EZH2 Down-Regulation Exacerbates Lipid Accumulation and Inflammation in in Vitro and in Vivo NAFLD

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
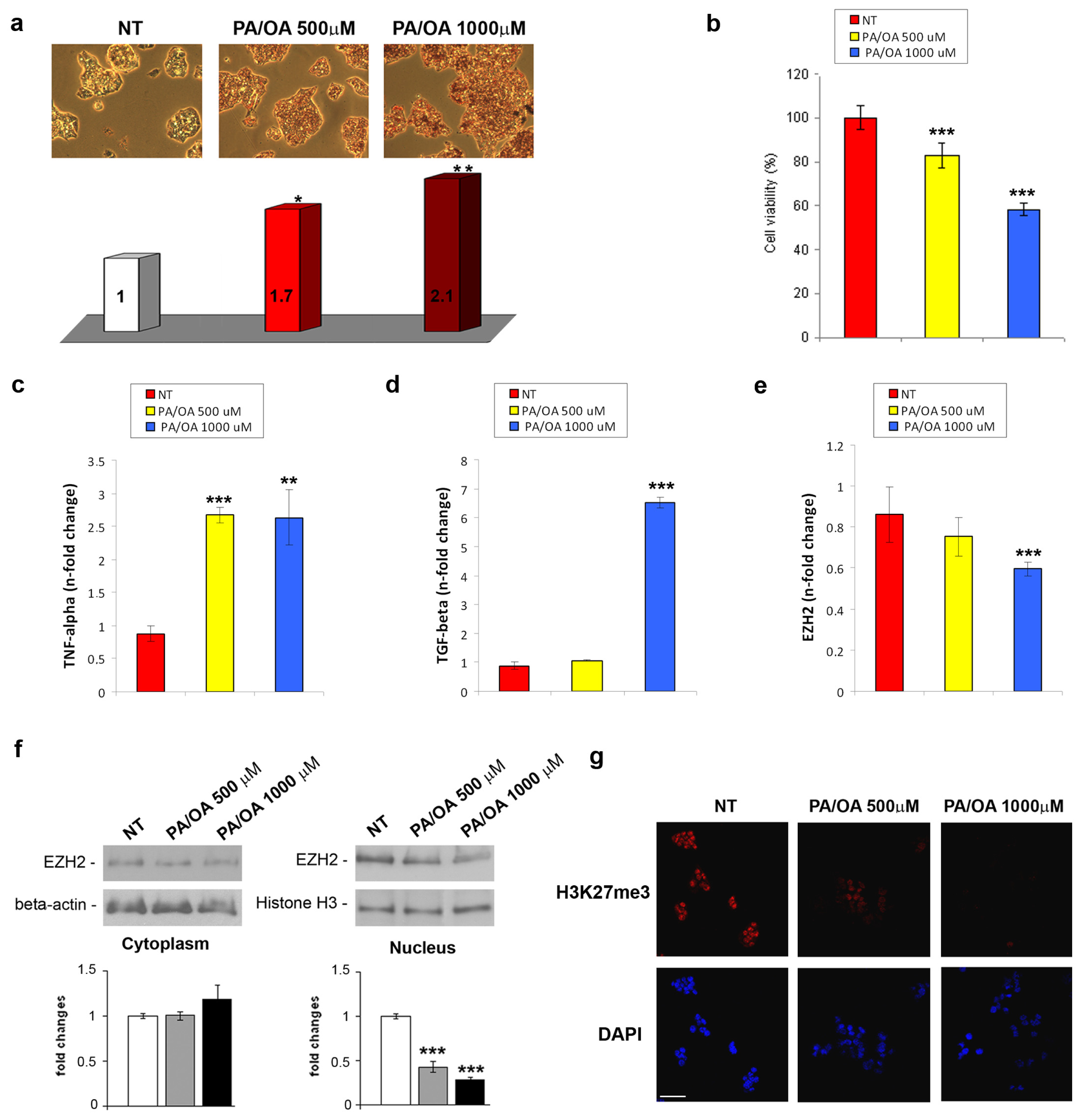
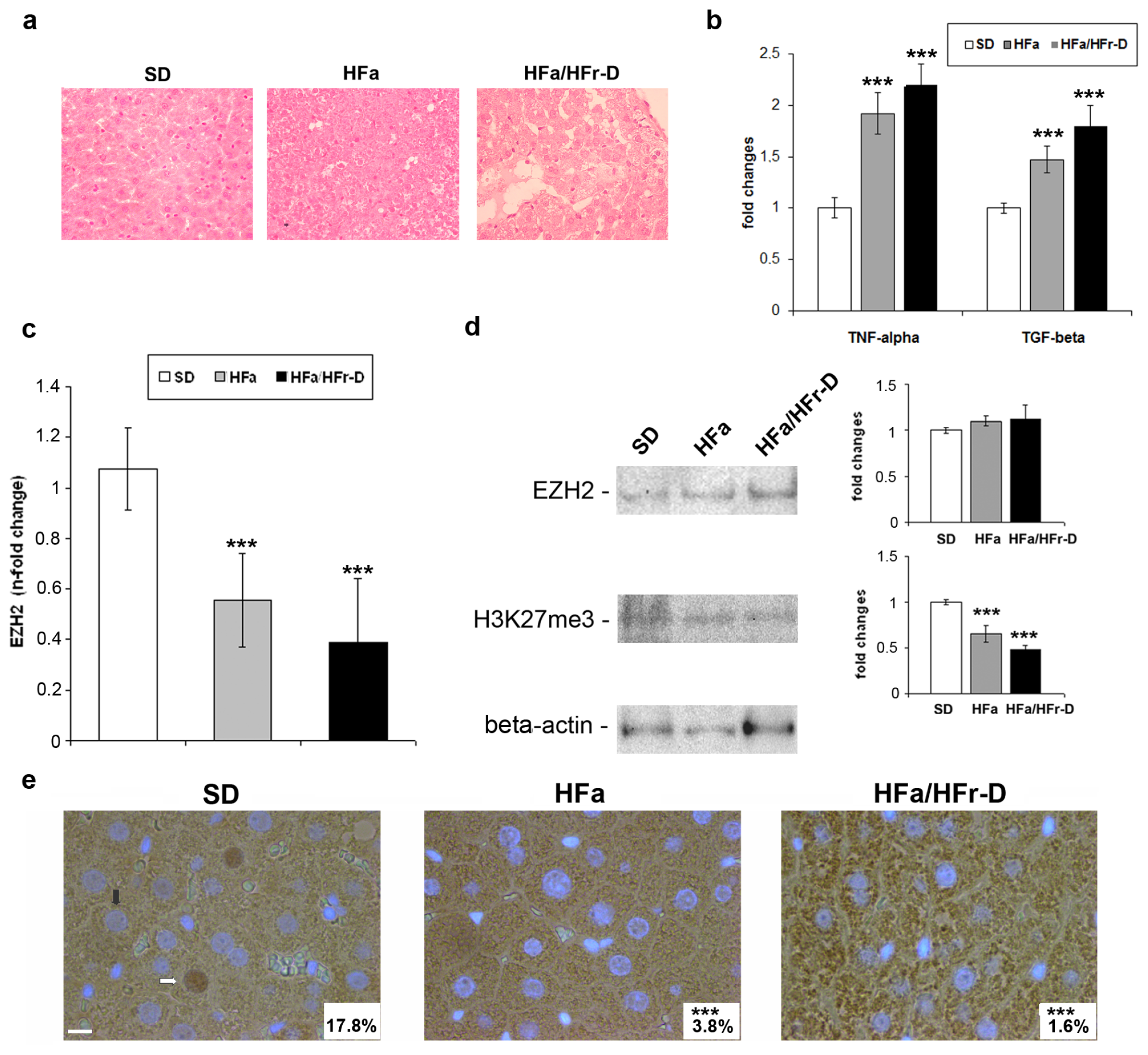
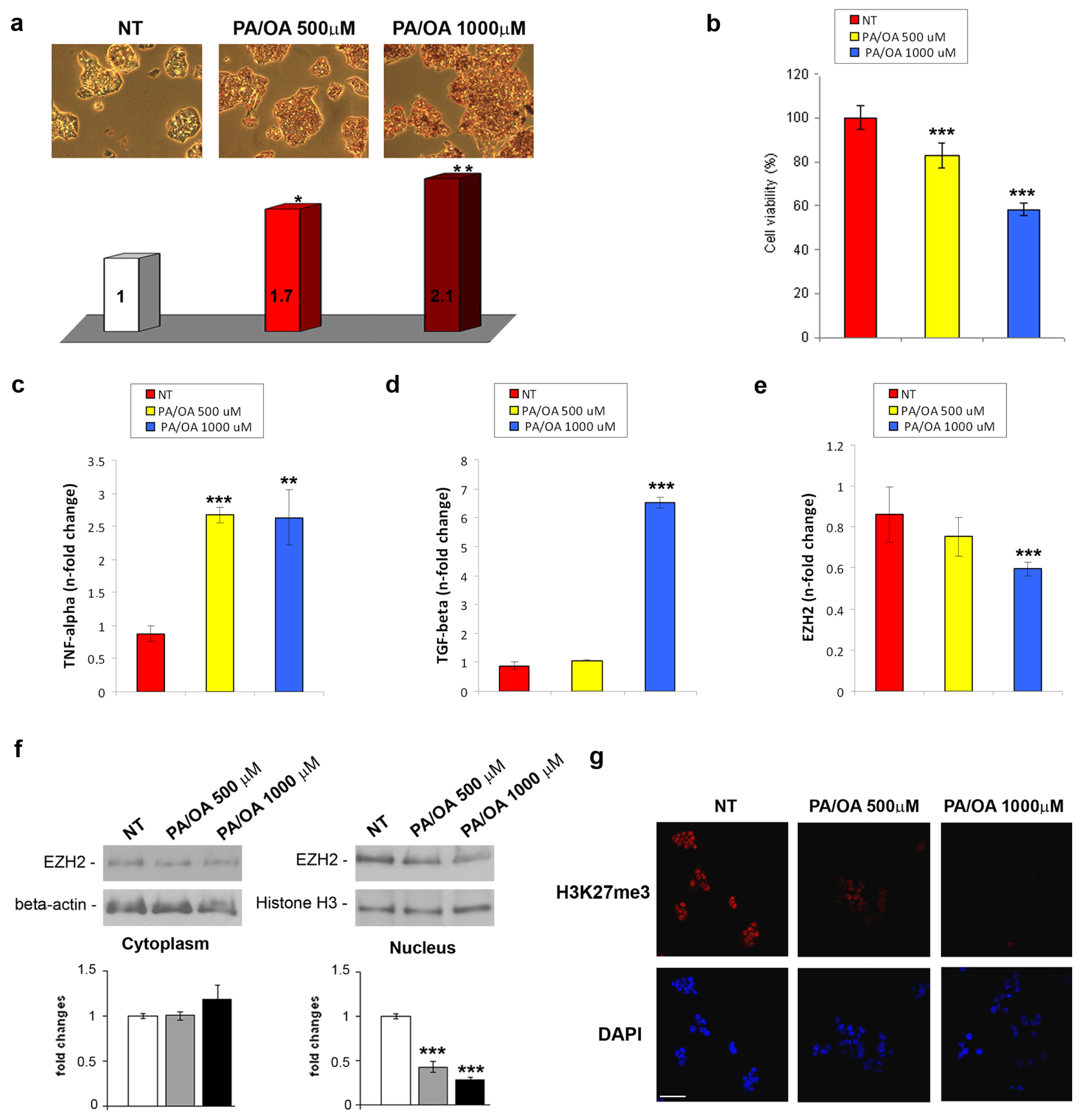
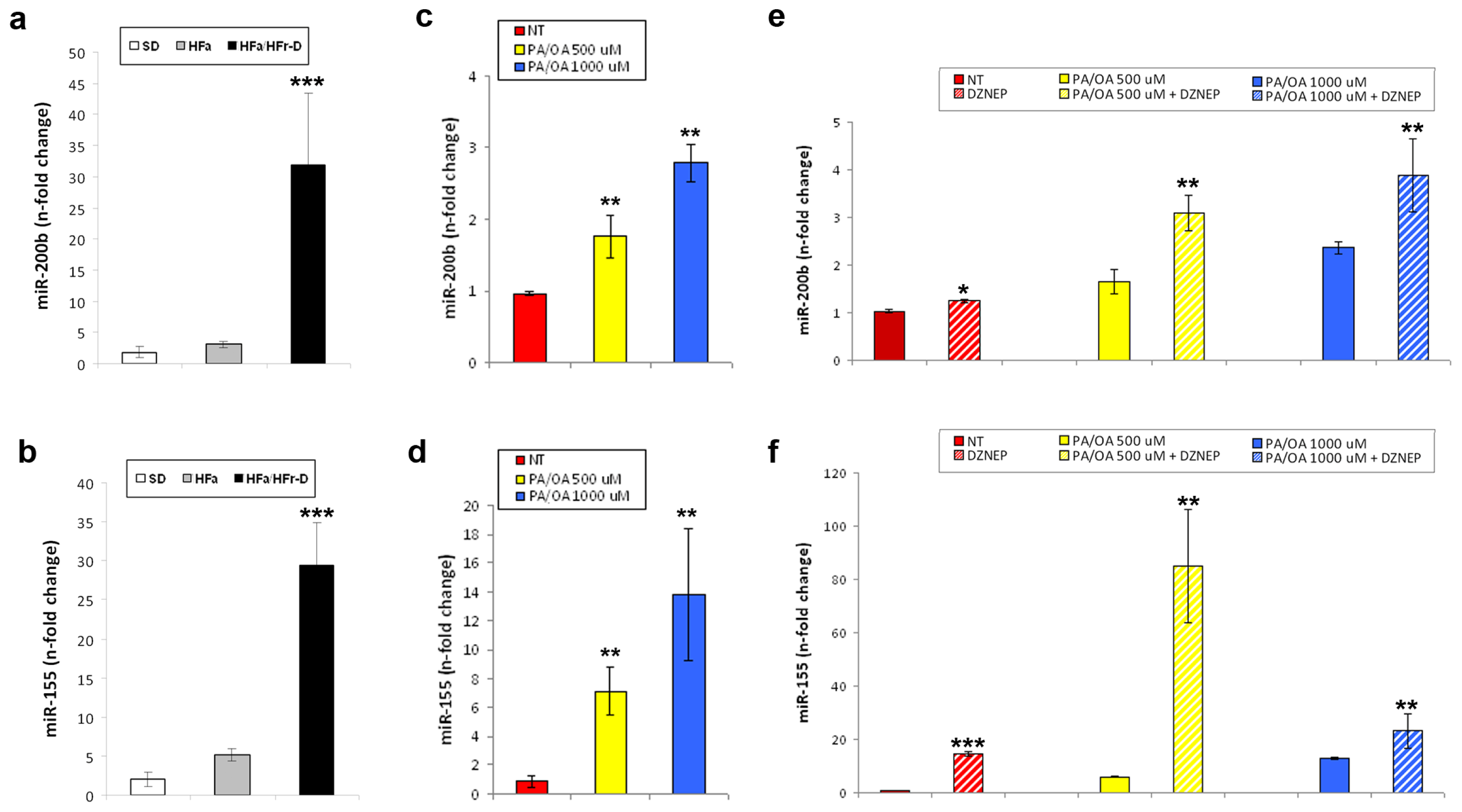
2.1. EZH2 Expression and Activity in in Vivo and in Vitro NAFLD
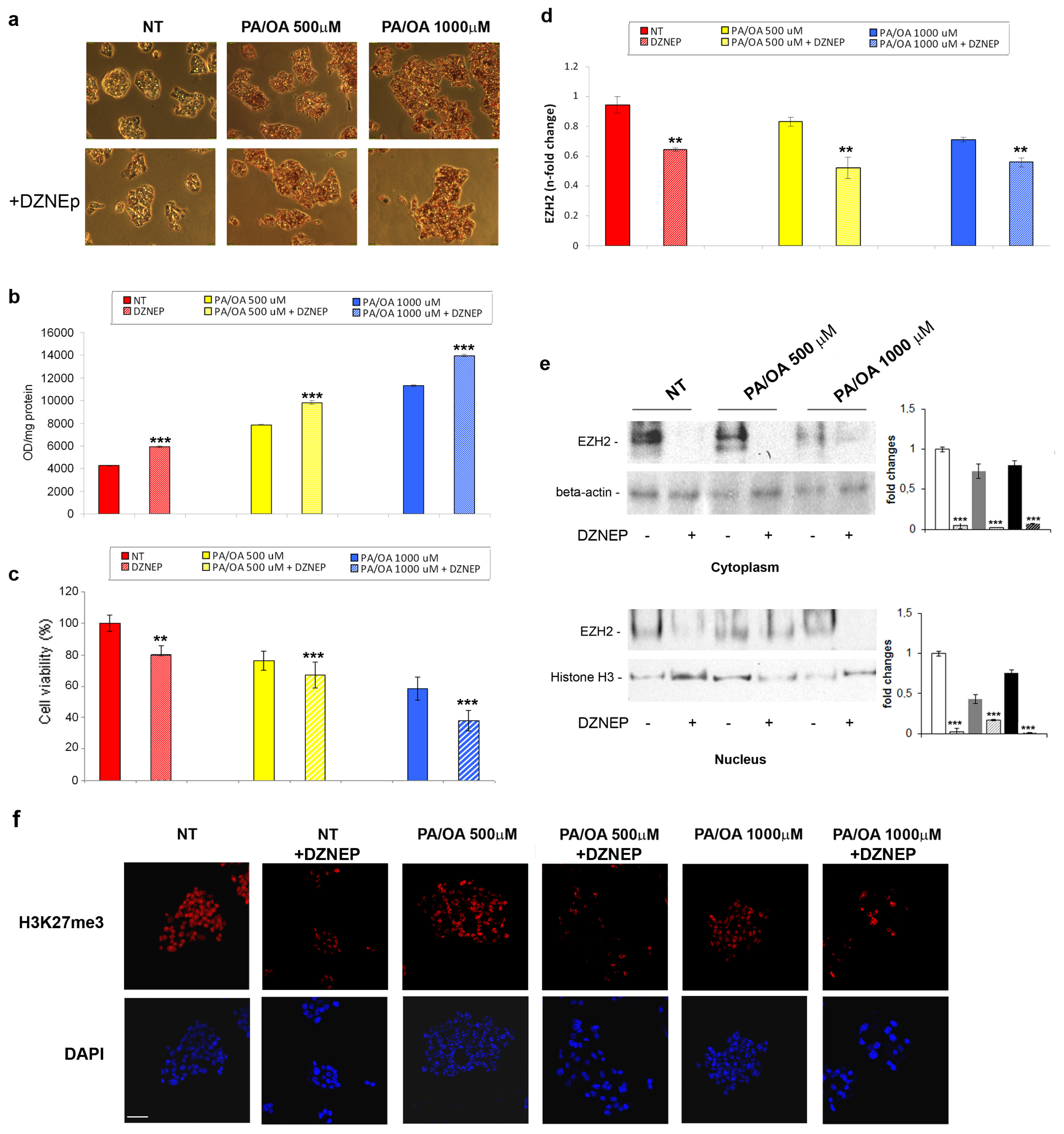
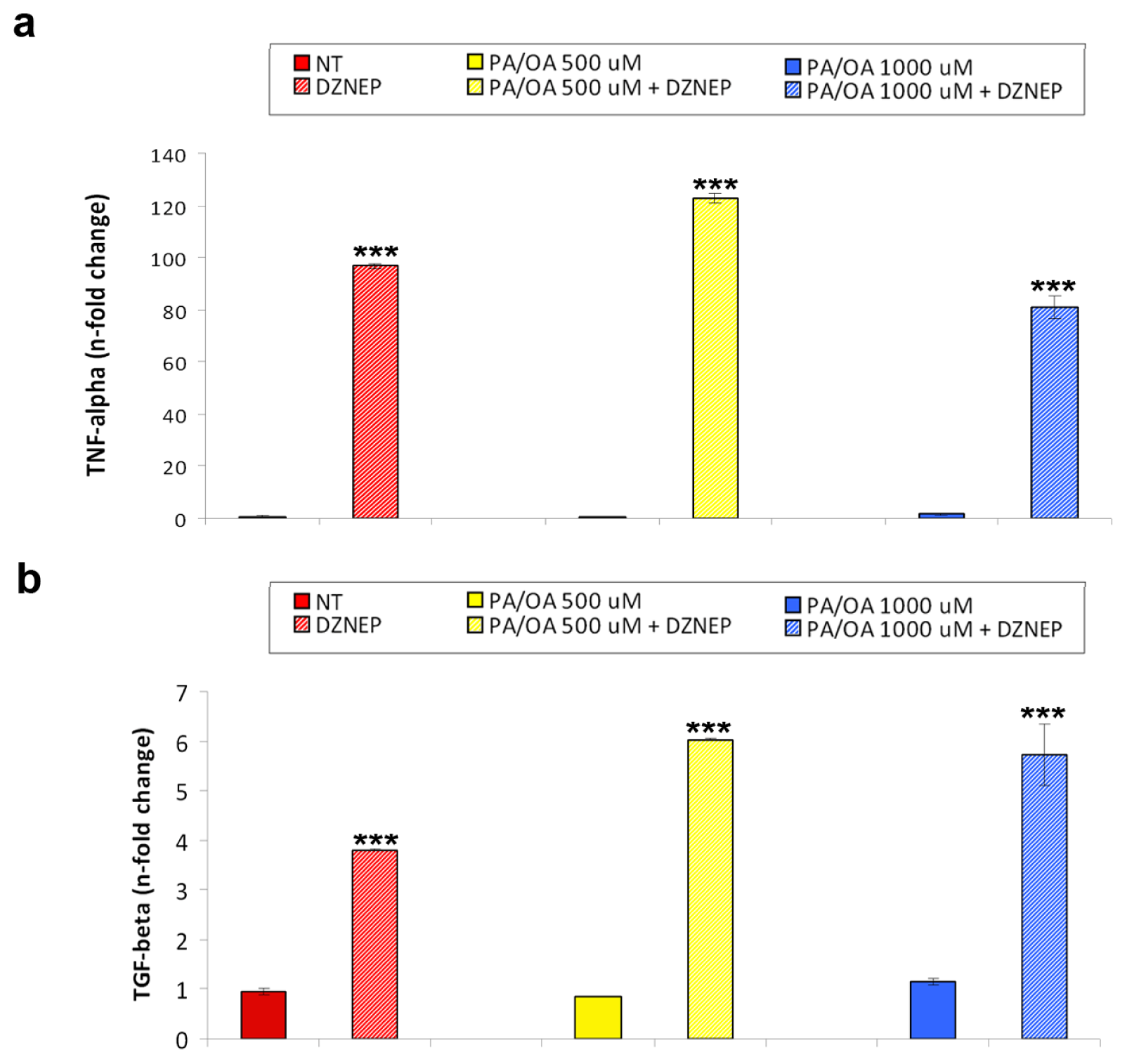
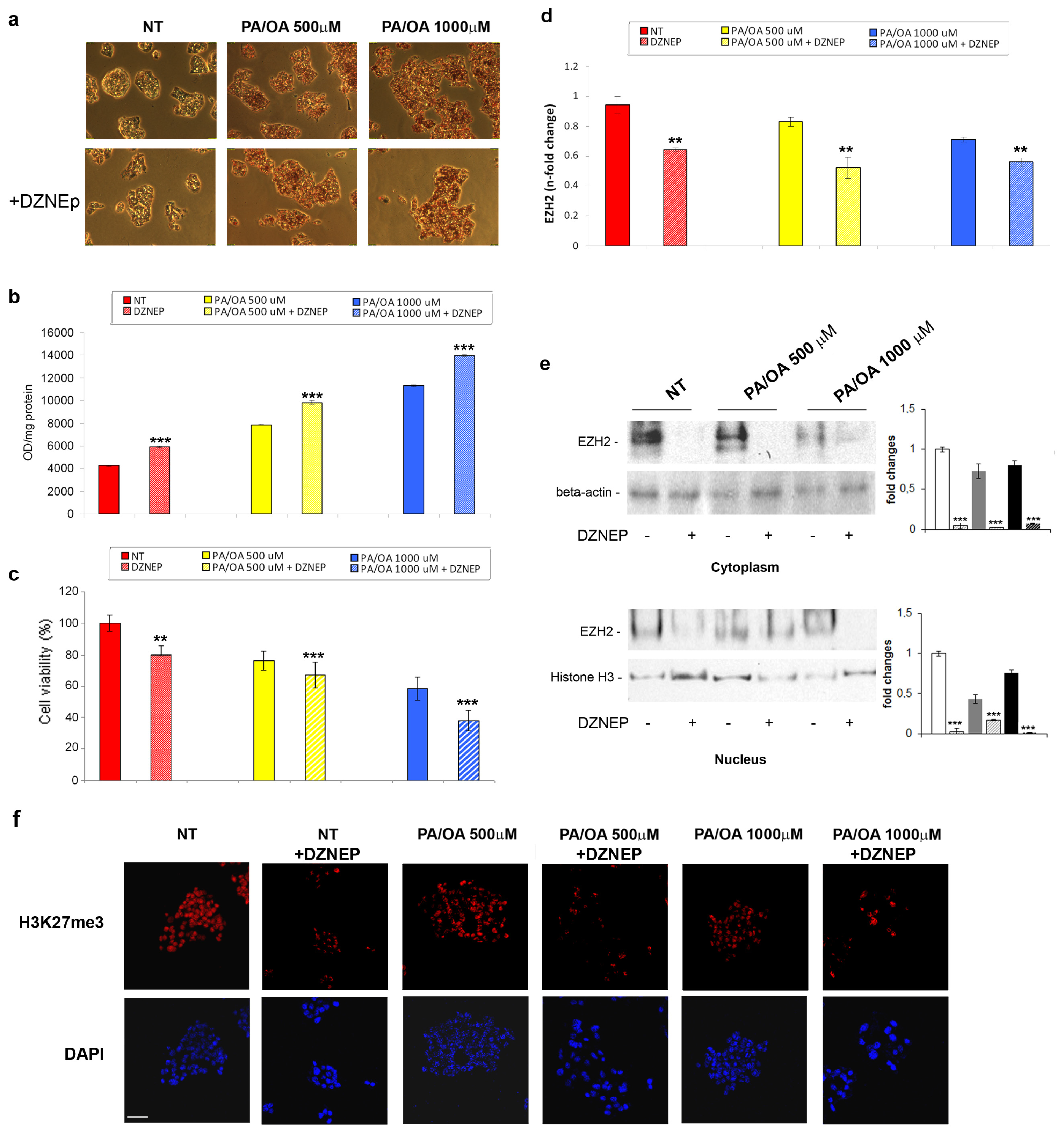
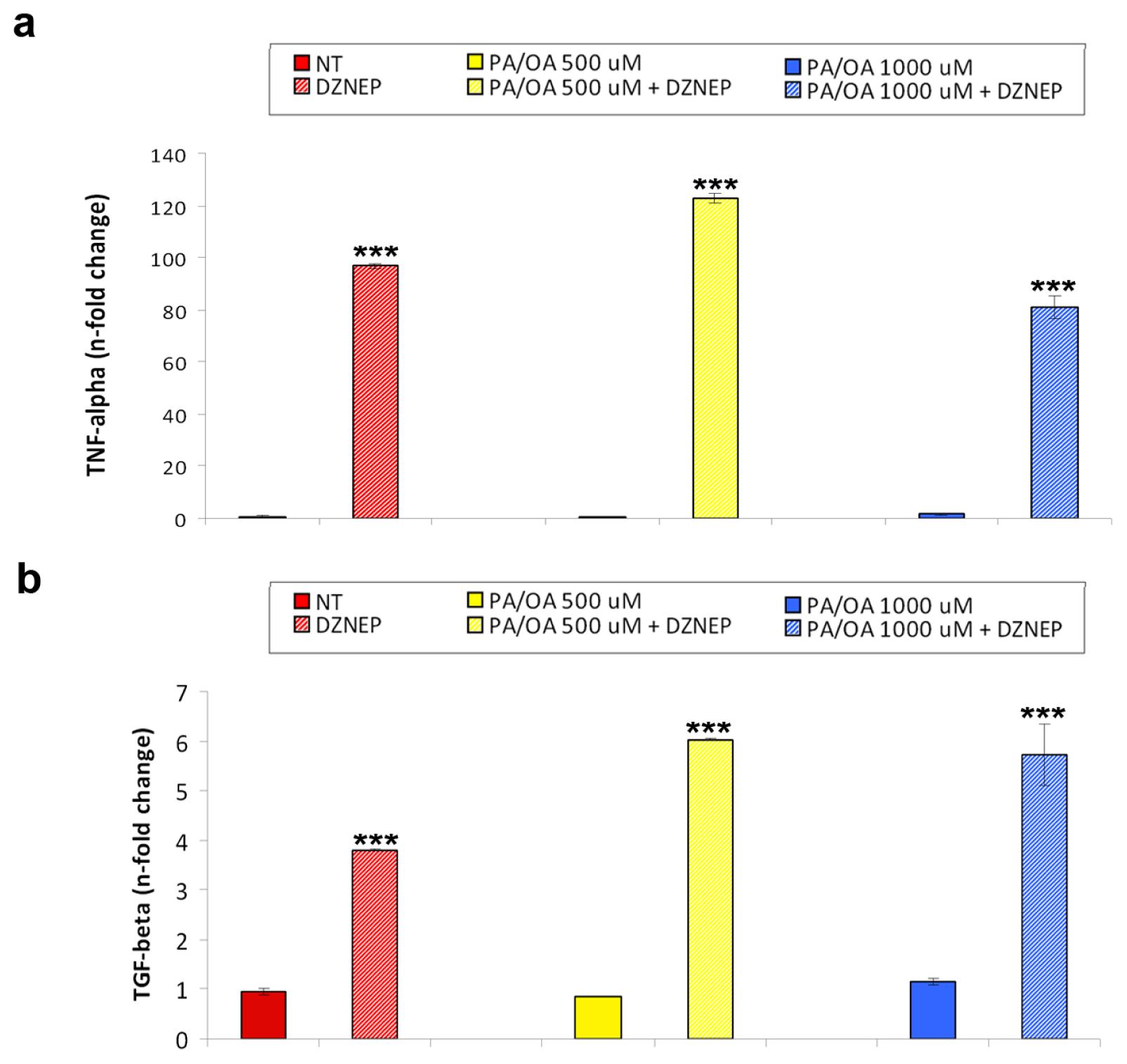
2.2. Effects of DZNep on Lipid Accumulation and Hepatic Expression of TNF-α and TGF-β in the Fatty HepG2 Cells
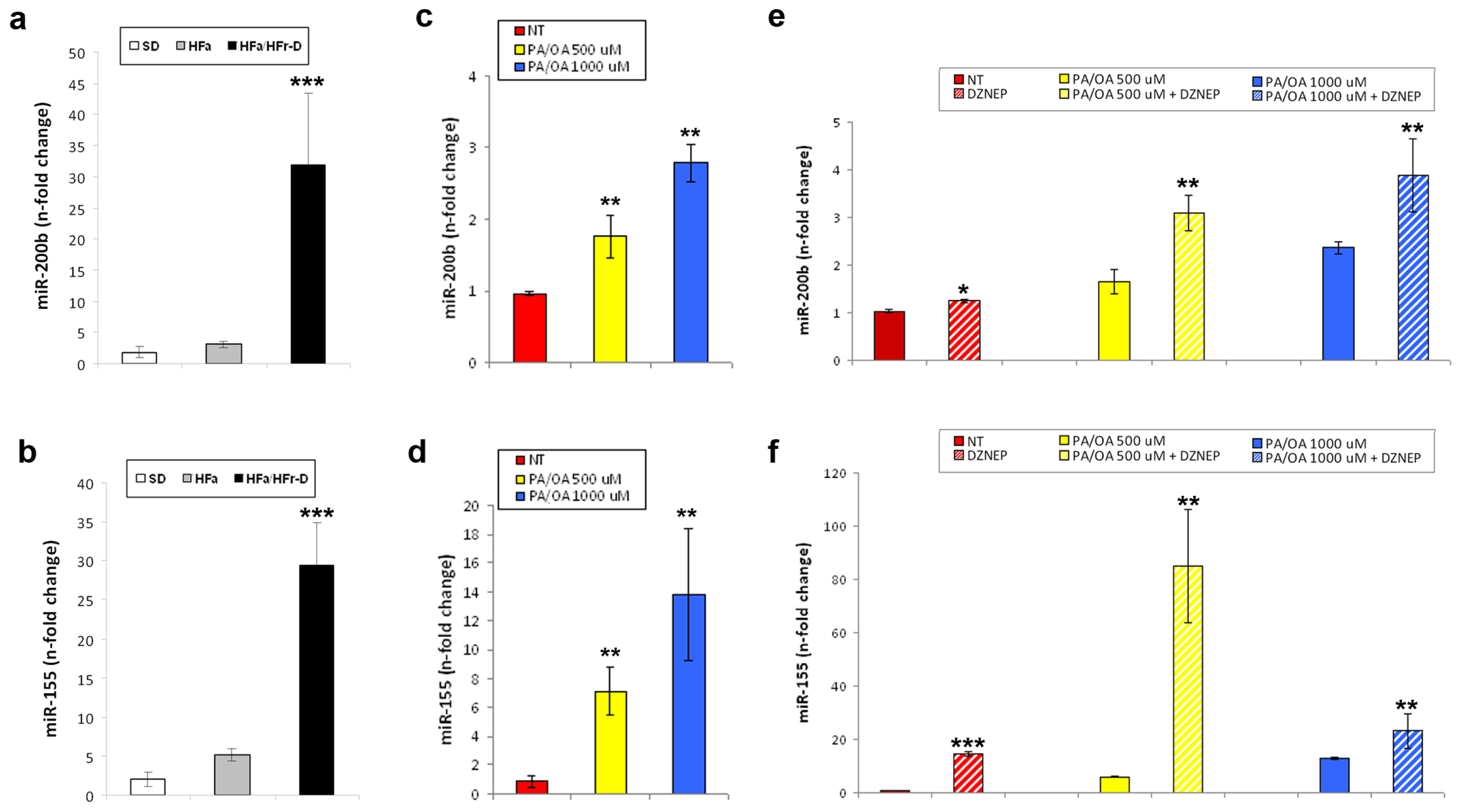
