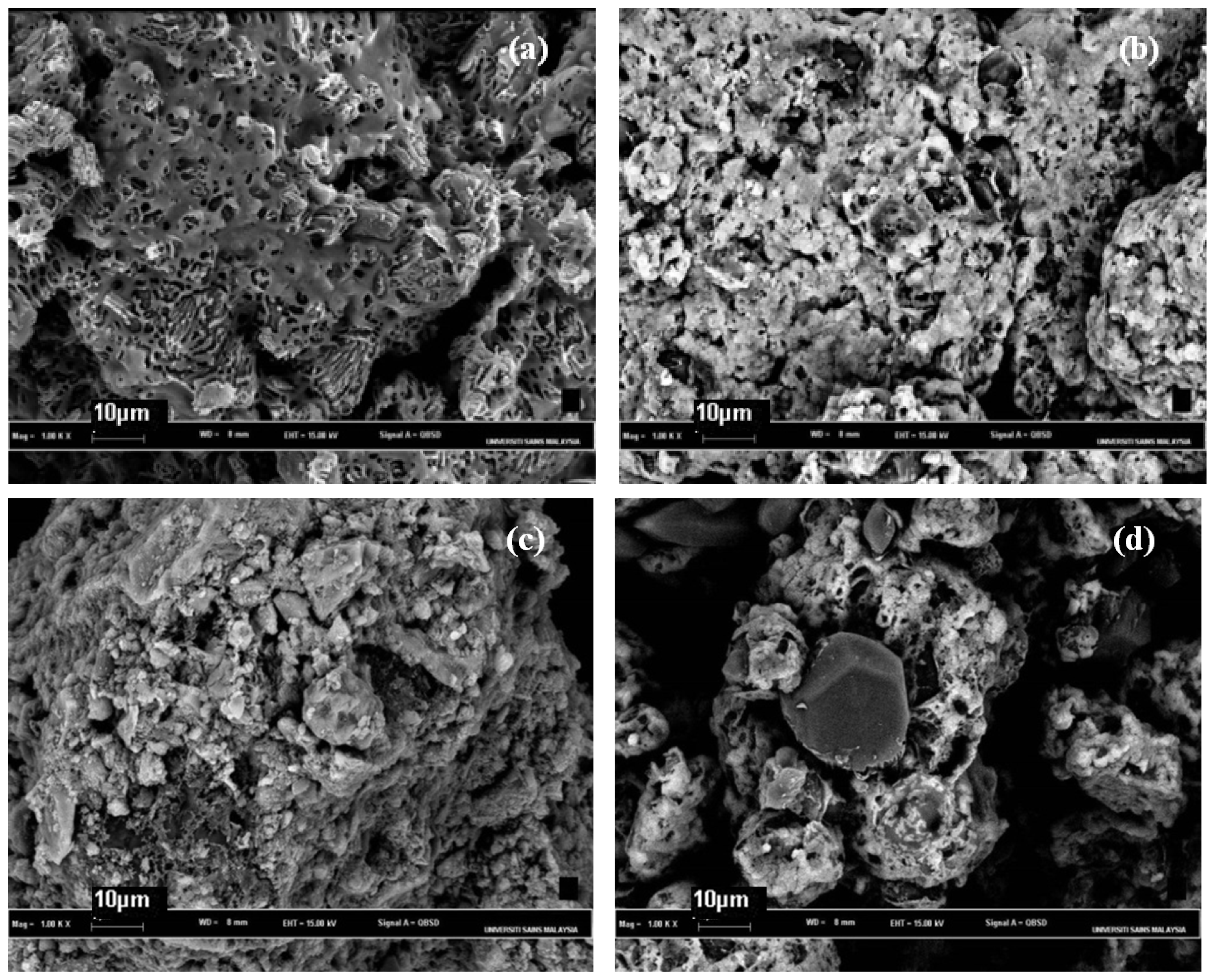
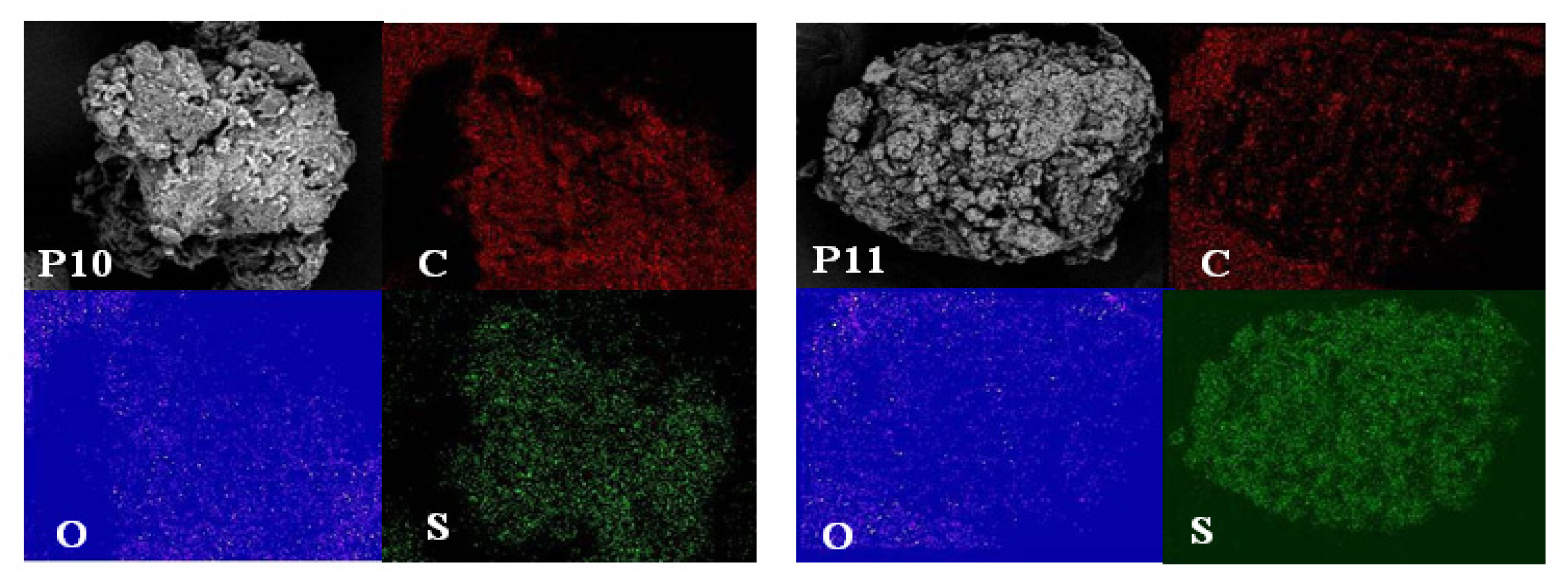
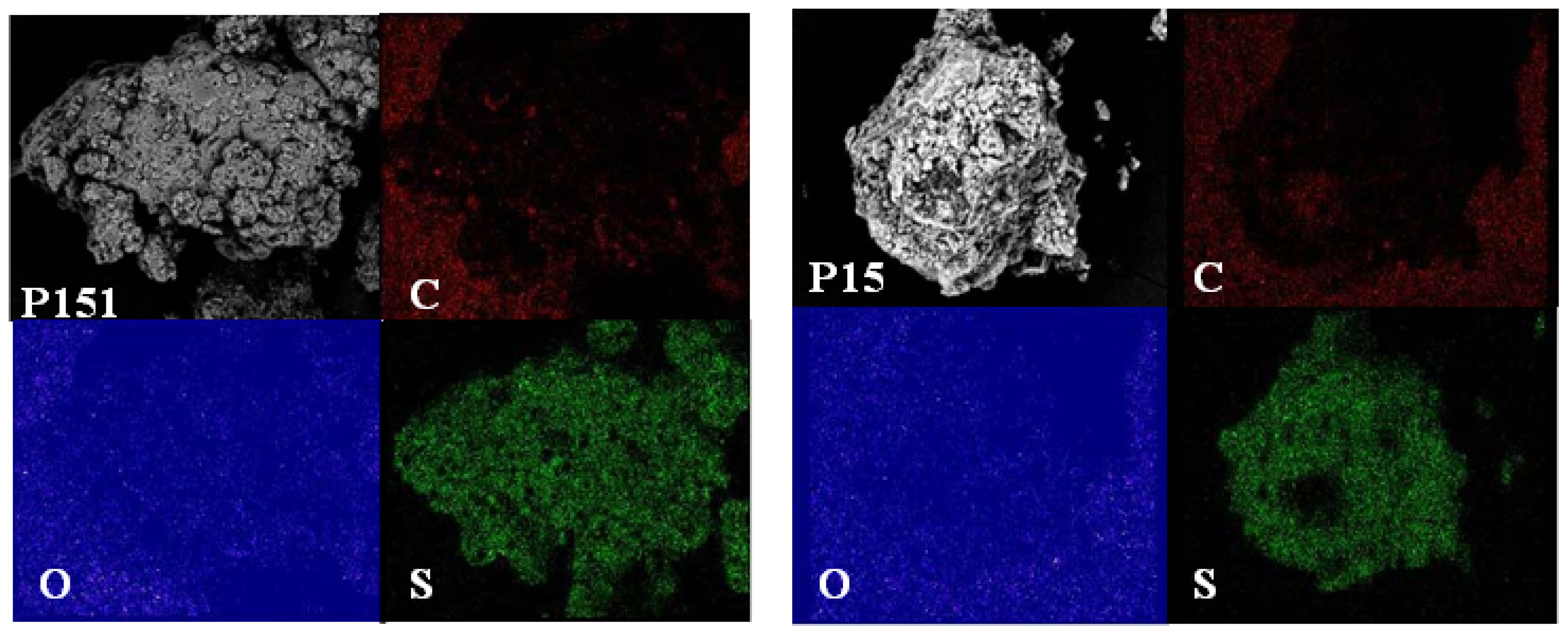
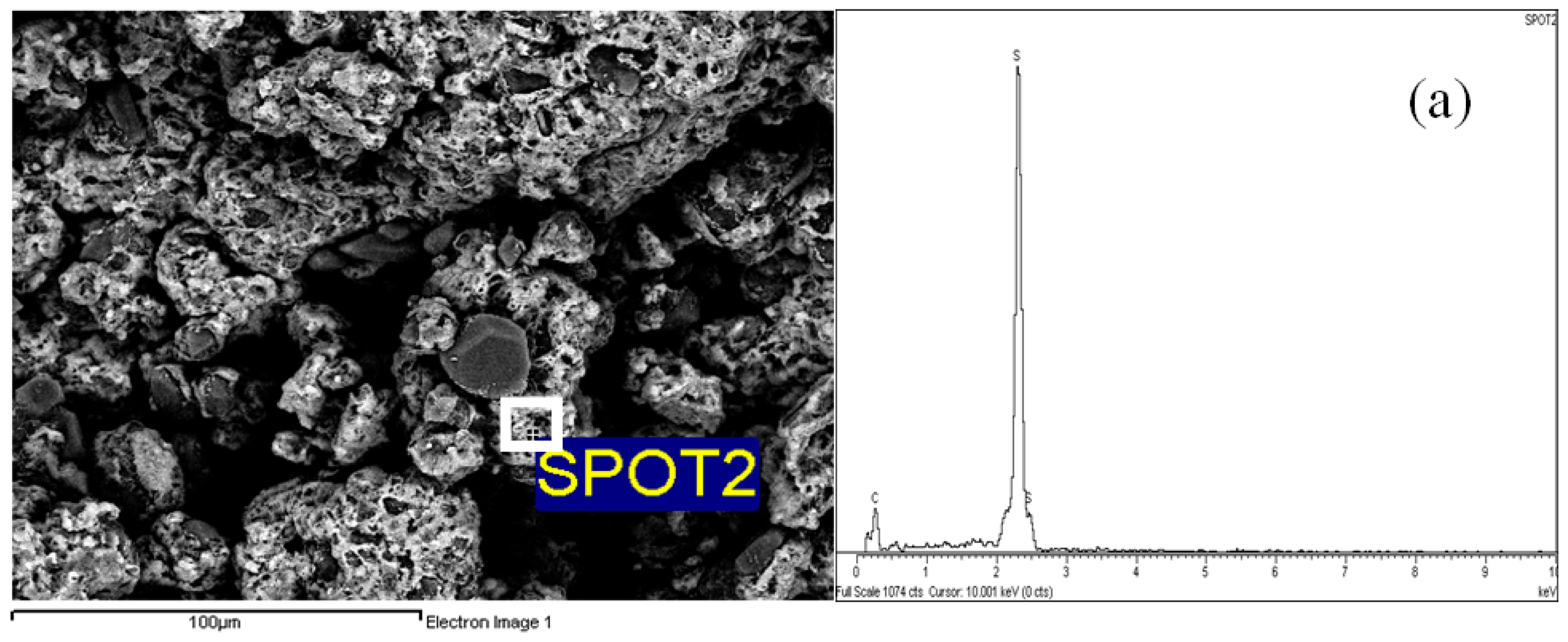
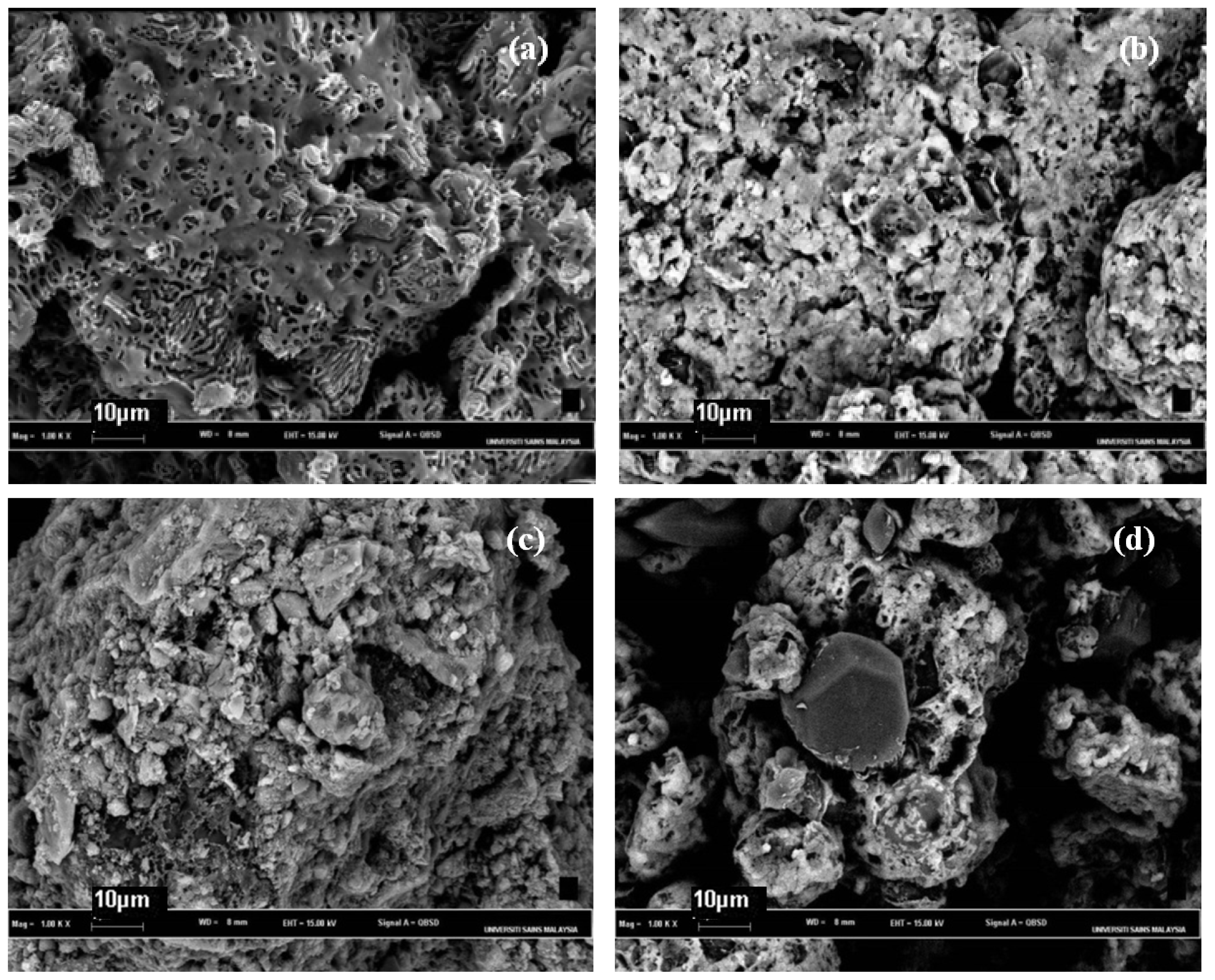
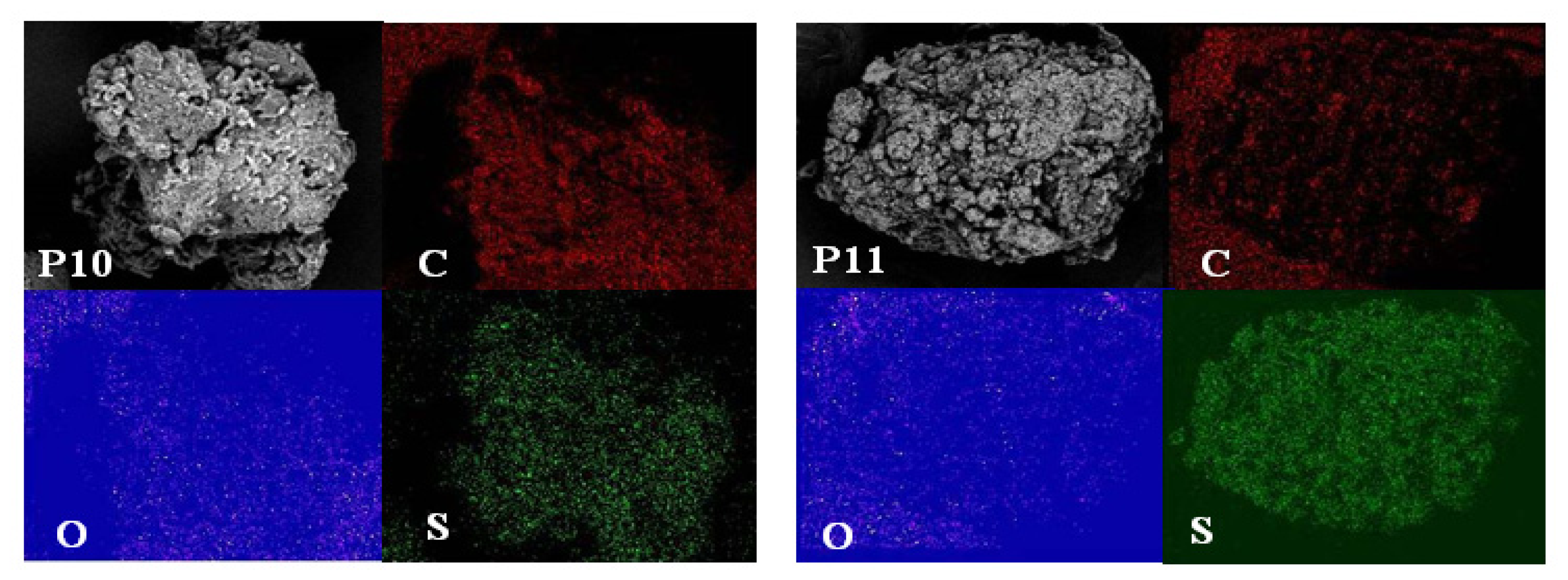
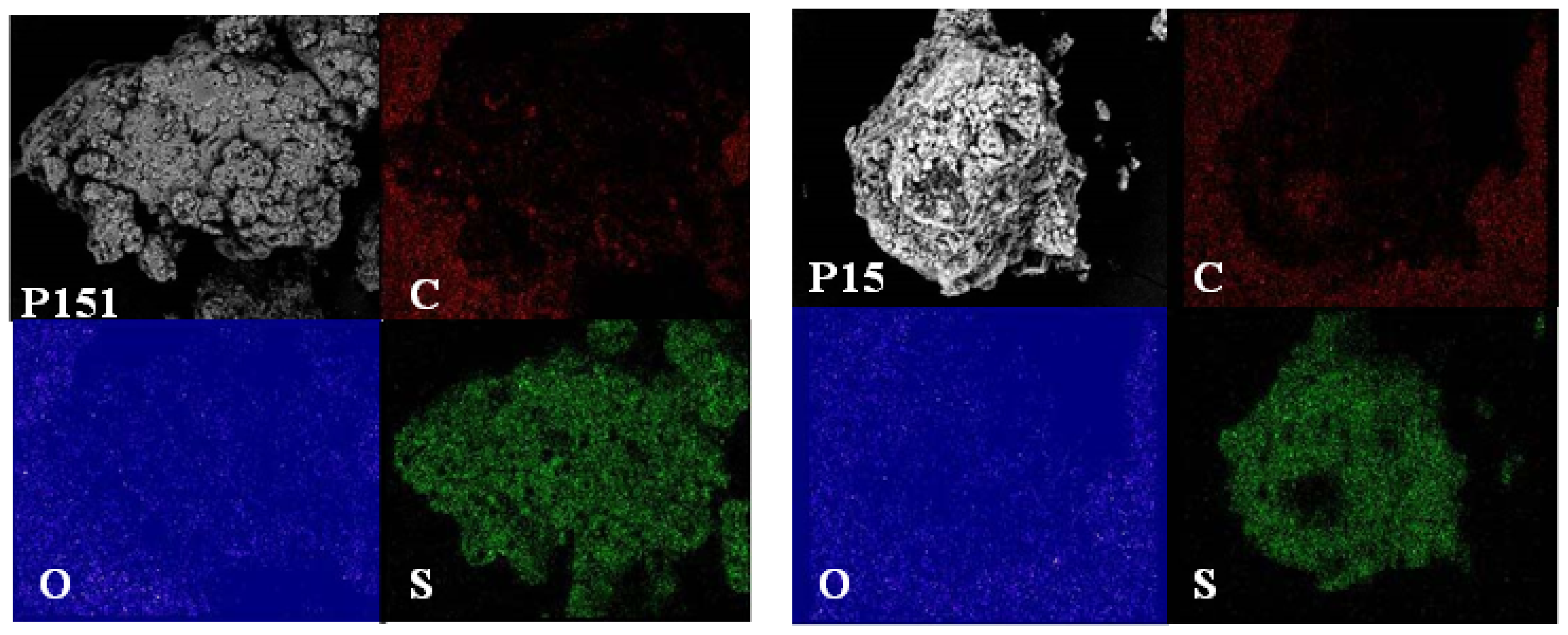
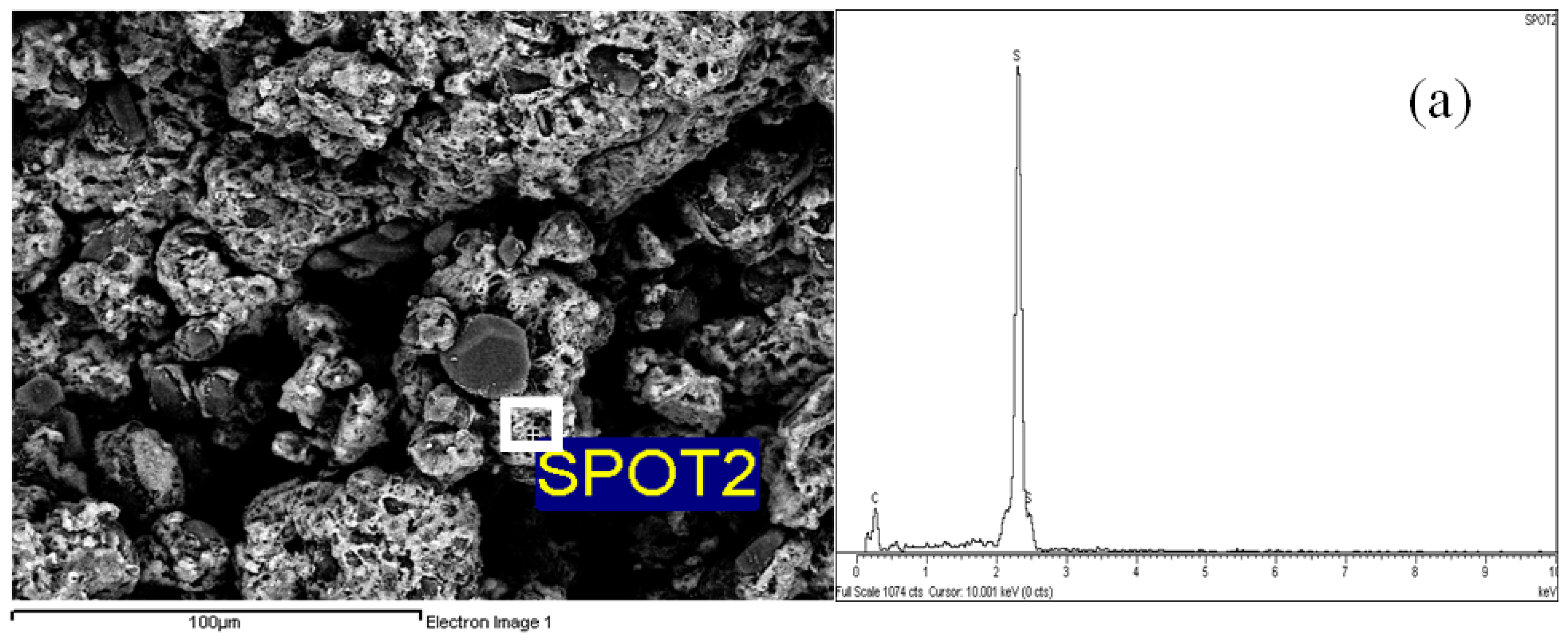
2.4. Morphological Aspects and Energy Dispersive X-ray (EDX) Micrographs
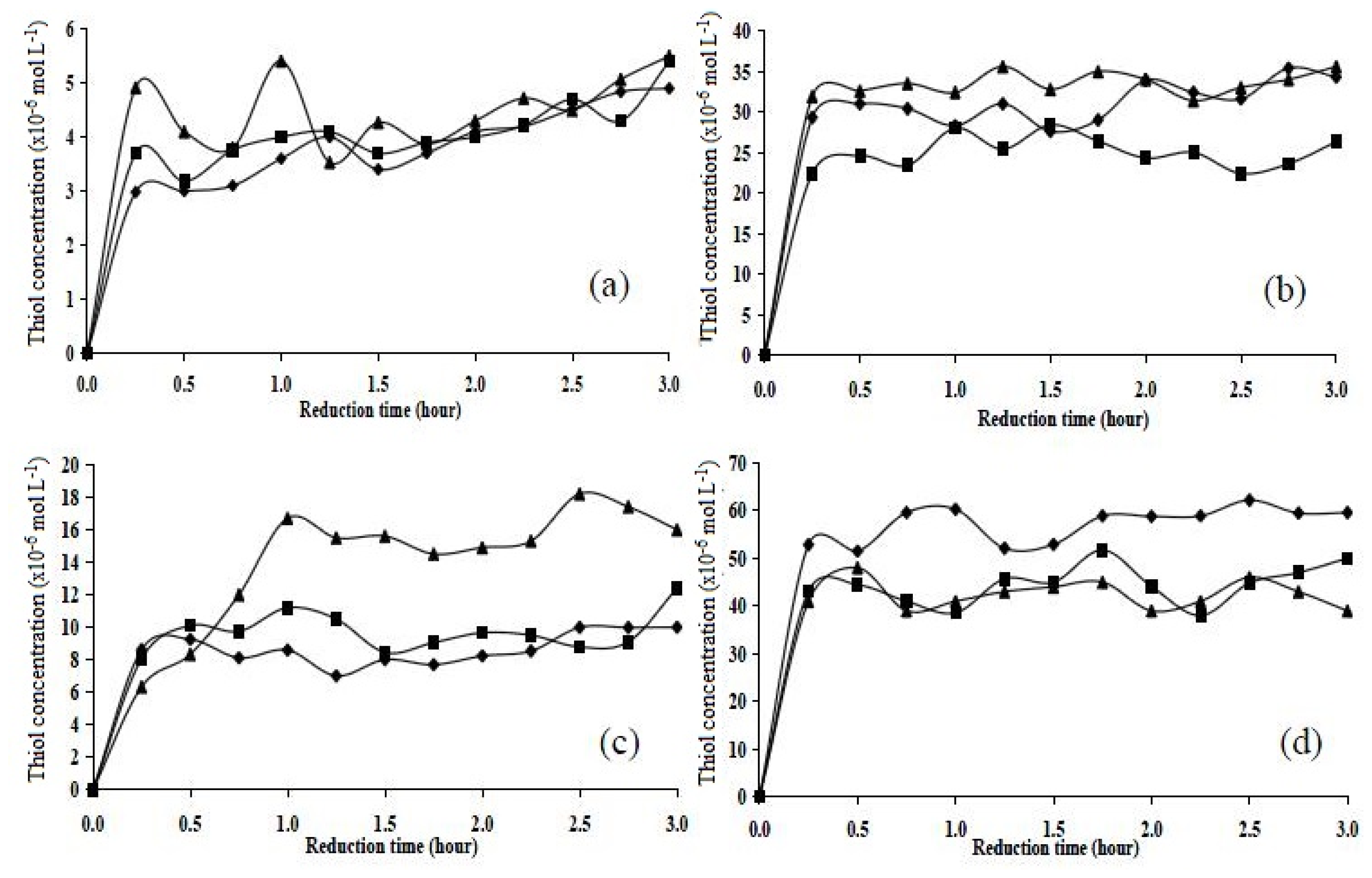
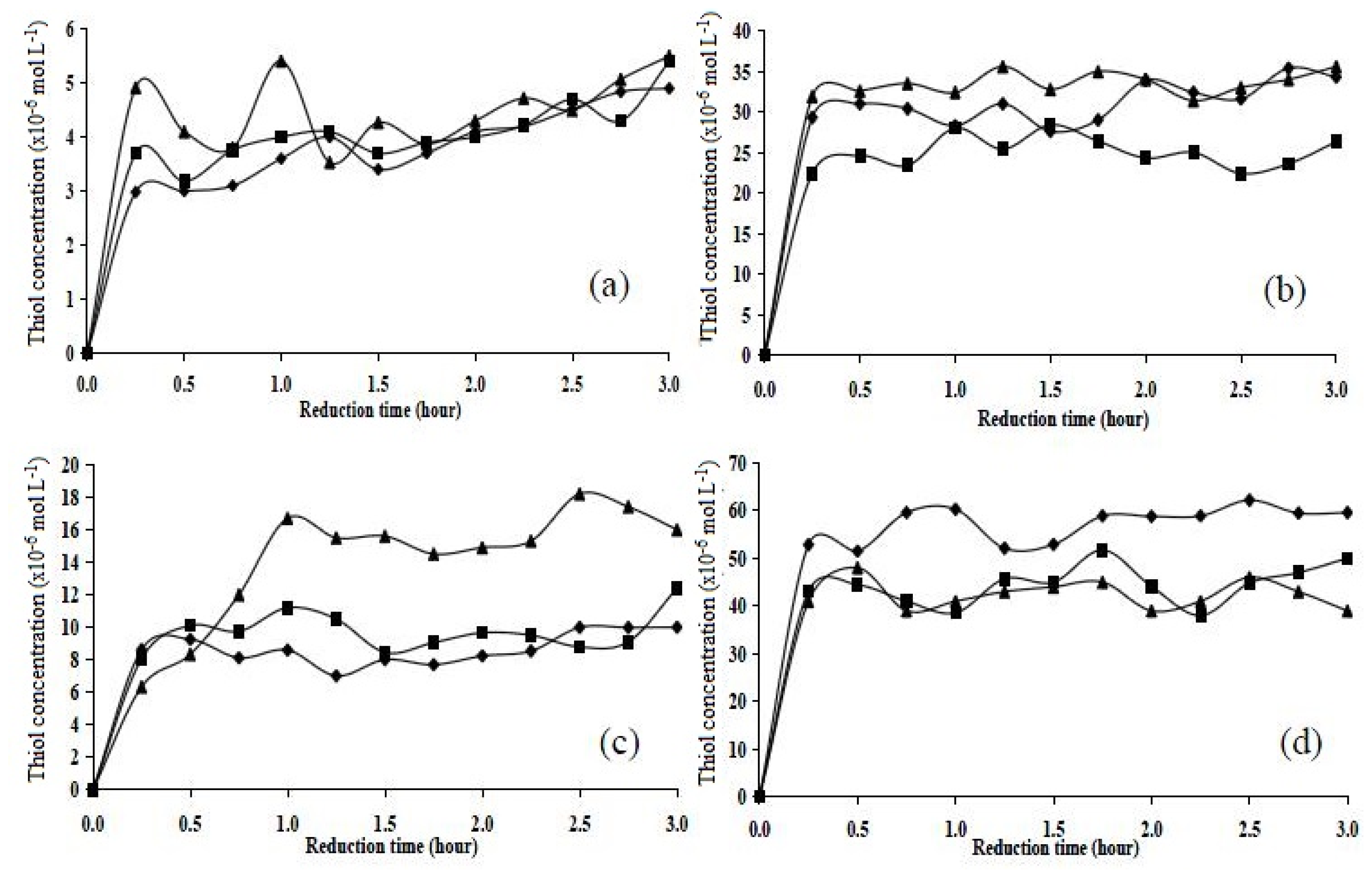
2.5. Chemical Reduction Studies
2.5.1. Chemical Reduction of Cystamine
