Cardiac Ablation of Rheb1 Induces Impaired Heart Growth, Endoplasmic Reticulum-Associated Apoptosis and Heart Failure in Infant Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
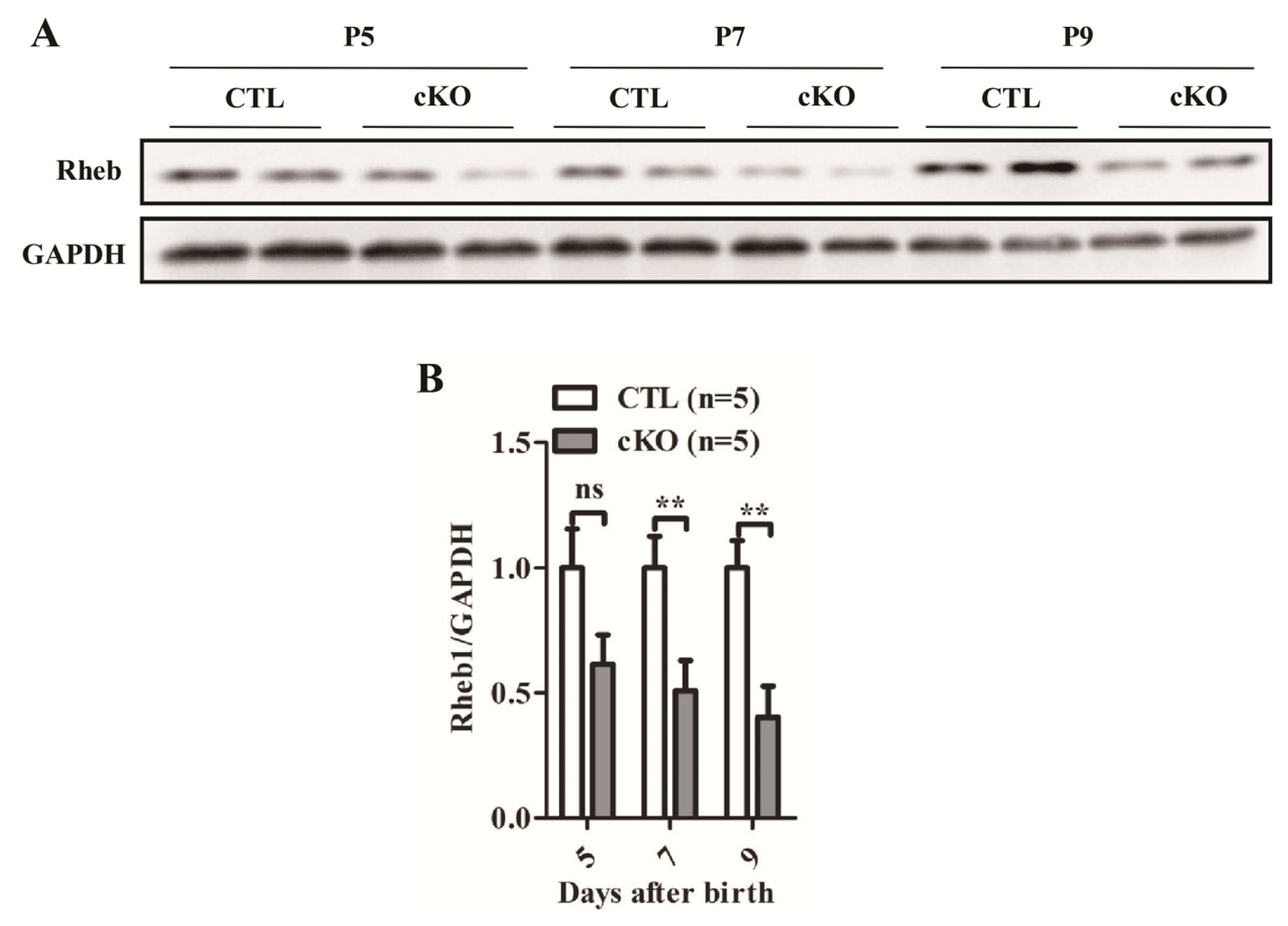
2.1.1. Development of Rheb1 cKO Mouse
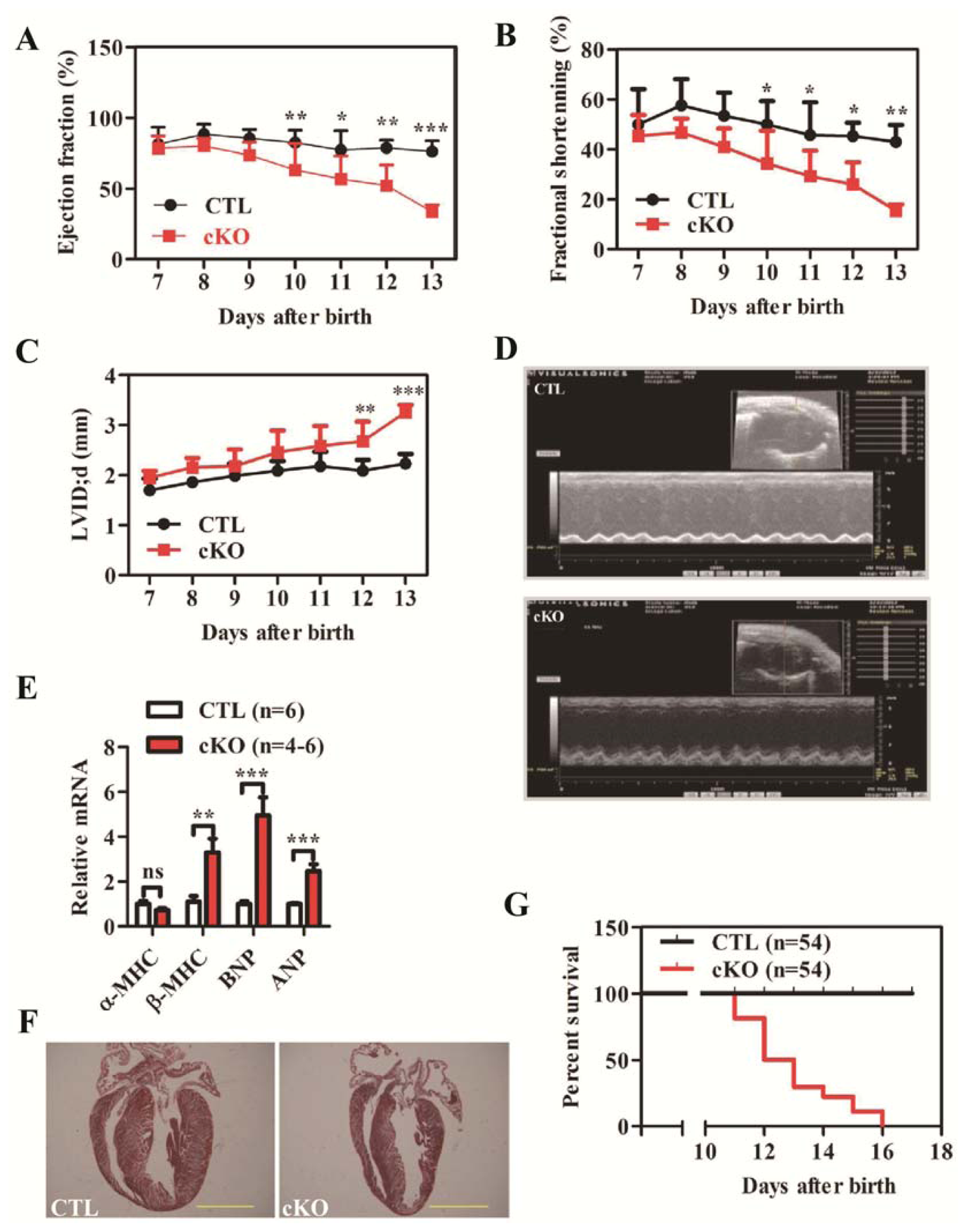
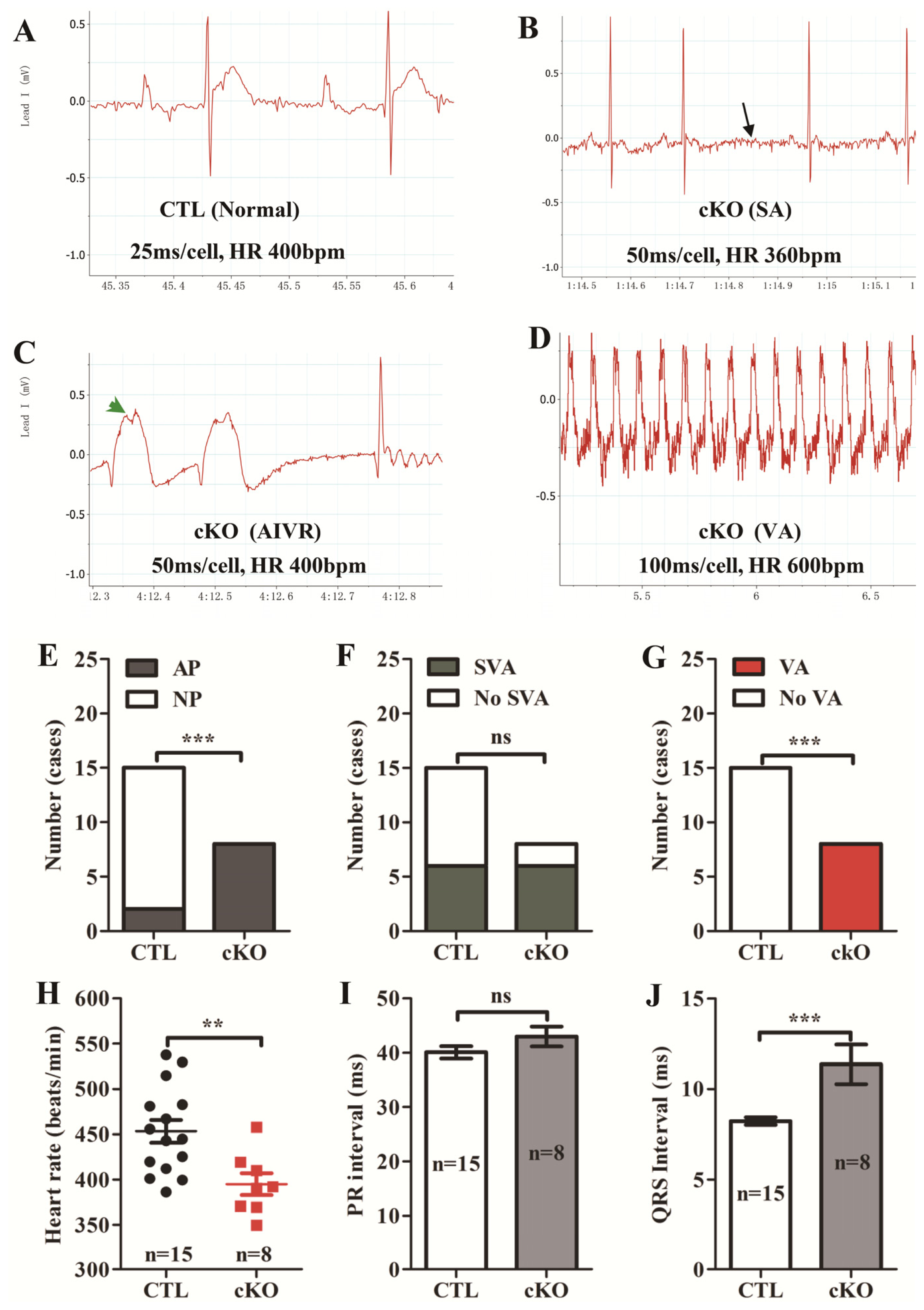
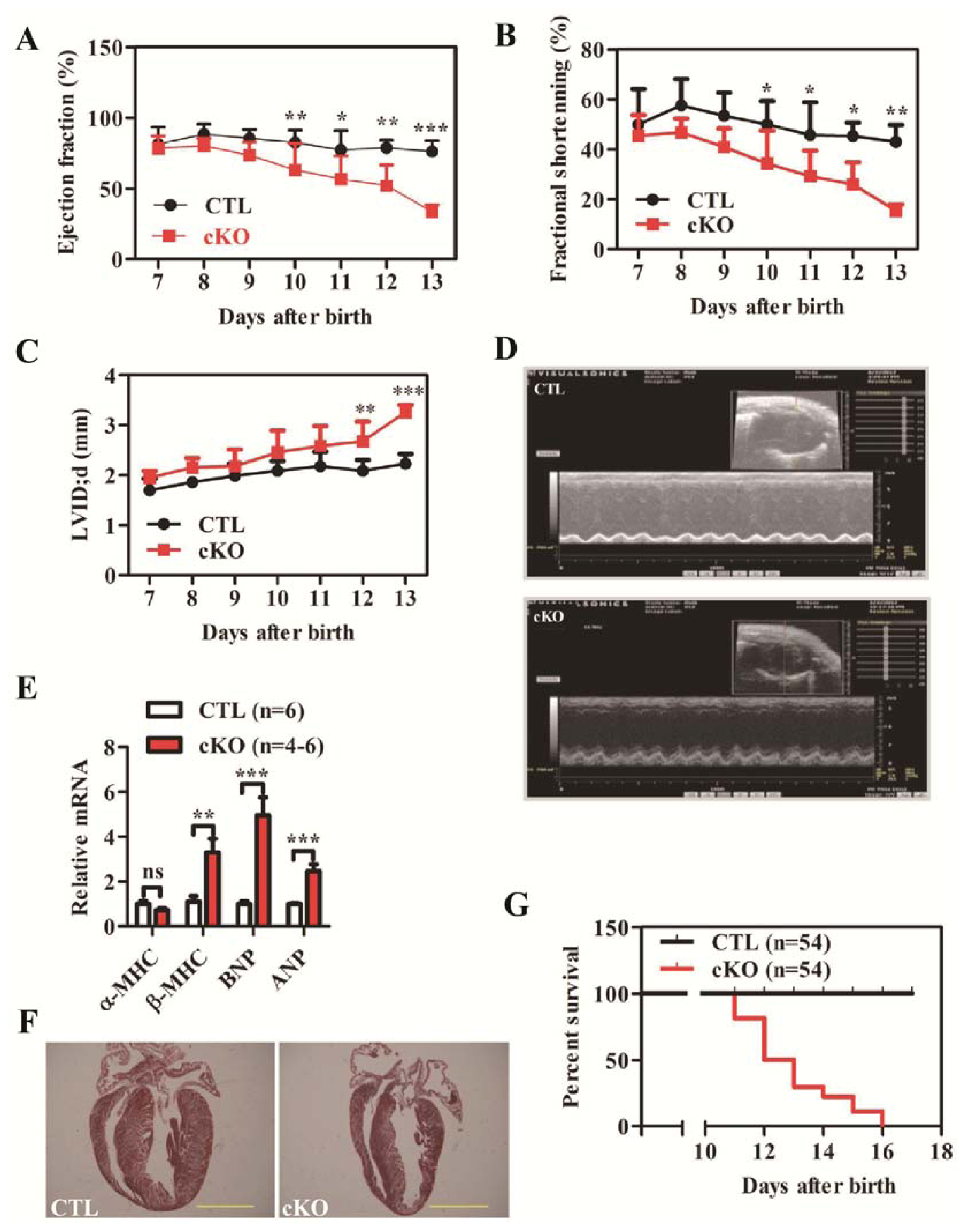
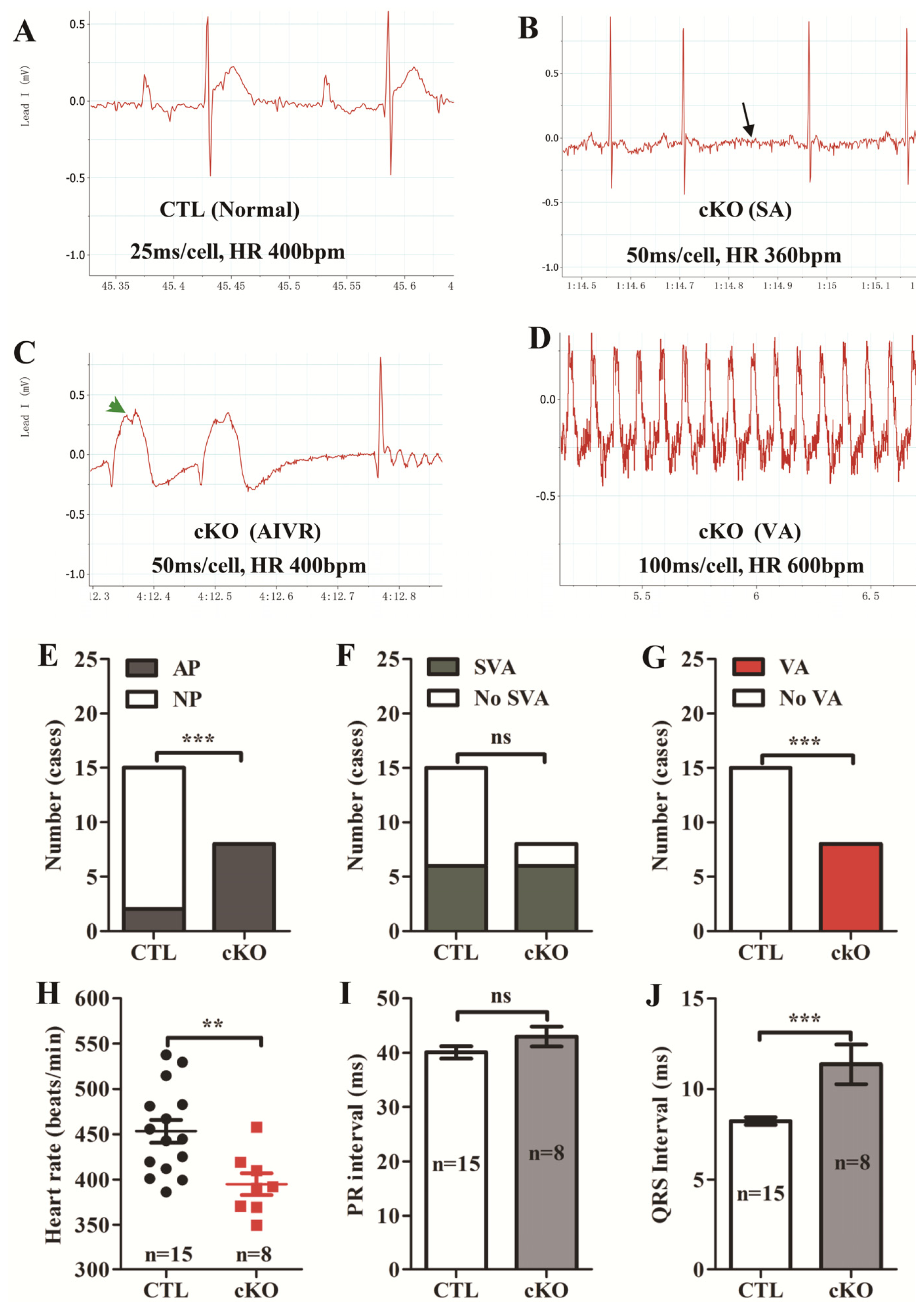
2.1.2. Loss of Rheb1 Causes Heart Failure, Malignant Arrhythmias and Premature Death at Infant Stage
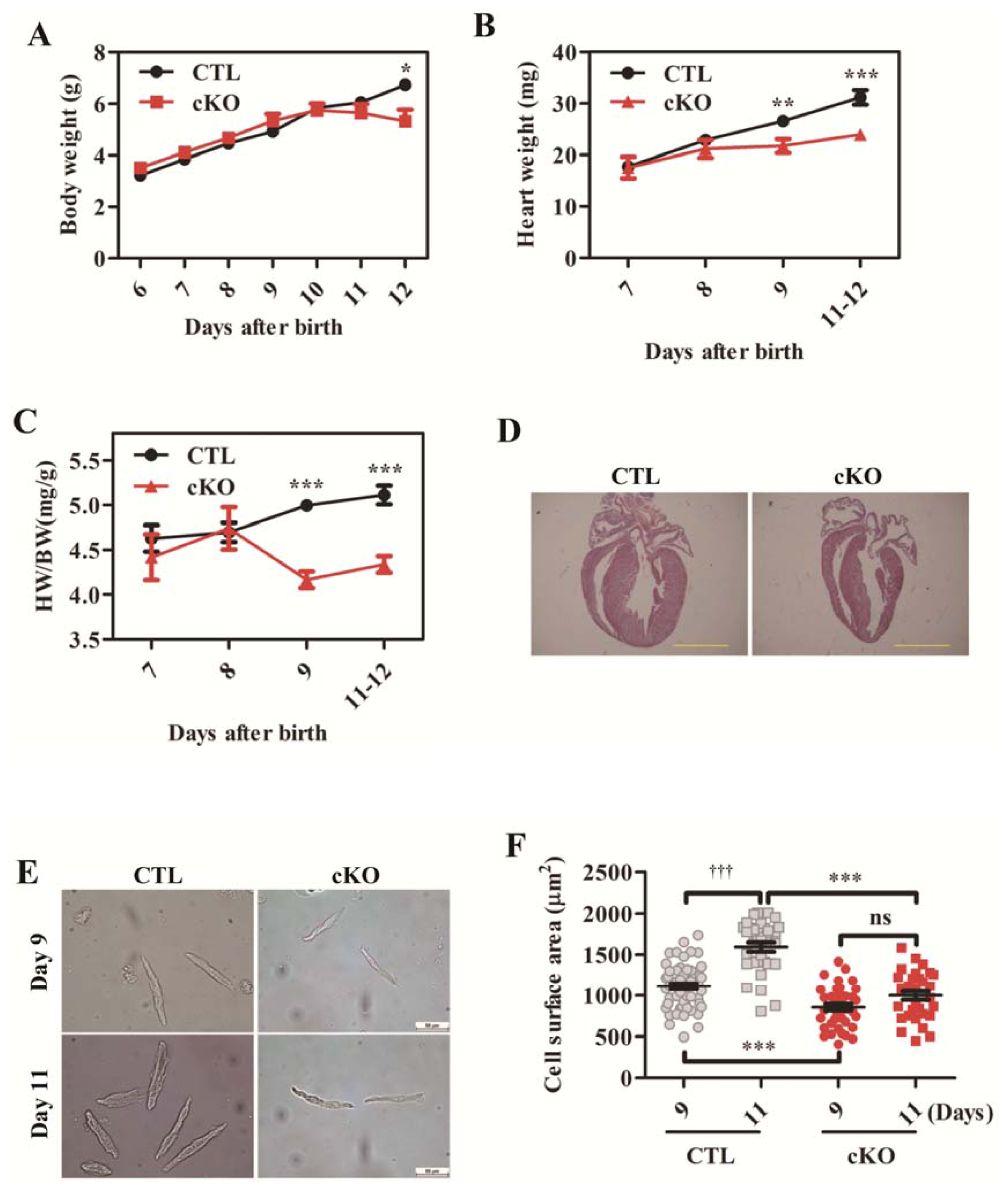
2.1.3. Cardiac Deletion of Rheb1 Results in Abnormal Heart Growth and Retarded Cardiomyocyte Size
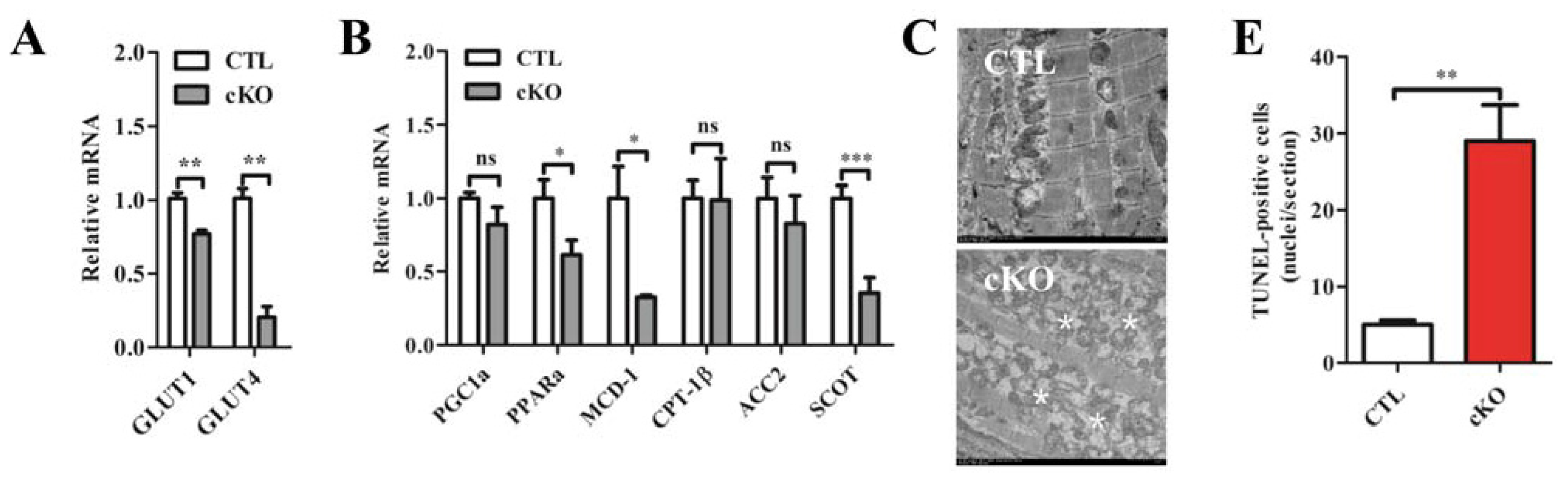
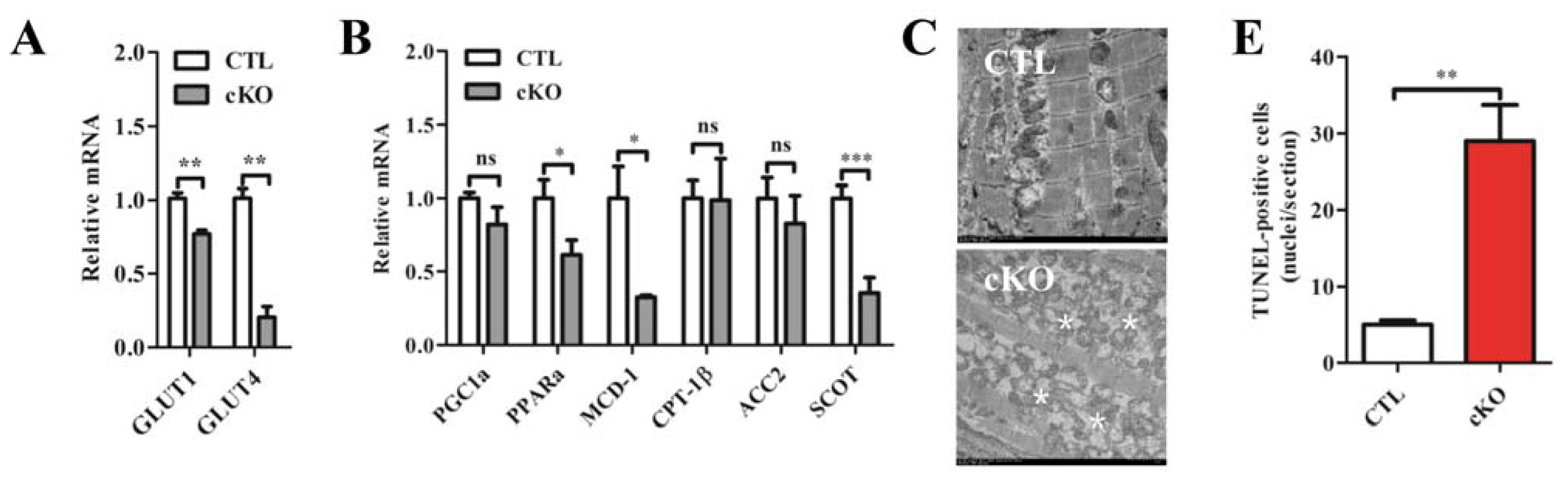
2.1.4. Rheb1 Deletion Results in a Disrupted Expression Profile of Metabolic Genes and Increased Cardiomyocyte Apoptosis, but Does not Affect Autophagy
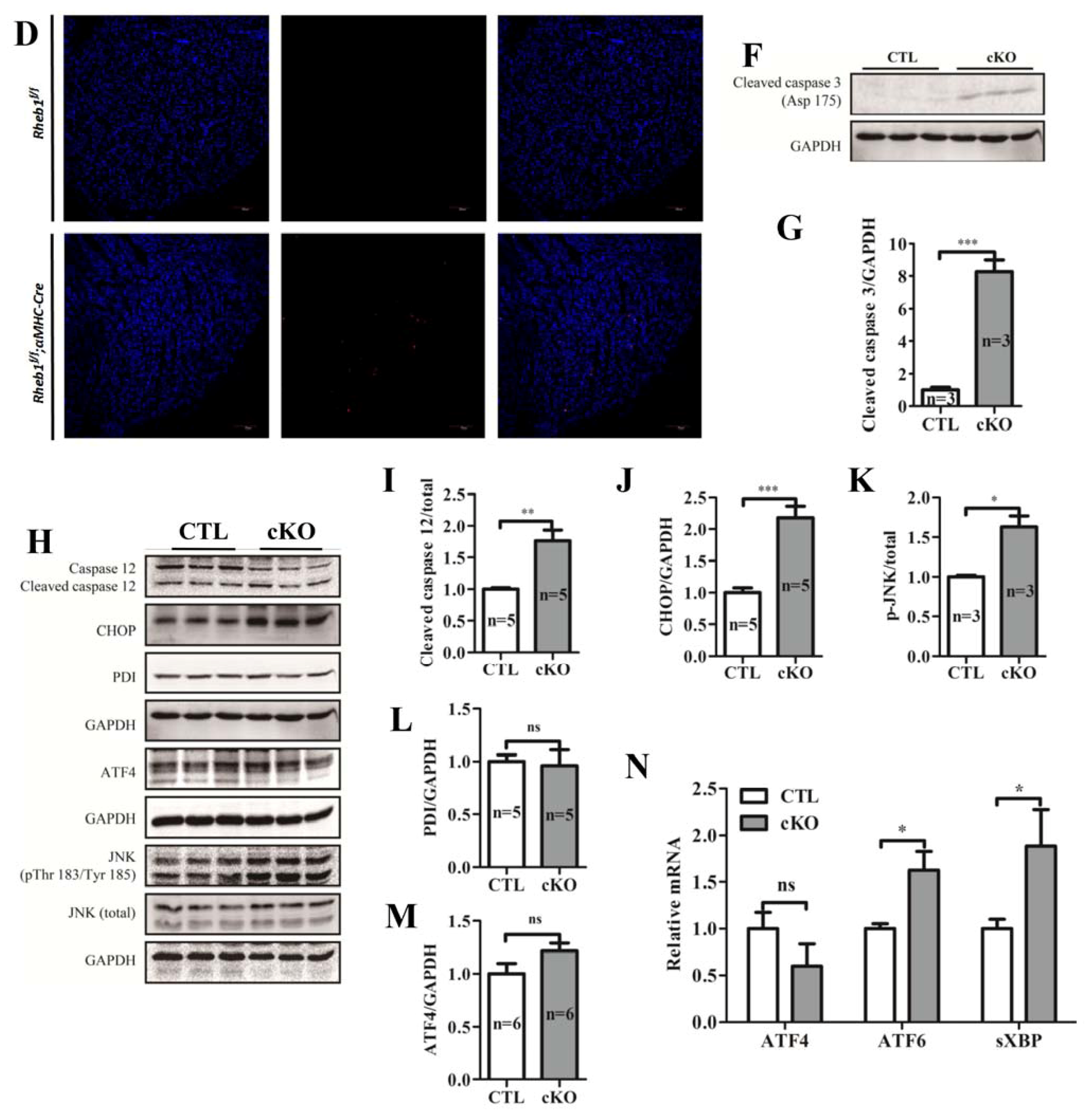
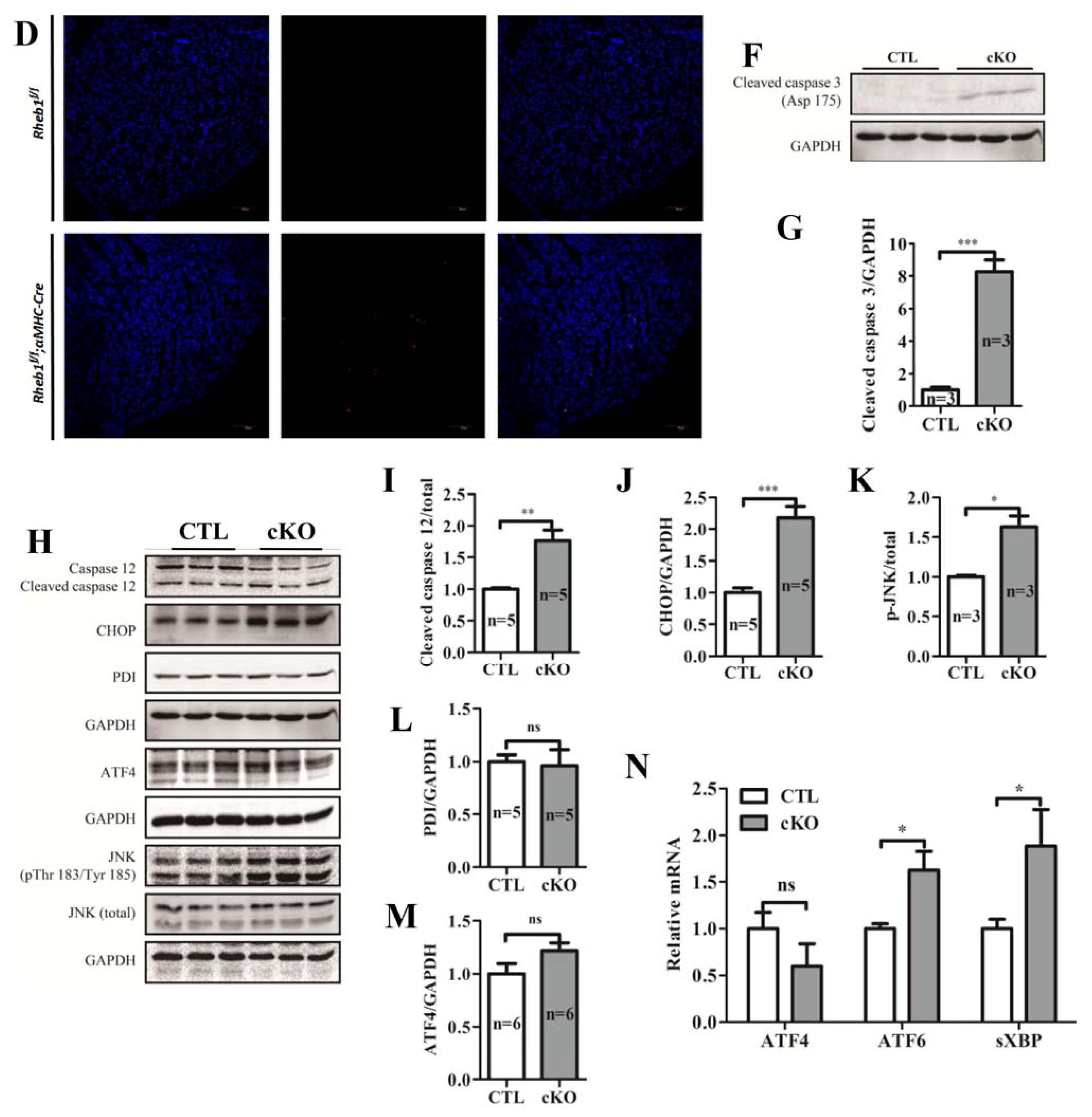
2.1.5. Knockout of Rheb1 Results in the Activation of Endoplasmic Reticulum (ER)-Associated Apoptosis Signaling Pathways
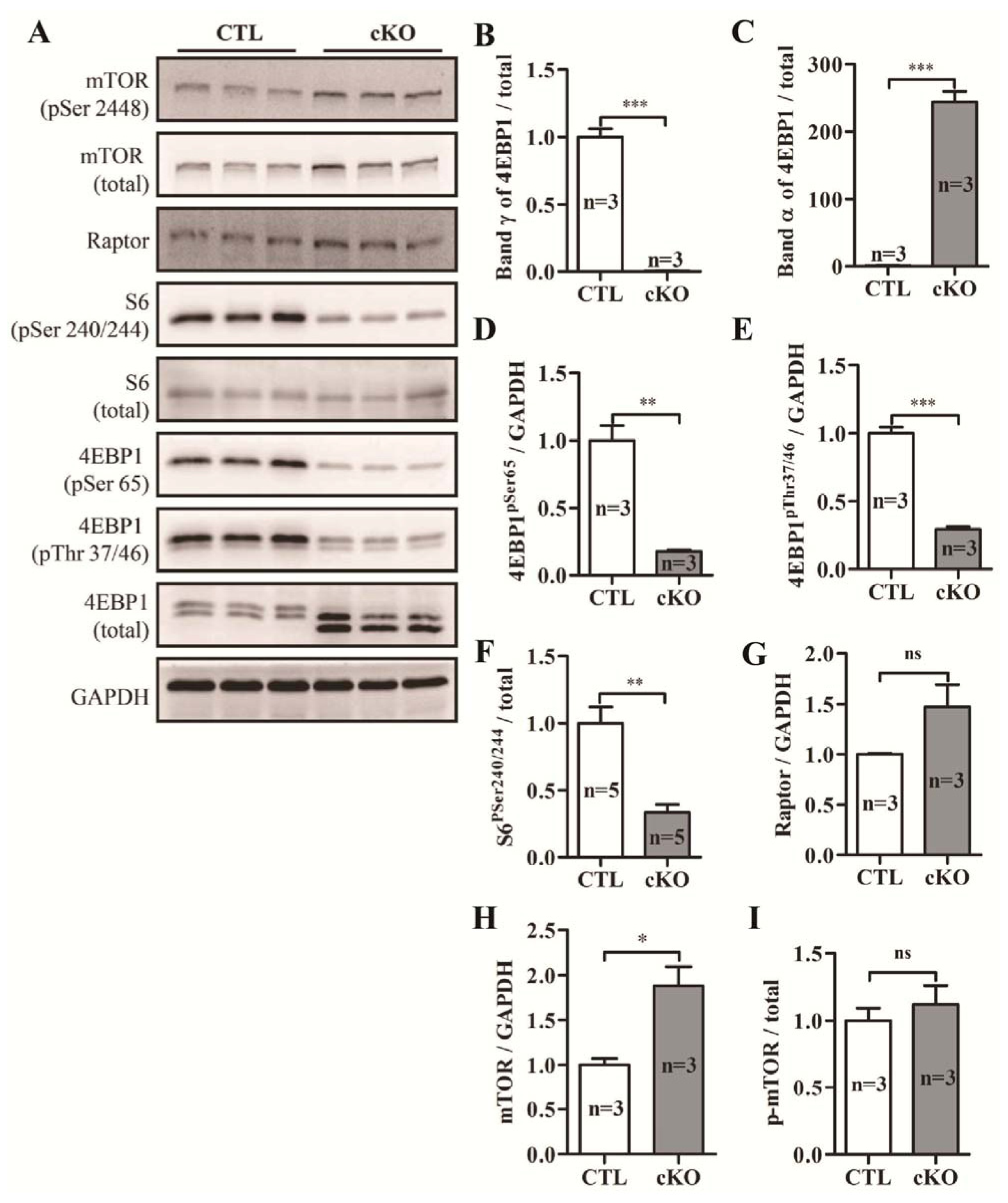
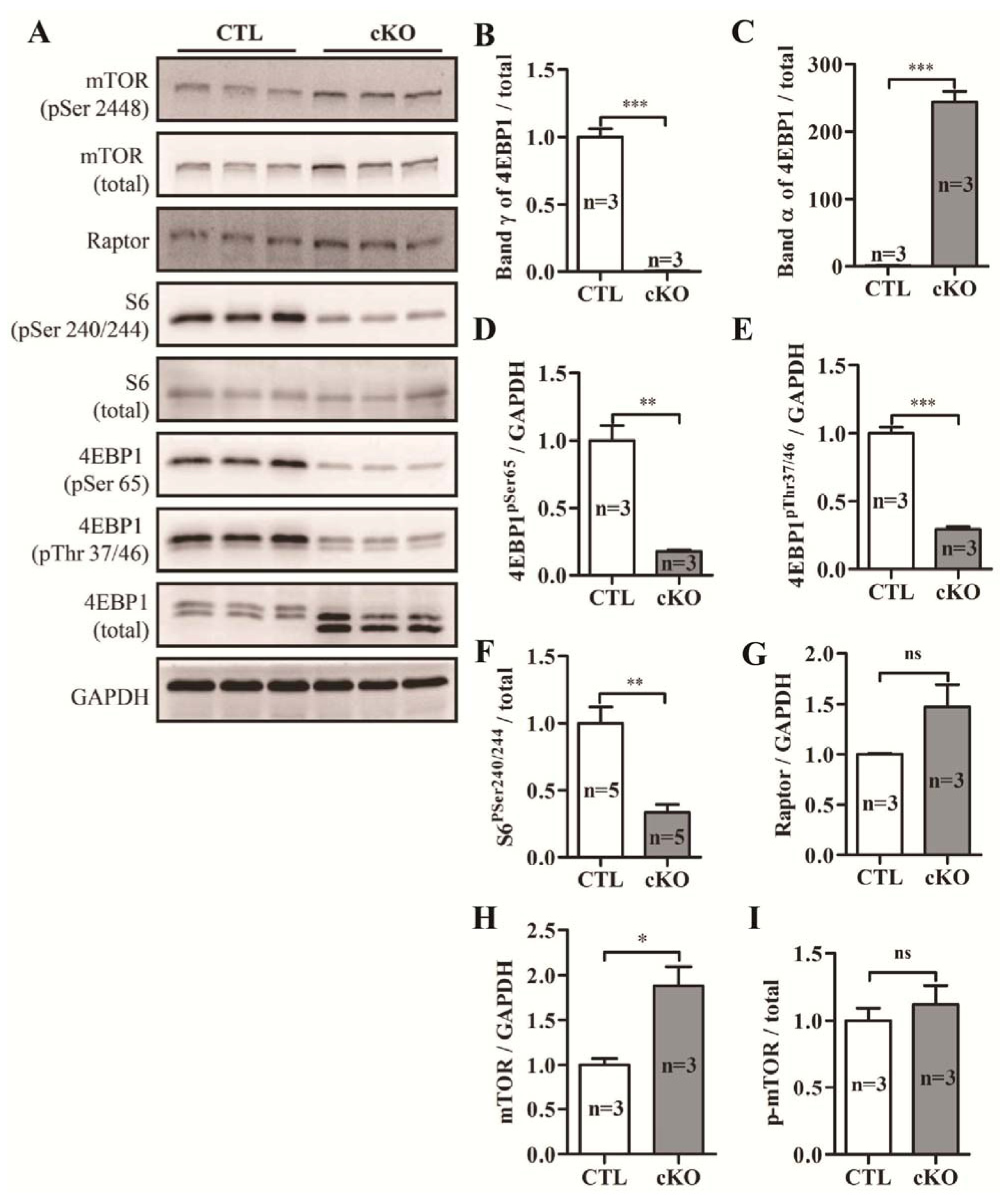
2.1.6. Knockout of Rheb1 Abolishes mTORC1 Signaling