2.3. Effects of DZNep on the Hepatic Expression of miR-200b and miR-155
3. Experimental Section
3.1. Animal Treatments and Histology
3.2. Histology, Immunohistochemistry and Immunofluorescence
3.3. ELISA
3.4. Cell Lines and Treatments
3.5. Oil-Red-O Staining
3.6. Cell Viability Assays
3.7. Real-Time RT-qPCR
3.8. Western Blotting
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bhala, N.; Jouness, R.I.; Bugianesi, E. Epidemiology and natural history of patients with NAFLD. Curr. Pharm. Des 2013, 19, 5169–5176. [Google Scholar]
- Farrell, G.C.; Wong, V.W.; Chitturi, S. NAFLD in Asia—As common and important as in the West. Nat. Rev. Gastroenterol 2013, 10, 307–318. [Google Scholar]
- Nobili, V.; Svegliati-Baroni, G.; Alisi, A.; Miele, L.; Valenti, L.; Vajro, P. A 360-degree overview of paediatric NAFLD: Recent insights. J. Hepatol 2013, 58, 1218–1229. [Google Scholar]
- Law, K.; Brunt, E.M. Nonalcoholic fatty liver disease. Clin. Liver Dis 2010, 14, 591–604. [Google Scholar]
- Larrain, S.; Rinella, M.E. A myriad of pathways to NASH. Clin. Liver Dis 2012, 16, 525–548. [Google Scholar]
- Naik, A.; Košir, R.; Rozman, D. Genomic aspects of NAFLD pathogenesis. Genomics 2013, 102, 84–95. [Google Scholar]
- Zimmer, V.; Lammert, F. Genetics and epigenetics in the fibrogenic evolution of chronic liver diseases. Best Pract. Res. Clin. Gastroenterol 2011, 25, 269–280. [Google Scholar]