2.5.2. Chemical Reduction of Disulphide Cross-Linked Polymers
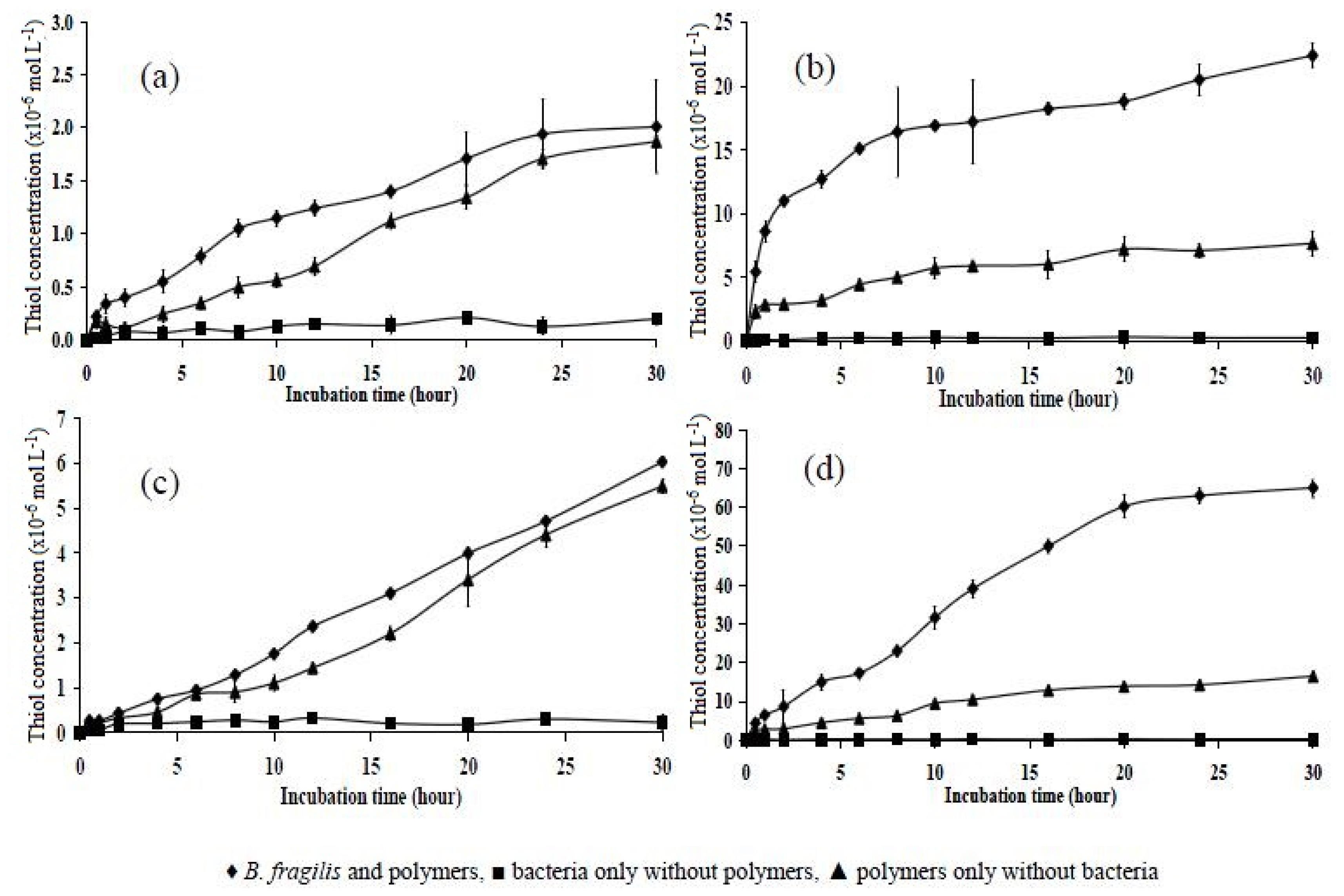
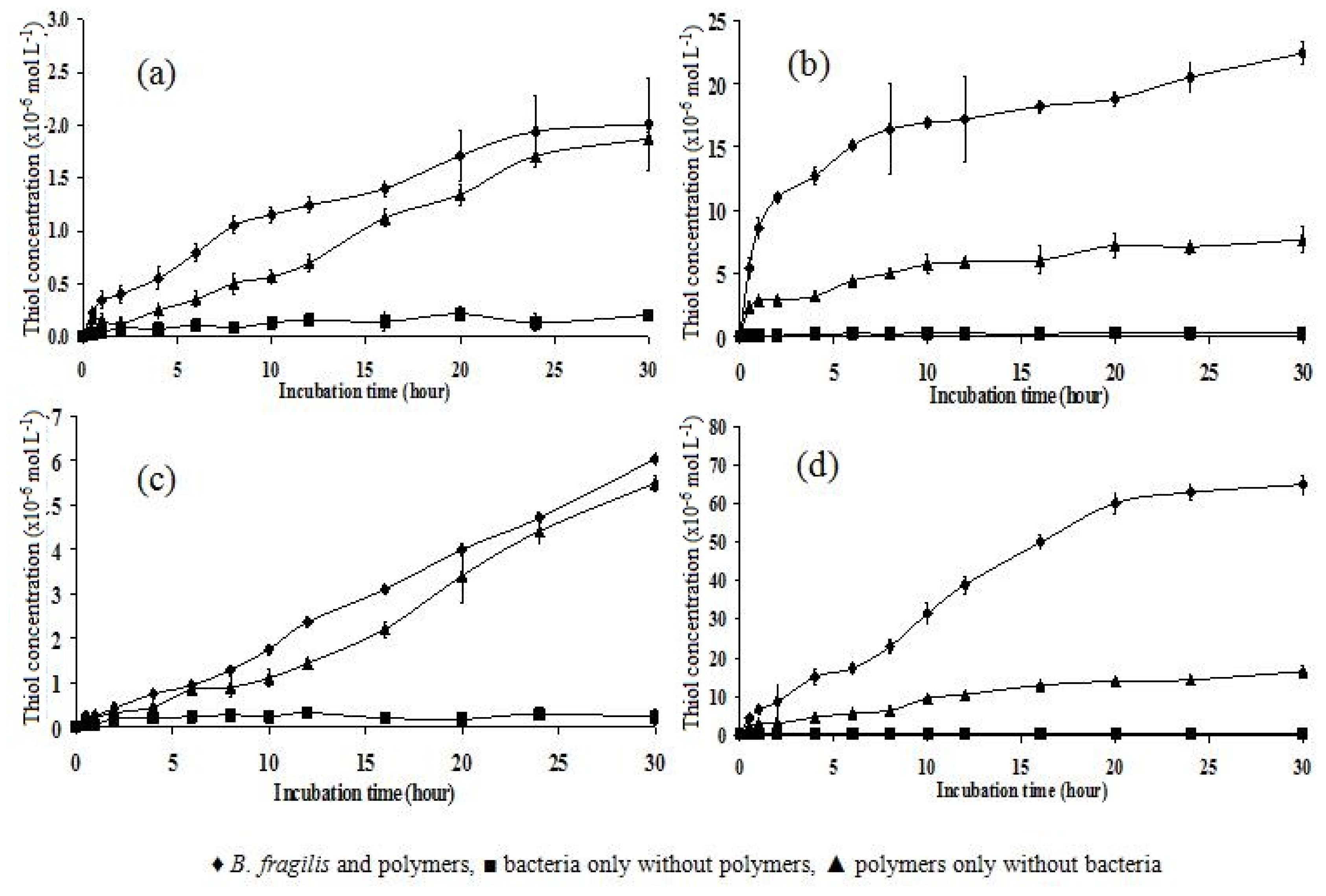
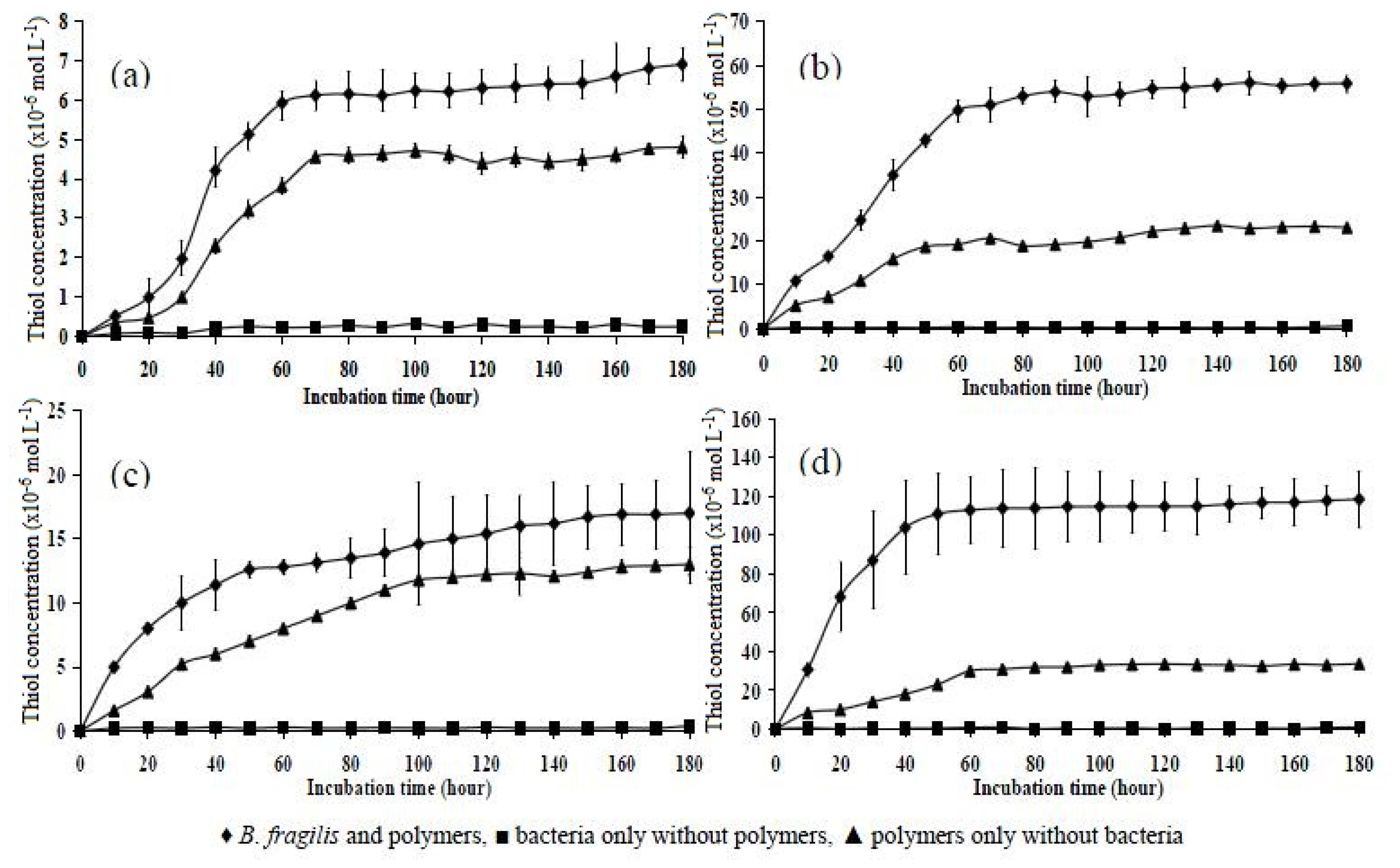
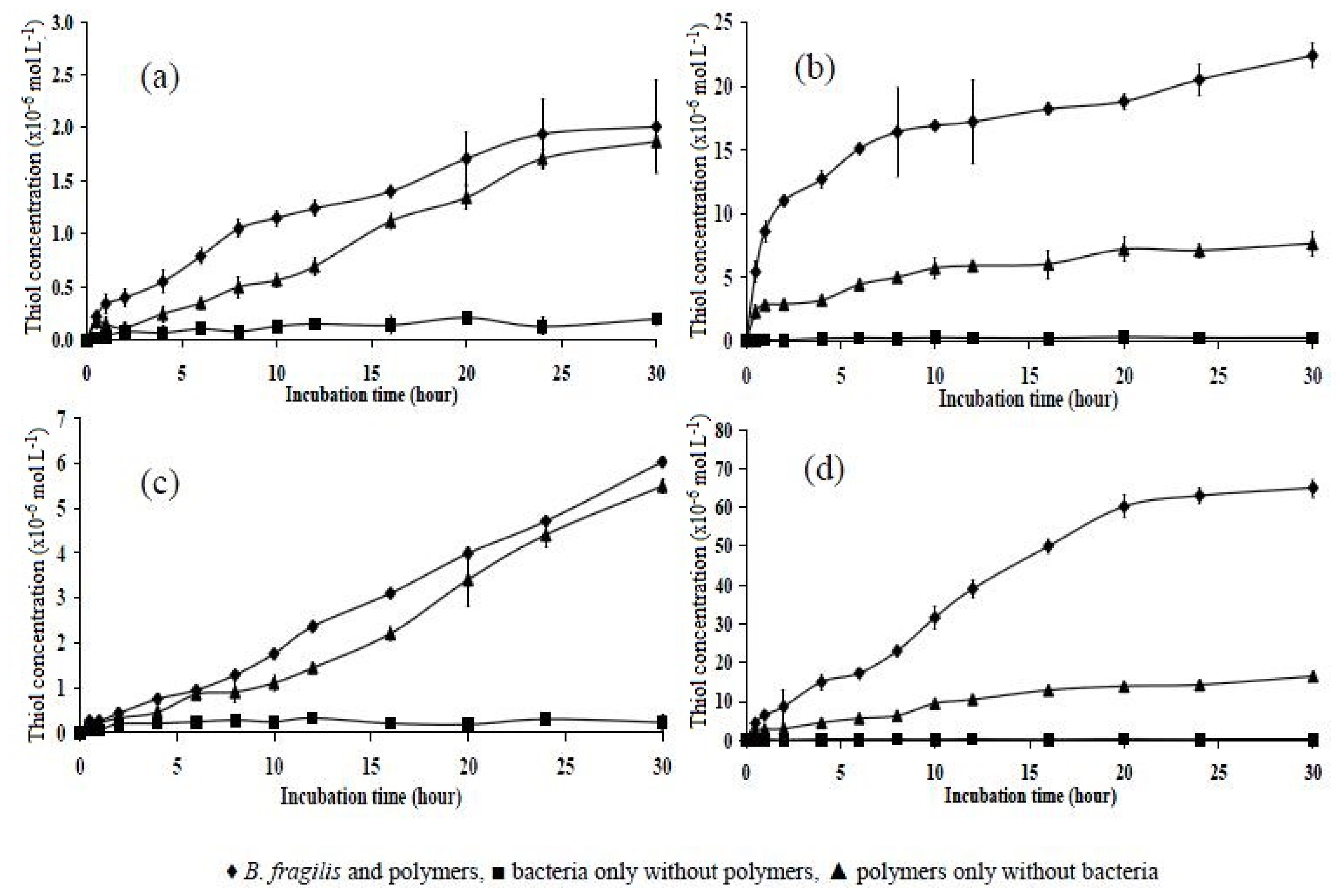
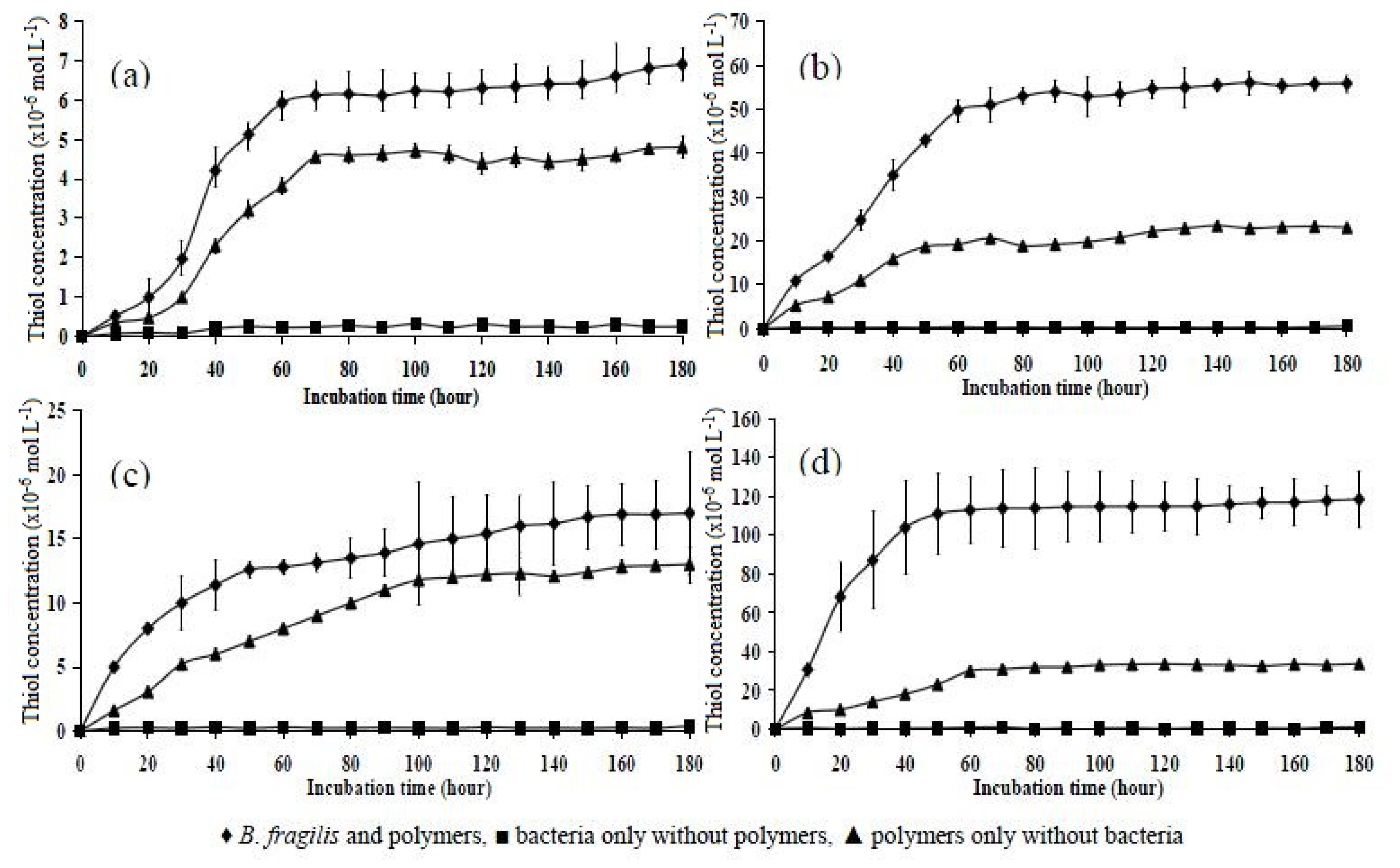
2.6. Incubation of Polymers with B. fragilis for 5, 30, and 180 h
3. Experimental Section
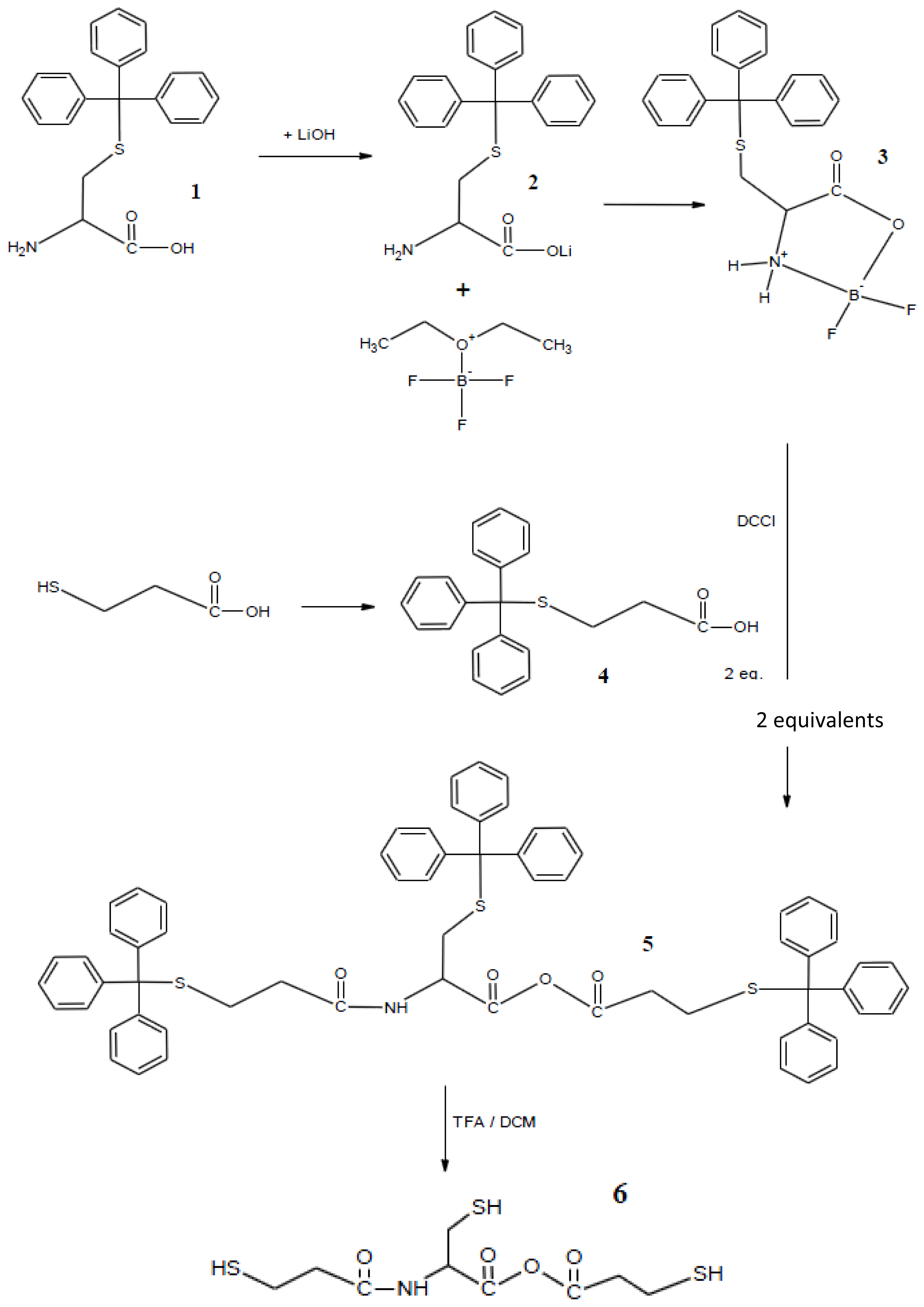
3.1. Synthesis of Monomers
3.1.1. Synthesis of (Triphenylmethylthio)-l-cysteine [Cys(Trt)-OH] (1)
3.1.2. Synthesis of Cys(Trt)–OH [Cys(Trt)-OLi]lithium Salt (2)