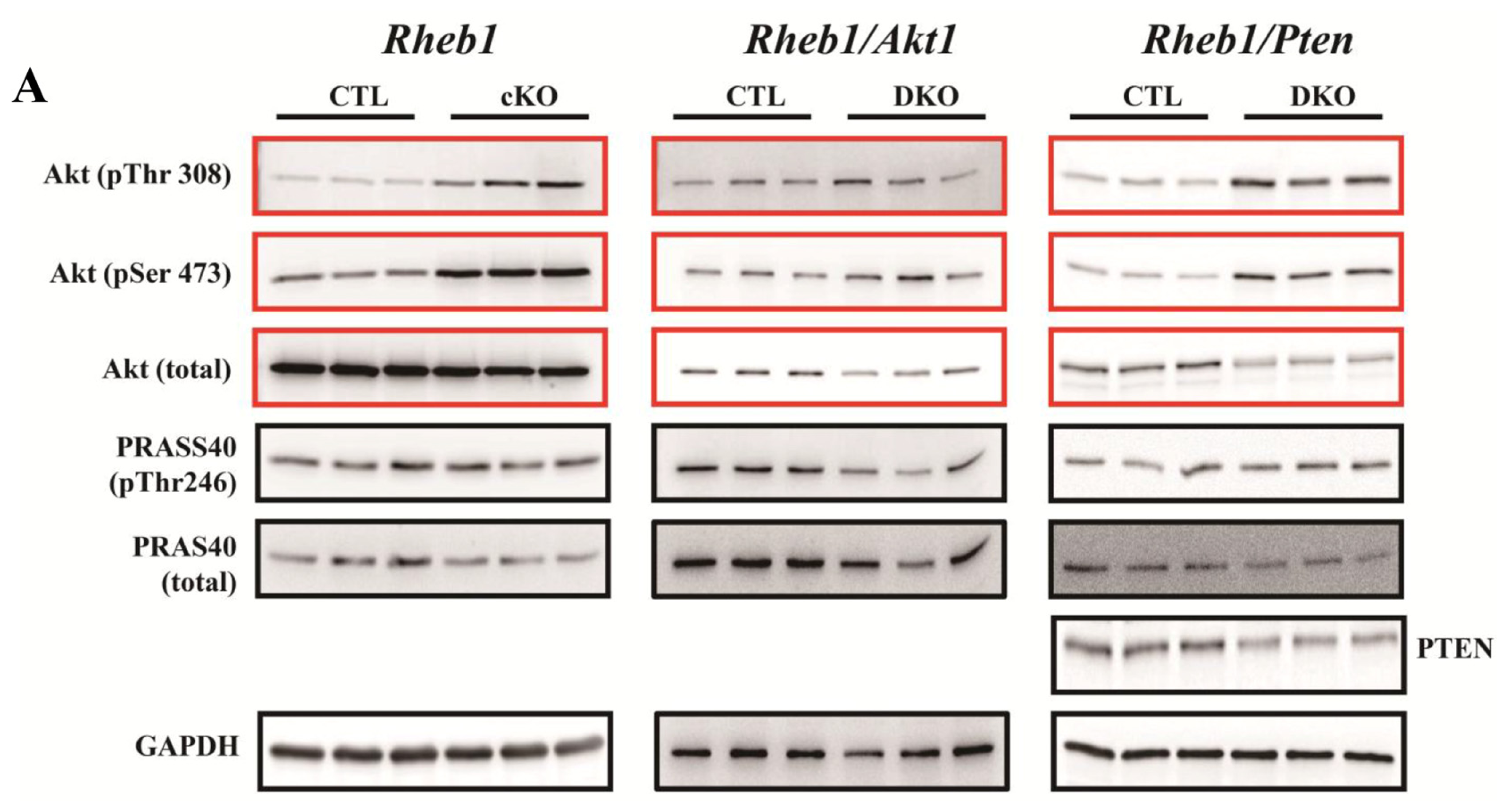
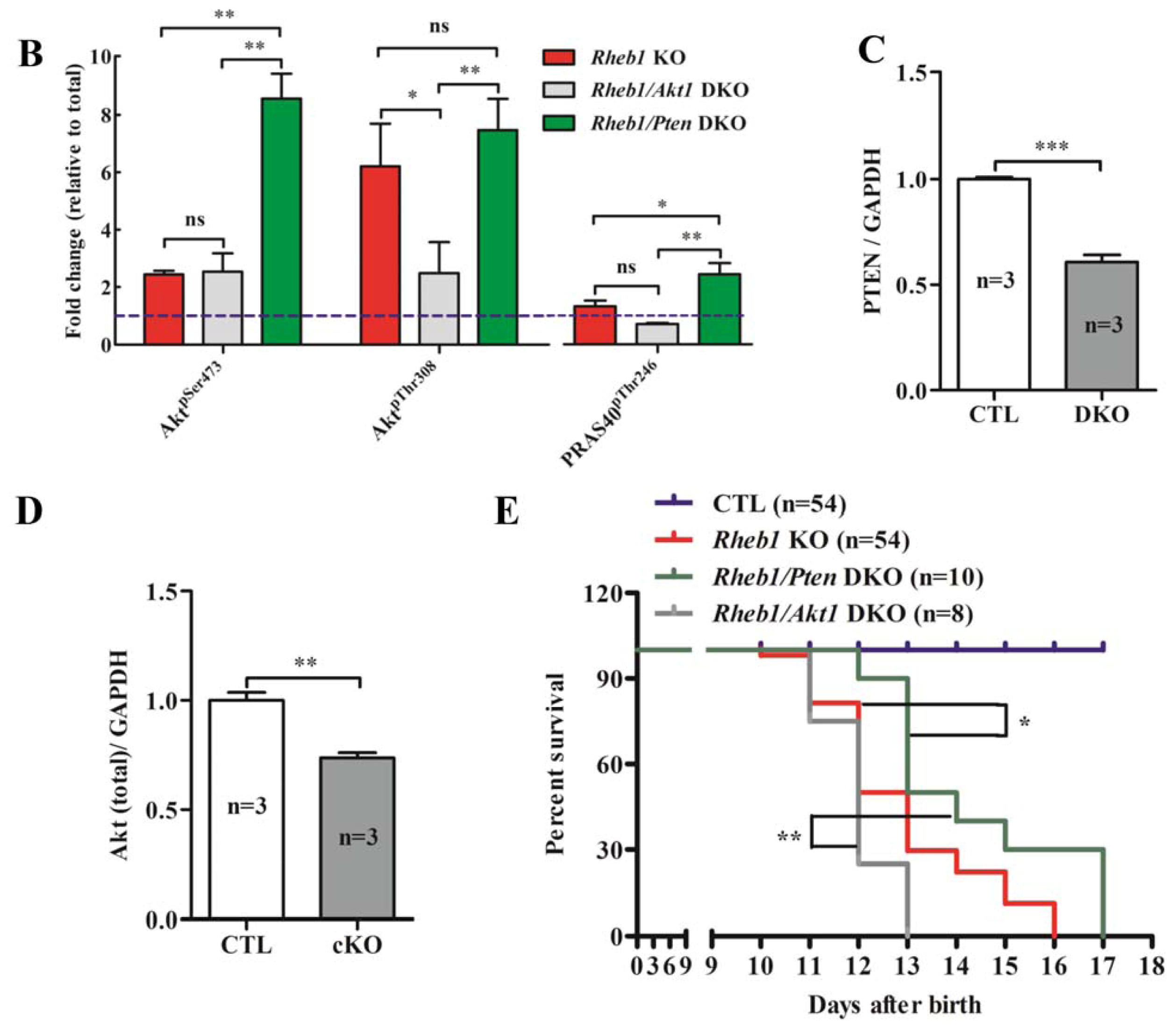
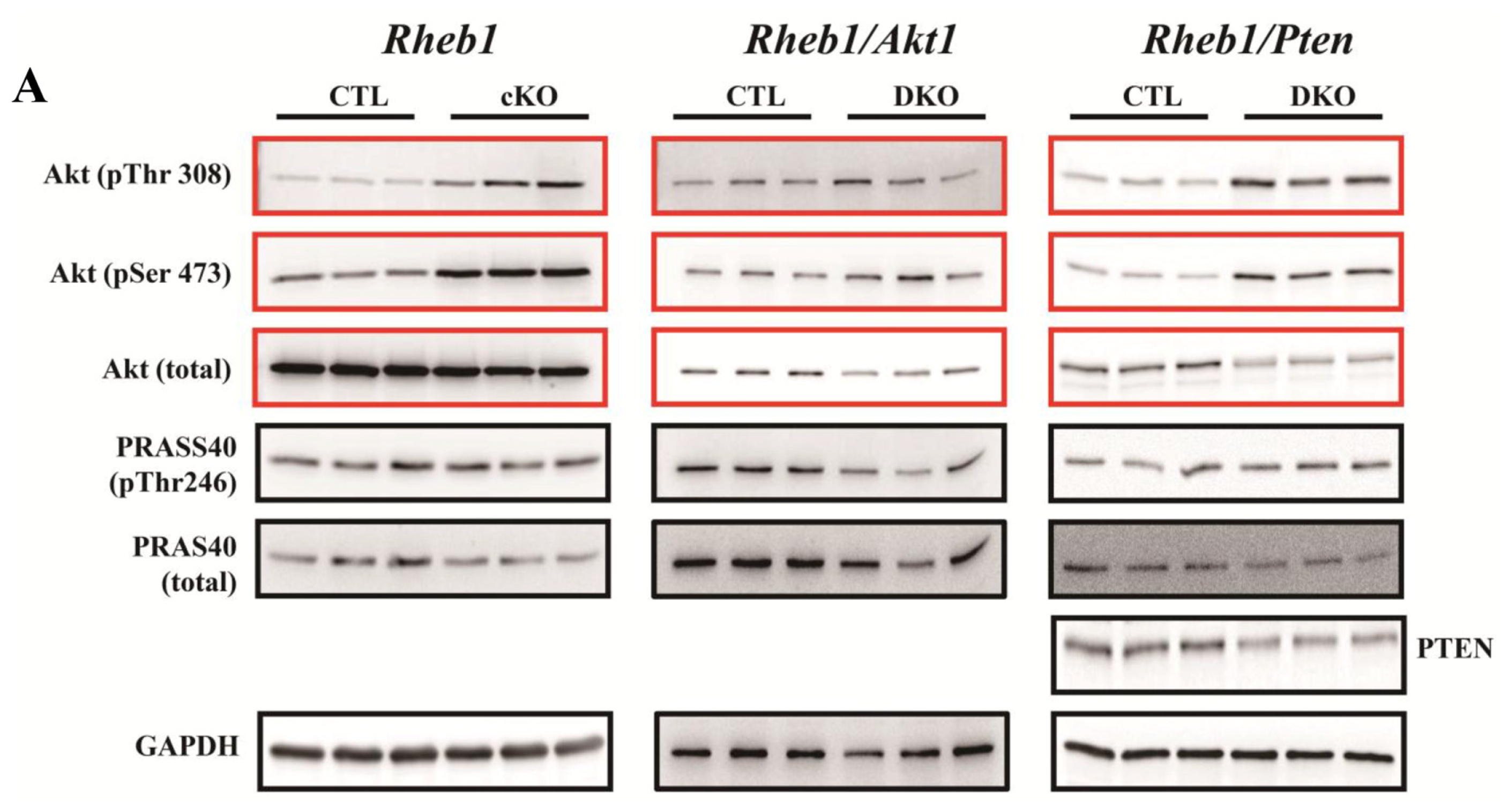
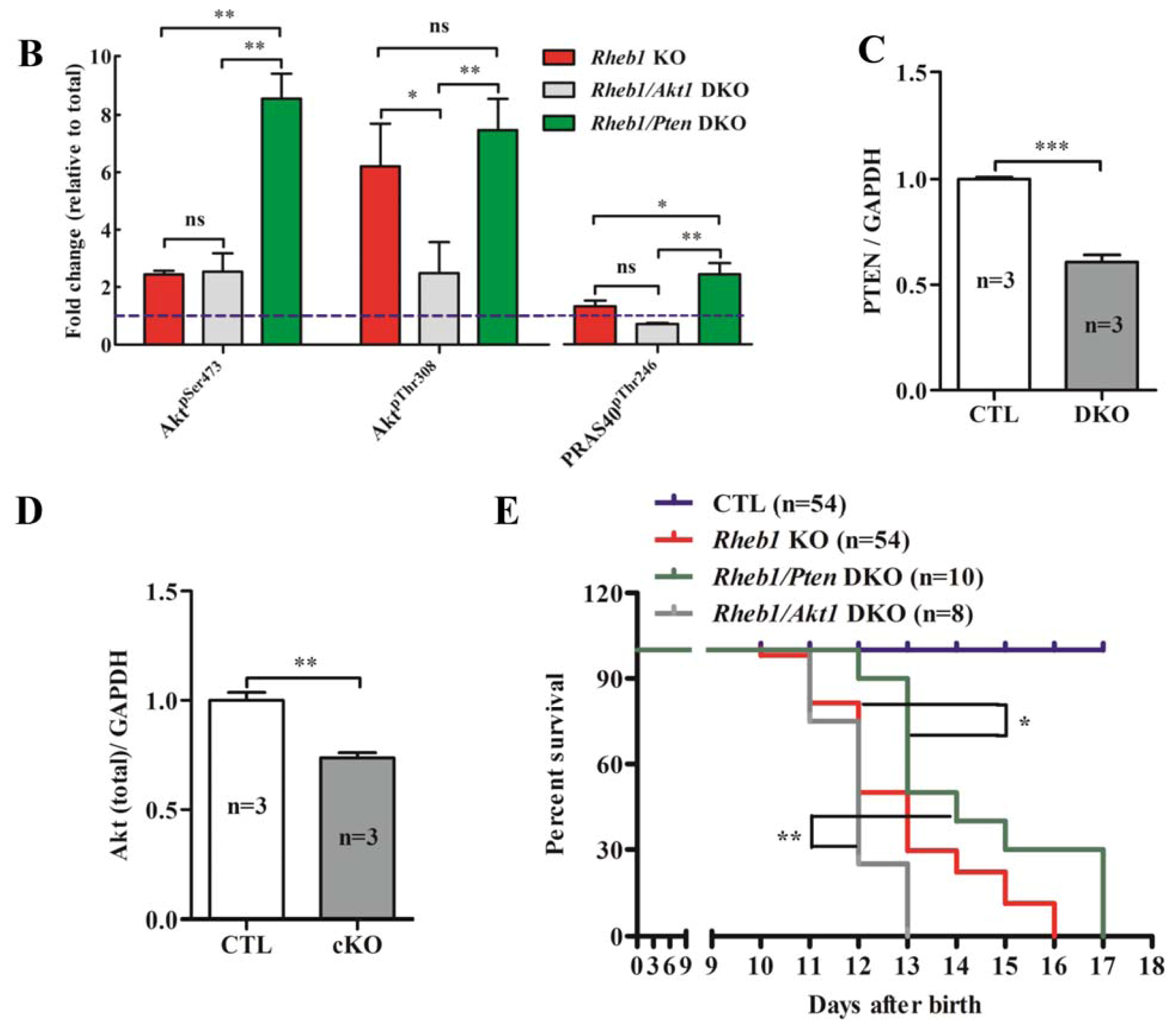
2.1.7. Removal of Pten Prolongs Survival of Rheb1 cKO Mice
2.2. Discussion
3. Experimental Section
3.1. Animal Model
3.2. Echocardiography Assessment of Cardiac Function
3.3. Western Blot Analyses
3.4. Histology and Immunofluorescence Staining
3.5. TUNEL Assay and Transmission Electron Microscopy (TEM) Study
3.6. Quantitative Real-Time PCR for Metabolism Relative Genes and Fetal Genes
3.7. Mouse Cardiomyocyte Isolation
3.8. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-14-24380-s001.pdfAcknowledgments
Conflicts of Interest
References
- Aspuria, P.-J.; Tamanoi, F. The Rheb family of GTP-binding proteins. Cell. Signal 2004, 16, 1105–1112. [Google Scholar]
- Inoki, K. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003, 17, 1829–1834. [Google Scholar]
- Wang, Y.; Huang, B.P.H.; Luciani, D.S.; Wang, X.; Johnson, J.D.; Proud, C.G. Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol 2008, 45, 812–820. [Google Scholar]
- Zou, J.; Zhou, L.; Du, X.-X.; Ji, Y.; Xu, J.; Tian, J.; Jiang, W.; Zou, Y.; Yu, S.; Gan, L.; et al. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 2011, 20, 97–108. [Google Scholar]