- Sookoian, S.; Pirola, C.J. DNA methylation and hepatic insulin resistance and steatosis. Curr. Opin. Clin. Nutr. Metab 2012, 15, 350–356. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar]
- Sato, F.; Tsuchiya, S.; Meltzer, S.J.; Shimizu, K. MicroRNAs and epigenetics. FEBS J 2011, 278, 1598–1609. [Google Scholar]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol 2013, 20, 274–281. [Google Scholar]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol 2013, 20, 259–266. [Google Scholar]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev 2006, 6, 846–856. [Google Scholar]
- Ciarapica, R.; Miele, L.; Giordano, A.; Locatelli, F.; Rota, R. Enhancer of zeste homolog 2 (EZH2) in pediatric soft tissue sarcomas: First implications. BMC Med 2011, 9, 63. [Google Scholar]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Dhanasekaran, S.M.; Asangani, I.A.; Prensner, J.R.; Kim, J.H.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 22, 1695–1699. [Google Scholar]
- Cai, M.Y.; Tong, Z.T.; Zheng, F.; Liao, Y.J.; Wang, Y.; Rao, H.L.; Chen, Y.C.; Wu, Q.L.; Liu, Y.H.; Guan, X.Y.; et al. EZH2 protein: A promising immunomarker for the detection of hepatocellular carcinomas in liver needle biopsies. Gut 2011, 60, 967–976. [Google Scholar]
- Hajósi-Kalcakosz, S.; Dezső, K.; Bugyik, E.; Bödör, C.; Paku, S.; Pávai, Z.; Halász, J.; Schlachter, K.; Schaff, Z.; Nagy, P. Enhancer of zeste homologue 2 (EZH2) is a reliable immunohistochemical marker to differentiate malignant and benign hepatic tumors. Diagn. Pathol 2012, 7, 86. [Google Scholar]