3.1.3. Synthesis of 2,2-Difluoro-4-tritylsulfanylmethyl-1,3,2-oxazoborolidin-5-one (3)
3.1.4. Synthesis of (Triphenylmethyl) Thiopropionic Acid (4)
3.1.5. Synthesis of 3-Tritylsulfanyl-N-2-(3-tritylsulfanylpropionamide)-3-tritylsulfanyl Propionic Anhydride (5)
3.1.6. Synthesis of 3-Mercapto-N-2-(3-mercaptopropionamide)-3-mercapto Propionic Anhydride (6)
3.2. Oxidation of Thiol Monomers
- P10 = trithiol monomer only
- P11 = 1.0 trithiol monomer:1.0 dithiol monomer
- P151 = 1.5 trithiol monomer:1.0 dithiol monomer
- P15 = 1.0 trithiol monomer:5.0 dithiol monomer
3.3. Detection of Thiol Using Sodium Nitroprusside, Na2Fe(CN)5NO
3.4. Physical Characterisation of the Synthesised Polymers
3.5. Raman Spectroscopy
3.6. Scanning Electron Microscope-Energy Dispersive X-ray (SEM–EDX)
3.7. Assay for Thiol
3.7.1. Preparation of SØrensen’s Phosphate Buffer
3.7.2. Determination of Thiol Content Using Ellman’s Reagent

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample cuvette | 50 μL Ellman’s reagent |
| 2.5 mL SØrensen’s phosphate buffer | |
| 250 μL sample solution | |
| Reference cuvette | 50 μL Ellman’s reagent |