- Goorden, S.M.; Hoogeveen-Westerveld, M.; Cheng, C.; van Woerden, G.M.; Mozaffari, M.; Post, L.; Duckers, H.J.; Nellist, M.; Elgersma, Y. Rheb is essential for murine development. Mol. Cell. Biol 2011, 31, 1672–1678. [Google Scholar]
- Wu, X.; Cao, Y.; Nie, J.; Liu, H.; Lu, S.; Hu, X.; Zhu, J.; Zhao, X.; Chen, J.; Chen, X.; et al. Genetic and pharmacological inhibition of Rheb1-mTORC1 signaling exerts cardioprotection against adverse cardiac remodeling in mice. Am. J. Pathol 2013, 182, 2005–2014. [Google Scholar]
- Tamai, T.; Yamaguchi, O.; Hikoso, S.; Takeda, T.; Taneike, M.; Oka, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Uno, Y.; et al. Ras homologue enriched in brain (Rheb)-dependent mTORC1 activation becomes indispensable for cardiac hypertrophic growth after early postnatal period. J. Biol. Chem 2013, 288, 10176–10187. [Google Scholar]
- Zhu, Y.; Pires, K.M.; Whitehead, K.J.; Olsen, C.D.; Wayment, B.; Zhang, Y.C.; Bugger, H.; Ilkun, O.; Litwin, S.E.; Thomas, G.; et al. Mechanistic target of rapamycin (Mtor) is essential for murine embryonic heart development and growth. PLoS One 2013, 8, e54221. [Google Scholar]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Ruegg, M.A.; et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 2011, 123, 1073–1082. [Google Scholar]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig 2010, 120, 2805–2816. [Google Scholar]
- Lopaschuk, G.D.; Spafford, M.A.; Marsh, D.R. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol 1991, 261, H1698–H1705. [Google Scholar]
- Fisher, D.J. Oxygenation and metabolism in the developing heart. Semin. Perinatol 1984, 8, 217–225. [Google Scholar]
- Ma, D.; Bai, X.; Zou, H.; Lai, Y.; Jiang, Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J. Biol. Chem 2010, 285, 8621–8627. [Google Scholar]
- Martinez, J.A.; Zhang, Z.; Svetlov, S.I.; Hayes, R.L.; Wang, K.K.; Larner, S.F. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis 2010, 15, 1480–1493. [Google Scholar]
- Vidal, R.; Caballero, B.; Couve, A.; Hetz, C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Curr. Mol. Med 2011, 11, 1–12. [Google Scholar]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar]
- Xu, X.; Liu, T.; Zhang, A.; Huo, X.; Luo, Q.; Chen, Z.; Yu, L.; Li, Q.; Liu, L.; Lun, Z.R.; et al. Reactive oxygen species-triggered trophoblast apoptosis is initiated by endoplasmic reticulum stress via activation of caspase-12, CHOP, and the JNK pathway in Toxoplasma gondii infection in mice. Infect. Immun 2012, 80, 2121–2132. [Google Scholar]
- Wang, Z.; Zhang, C.; Hong, Z.; Chen, H.; Chen, W.; Chen, G. C/EBP homologous protein (CHOP) mediates neuronal apoptosis in rats with spinal cord injury. Exp. Ther. Med 2013, 5, 107–111. [Google Scholar]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem 2012, 151, 217–219. [Google Scholar]
- Sari, F.R.; Widyantoro, B.; Thandavarayan, R.A.; Harima, M.; Lakshmanan, A.P.; Zhang, S.; Muslin, A.J.; Suzuki, K.; Kodama, M.; Watanabe, K. Attenuation of CHOP-mediated myocardial apoptosis in pressure-overloaded dominant negative p38alpha mitogen-activated protein kinase mice. Cell. Physiol. Biochem 2011, 27, 487–496. [Google Scholar]
- Lakshmanan, A.P.; Thandavarayan, R.A.; Palaniyandi, S.S.; Sari, F.R.; Meilei, H.; Giridharan, V.V.; Soetikno, V.; Suzuki, K.; Kodama, M.; Watanabe, K. Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci 2011, 44, 627–634. [Google Scholar]
- Ariyama, Y.; Tanaka, Y.; Shimizu, H.; Shimomura, K.; Okada, S.; Saito, T.; Yamada, E.; Oyadomari, S.; Mori, M. The role of CHOP messenger RNA expression in the link between oxidative stress and apoptosis. Metabolism 2008, 57, 1625–1635. [Google Scholar]