| 2.75 mL SØrensen’s phosphate buffer |
3.7.3. Application of Beer-Lambert Equation
3.8. Chemical Reduction
3.8.1. Reduction of Cystamine by Zinc/Acetic Acid
3.8.2. Reduction of Disulphide Cross-Linked Polymers by Zinc/Acetic Acid
3.9. Dissolution Studies
3.9.1. Bacteria
3.9.2. Preparation of Pre-Reduced, Anaerobically Sterile (Pras) Media
3.9.3. Preparation of Bacterial Pellets
3.9.4. Incubation of Polymers in Bacterial Cultures
3.9.5. Detection of the Reduction of Disulphides Using Ellman’s Reagent
3.9.6. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-14-24670-s001.pdfAcknowledgments
Conflicts of Interest
References
- Lloyd, A.W.; Martin, G.P.; Soozandehfar, S.H. Azopolymers: A means of colon specific drug delivery. Int. J. Pharm 1994, 106, 255–260. [Google Scholar]
- Leopold, C.S.; Eikeler, D. Eudragit E as coating material for the pH-controlled drug release in the topical of inflammatory bowel disease (IBD). J. Drug Target 1998, 6, 85–94. [Google Scholar]
- Chourasia, M.K.; Jain, S.K. Polysaccharides for colon targeted drug delivery. Drug Deliv 2004, 11, 129–148. [Google Scholar]
- Bragger, J.L.; Lloyd, A.W.; Soozandehfar, S.H.; Bloomfield, S.F.; Marriott, C.; Martin, G.P. Investigations into the azo reducing activity of a common colonic microorganism. Int. J. Pharm 1997, 157, 61–71. [Google Scholar]
- Dollendorf, C.; Hetzer, M.; Ritter, H. Polymeric redox-responsive delivery systems bearing ammonium salts cross-linked via disulfides. Beilstein J. Organ. Chem 2013, 9, 1652–1662. [Google Scholar]
- Schact, E.; Wilding, I.R. Process for the preparation of azo- and/or disulphide-containing polymers PCT:WO91/11175, 8 August 1991.