- Jager, R.; Bertrand, M.J.; Gorman, A.M.; Vandenabeele, P.; Samali, A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol. Cell 2012, 104, 259–270. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol 2012, 13, 89–102. [Google Scholar]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J 2010, 24, 296–308. [Google Scholar]
- Guo, F.J.; Liu, Y.; Zhou, J.; Luo, S.; Zhao, W.; Li, X.; Liu, C. XBP1S protects cells from ER stress-induced apoptosis through Erk1/2 signaling pathway involving CHOP. Histochem. Cell Biol 2012, 138, 447–460. [Google Scholar]
- Manning, B.D. Balancing Akt with S6K: Implications for both metabolic diseases and tumorigenesis. J. Cell Biol 2004, 167, 399–403. [Google Scholar]
- Di, R.; Wu, X.; Chang, Z.; Zhao, X.; Feng, Q.; Lu, S.; Luan, Q.; Hemmings, B.A.; Li, X.; Yang, Z. S6K inhibition renders cardiac protection against myocardial infarction through PDK1 phosphorylation of Akt. Biochem. J 2012, 441, 199–207. [Google Scholar]
- Haq, S.; Choukroun, G.; Lim, H.; Tymitz, K.M.; del Monte, F.; Gwathmey, J.; Grazette, L.; Michael, A.; Hajjar, R.; Force, T.; et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 2001, 103, 670–677. [Google Scholar]