- Liu, H.; Wang, H.; Yang, W.; Cheng, Y. Disulfide cross-linked low generation dendrimers with high gene transfection efficacy, low cytotoxicity and low cost. J. Am. Chem. Soc 2013, 134, 17680–17687. [Google Scholar]
- Harada, A.; Matsuki, R.; Ichimura, S.; Yuba, E.; Kono, K. Intracellular environment-resposive stabilization of polymer vesicles formed from head-tail type polycations composed of a polyamidoamine dendron and poly(l-lysine). Molecules 2013, 18, 12168–12179. [Google Scholar]
- Wilkes, G.L. An overview of the basic rheological behaviour of polymer fluids with an emphasis of polymer melts. J. Chem. Edu 1981, 58, 880–892. [Google Scholar]
- Le, Q.G. Novel polymers for targeting drugs to the colon. Ph.D. Thesis, King’s College London, London, UK, February 1998. [Google Scholar]
- Sahudin, S. Novel disulphide-containing crosslinked polymer for colon-specific drug delivery. Ph.D. Thesis, King’s College London, London, UK, October 2001. [Google Scholar]
- Satav, S.S.; Karmalkar, R.N.; Kulkarni, M.G.; Mulpuri, N.; Sastry, G.N. Formation of linear polymers with pendant vinyl groups via inclusion complex mediated polymerization of divinyl monomers. J. Am. Chem. Soc 2006, 128, 7752–7753. [Google Scholar]
- Tam, J.P.; Wu, C.R.; Liu, W.; Zhang, J.W. Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. J. Am. Chem. Soc 1991, 113, 6657–6662. [Google Scholar]
- Bulaj, G. Formation of disulfide bonds in proteins and peptides. Biotechnol. Adv 2005, 23, 87–92. [Google Scholar]
- Sim, J.H.; Yamada, K.; Lee, S.H.; Yokokura, S.; Sato, H. Synthesis and characterization of triphenylamine derivatives by oxidative polymerization. Synth. Met 2007, 157, 940–944. [Google Scholar]
- Sim, J.H.; Ueno, E.; Natori, I.; Ha, J.; Sato, H. Oxidation polymerization of N-butyl-N,N diphenylamine (BDPA) and N-4-butylphenyl-N,N-diphenylamine (BTPA). Synth. Met 2008, 158, 345–349. [Google Scholar]
- Wallace, T.J.; Mahon, J.J. Reactions of thiols with sulfoxides. II. Kinetics and mechanistic implications. J. Am. Chem. Soc 1964, 86, 4099–4103. [Google Scholar]
- Gray, W.R.; Luque, F.A.; Galyean, R.; Atherthon, E.; Sheppard, R.C.; Stone, B.L.; Reyes, A.; Alford, J.; McIntosh, M.; Olivera, B.M.; et al. Conotoxin GI: Disulfide bridges, synthesis and preparation of iodinated derivatives. Biochemistry 1981, 23, 2796–2802. [Google Scholar]
- Snow, J.T.; Finley, J.W.; Friedman, M. Oxidation of sulfhydryl groups to disulfides by sulfoxides. Biochem. Biophys. Res. Commun 1975, 64, 441–447. [Google Scholar]
- Diaz, F.R.; Sanchez, C.O.; del Valle, M.A.; Radic, D.; Bernede, J.C.; Tregouet, Y.; Molinie, P. Synthesis, characterization and electrical properties of poly[bis-(2-aminophenyl)disulfide] and poly[bis-(2-aminophenyl)diselenide] Part II. XPS, ESR, SEM and conductivity study. Synth. Met 2000, 110, 71–77. [Google Scholar]
- Scheiba, F.; Benker, N.; Kunz, U.; Rzoth, C.; Fuess, H. Electron microscopy techniques for the analysis of the polymer electrolyte distribution in proton exchange membrane fuel cells. J. Power Sources 2008, 177, 272–280. [Google Scholar]
- Kimura, Y.; Makita, Y.; Kumagai, T.; Yamane, H.; Kitao, T. Degradation of azo-containing polyurethane by the action of intestinal flora: Its mechanism and application as drug delivery system. Polymers 1992, 33, 5294–5299. [Google Scholar]
- Otaka, A.; Koide, T.; Shide, A.; Fujii, N. Application of dimethylsulphoxide (DMSO)/trifluoroacetic acid (TFA) oxidation on the synthesis of cystine-coating peptide. Tet. Lett 1991, 32, 1223–1226. [Google Scholar]
- March, J. Advanced organic chemistry: Reactions, mechanisms and structures, 4th ed; John Wiley and Sons Inc: New York, NY, USA, 1992. [Google Scholar]
- Holdeman, L.V.; Moore, W.E.C. Anaerobe laboratory manual; Virginia Polytechnic Institute and State University: Blacksburg, VA, USA, 1973. [Google Scholar]
- Andreas, D.; Steffen, D.; Markus, B.; Stefan, S.; Matthias, O.; Sebastian, S.; Patrick, S.; Alexandra, M.; Masayuki, W.; Tadashi, T.; et al. Protein tyrosine nitration and thiol oxidation by peroxynitrite—Strategies to prevent these oxidative modifications. Int. J. Mol. Sci 2013, 14, 7542–7570. [Google Scholar]
- SPSS. Available online: http://www-01.ibm.com/software/analytics/spss/ (accessed on 7 September 2013).


| Polymer | Percentage yield | Solubility | Appearance |
|---|---|---|---|
| P10 | 80%–85% | Insoluble | Rugged white solid |
| P11 | 87%–91% | Insoluble | White solid |
| P151 | 86%–88% | Insoluble | White solid |
| P15 | 91%–93% | Soluble in DMSO | Yellowish white powder |
| Time (min) | Thiol concentration (mol L−1 × 10−5) | % Reduction |
|---|---|---|
| 10 | 2.84 | 49 |
| 20 | 5.82 | 101 |
| 30 | 5.81 | 101 |
| 40 | 5.88 | 102 |
| Polymer | 5 h | 30 h | 180 h |
|---|---|---|---|
| P10 | 1.20 ± 0.126 | 2.01 ± 0.435 | 6.90 ± 0.416 |
| P151 | 2.50 ± 0.146 | 6.03 ± 0.106 | 17.10 ± 0.480 |
| P11 | 12.50 ± 0.757 | 22.40 ± 0.928 | 55.92 ± 1.980 |
| P15 | 25.40 ± 1.120 | 65.00 ± 2.419 | 118.60 ± 1.450 |
| Statistical analysis | p < 0.05 | p < 0.05 | p < 0.05 |
| Tukey’s HSD test (significant) | All significantly different | All significantly different | All significantly different |
| Incubation medium | P10 | P151 | P11 | P15 |
|---|---|---|---|---|
| Bacteria only (1) | 0.247 ± 0.131 | 0.401 ± 0.096 | 0.624 ± 0.082 | 0.848 ± 0.234 |
| Polymer only (2) | 4.800 ± 0.289 | 13.012 ± 1.402 | 23.120 ± 0.531 | 33.701 ± 0.696 |
| Bacteria + polymer (3) | 6.900 ± 0.416 | 17.100 ± 0.480 | 55.920 ± 1.980 | 118.600 ± 1.450 |
| Statistical analysis | p < 0.05 | p < 0.05 | p < 0.05 | p < 0.05 |
| Dunnett’s test (2-sided) (significant) | 1 & 3 2 & 3 | 1 & 3 2 & 3 | 1 & 3 2 & 3 | 1 & 3 2 & 3 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lim, V.; Peh, K.K.; Sahudin, S. Synthesis, Characterisation, and Evaluation of a Cross-Linked Disulphide Amide-Anhydride-Containing Polymer Based on Cysteine for Colonic Drug Delivery. Int. J. Mol. Sci. 2013, 14, 24670-24691. https://doi.org/10.3390/ijms141224670
Lim V, Peh KK, Sahudin S. Synthesis, Characterisation, and Evaluation of a Cross-Linked Disulphide Amide-Anhydride-Containing Polymer Based on Cysteine for Colonic Drug Delivery. International Journal of Molecular Sciences. 2013; 14(12):24670-24691. https://doi.org/10.3390/ijms141224670
Chicago/Turabian StyleLim, Vuanghao, Kok Khiang Peh, and Shariza Sahudin. 2013. "Synthesis, Characterisation, and Evaluation of a Cross-Linked Disulphide Amide-Anhydride-Containing Polymer Based on Cysteine for Colonic Drug Delivery" International Journal of Molecular Sciences 14, no. 12: 24670-24691. https://doi.org/10.3390/ijms141224670