- Nagoshi, T.; Matsui, T.; Aoyama, T.; Leri, A.; Anversa, P.; Li, L.; Ogawa, W.; del Monte, F.; Gwathmey, J.K.; Grazette, L.; et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J. Clin. Investig 2005, 115, 2128–2138. [Google Scholar]
- Sussman, M.A.; Volkers, M.; Fischer, K.; Bailey, B.; Cottage, C.T.; Din, S.; Gude, N.; Avitabile, D.; Alvarez, R.; Sundararaman, B.; et al. Myocardial AKT: The omnipresent nexus. Physiol. Rev 2011, 91, 1023–1070. [Google Scholar]
- Qi, X.; Yang, G.; Yang, L.; Lan, Y.; Weng, T.; Wang, J.; Wu, Z.; Xu, J.; Gao, X.; Yang, X. Essential role of Smad4 in maintaining cardiomyocyte proliferation during murine embryonic heart development. Dev. Biol 2007, 311, 136–146. [Google Scholar]
- Pochynyuk, O.; Stockand, J.D.; Staruschenko, A. Ion channel regulation by Ras, Rho, and Rab small GTPases. Exp. Biol. Med 2007, 232, 1258–1265. [Google Scholar]
- Respress, J.L.; Wehrens, X.H. Transthoracic echocardiography in mice. J. Vis. Exp 2010, 39, 1738. [Google Scholar]
- Zhou, Y.Y.; Wang, S.Q.; Zhu, W.Z.; Chruscinski, A.; Kobilka, B.K.; Ziman, B.; Wang, S.; Lakatta, E.G.; Cheng, H.; Xiao, R.P. Culture and adenoviral infection of adult mouse cardiac myocytes: Methods for cellular genetic physiology. Am. J. Physiol. Heart Circ. Physiol 2000, 279, H429–H436. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cao, Y.; Tao, L.; Shen, S.; Xiao, J.; Wu, H.; Li, B.; Wu, X.; Luo, W.; Xiao, Q.; Hu, X.; et al. Cardiac Ablation of Rheb1 Induces Impaired Heart Growth, Endoplasmic Reticulum-Associated Apoptosis and Heart Failure in Infant Mice. Int. J. Mol. Sci. 2013, 14, 24380-24398. https://doi.org/10.3390/ijms141224380
Cao Y, Tao L, Shen S, Xiao J, Wu H, Li B, Wu X, Luo W, Xiao Q, Hu X, et al. Cardiac Ablation of Rheb1 Induces Impaired Heart Growth, Endoplasmic Reticulum-Associated Apoptosis and Heart Failure in Infant Mice. International Journal of Molecular Sciences. 2013; 14(12):24380-24398. https://doi.org/10.3390/ijms141224380
Chicago/Turabian StyleCao, Yunshan, Lichan Tao, Shutong Shen, Junjie Xiao, Hang Wu, Beibei Li, Xiangqi Wu, Wen Luo, Qi Xiao, Xiaoshan Hu, and et al. 2013. "Cardiac Ablation of Rheb1 Induces Impaired Heart Growth, Endoplasmic Reticulum-Associated Apoptosis and Heart Failure in Infant Mice" International Journal of Molecular Sciences 14, no. 12: 24380-24398. https://doi.org/10.3390/ijms141224380