- Au, S.L.; Wong, C.C.; Lee, J.M.; Fan, D.N.; Tsang, F.H.; Ng, I.O.; Wong, C.M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar]
- Ruemmele, F.M.; Garnier-Lengliné, H. Why are genetics important for nutrition? Lessons from epigenetic research. Ann. Nutr. Metab 2012, 60, 38–43. [Google Scholar]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar]
- Ceccarelli, S.; Panera, N.; Gnani, D.; Nobili, V. Dual role of microRNAs in NAFLD. Int. J. Mol. Sci 2013, 14, 8437–8455. [Google Scholar]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol 2009, 51, 176–186. [Google Scholar]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar]
- Alisi, A.; Pastore, A.; Ceccarelli, S.; Panera, N.; Gnani, D.; Bruscalupi, G.; Massimi, M.; Tozzi, G.; Piemonte, F.; Nobili, V. Emodin prevents intrahepatic fat accumulation, inflammation and redox status imbalance during diet-induced hepatosteatosis in rats. Int. J. Mol. Sci 2012, 13, 2276–2289. [Google Scholar]
- Yao, H.R.; Liu, J.; Plumeri, D.; Cao, Y.B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res 2011, 3, 284–291. [Google Scholar]
- Tilg, H. The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis 2010, 28, 179–185. [Google Scholar]
- French, B.A.; Oliva, J.; Bardag-Gorce, F.; Li, J.; Zhong, J.; Buslon, V.; French, S.W. Mallory-Denk bodies form when EZH2/H3K27me3 fails to methylate DNA in the nuclei of human and mice liver cells. Exp. Mol. Pathol 2012, 92, 318–326. [Google Scholar]
- Hayden, A.; Johnson, P.; Packham, G.; Crabb, S.J. S-adenosylhomocysteine hydrolase inhibition by 3-deazaneplanocin A analogues induces anti-cancer effects in breast cancer cell lines and synergy with both histone deacetylase and HER2 inhibition. Breast Cancer Res. Treat 2011, 127, 109–119. [Google Scholar]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar]
- Zeybel, M.; Walsh, M.J.; Chu, D.C.; Tsukamoto, H.; Mann, D.A.; Mann, J. DZNep Attenuates Liver Fibrosis by Blocking EZH2 and H3K27me3. Proceedings of 63rd Annual Meeting of the American-Association-for-the-Study-of-Liver-Diseases (AASLD), Boston, MA, USA, 9–13 November 2012; 56, p. 772A.
- Benetatos, L.; Voulgaris, E.; Vartholomatos, G.; Hatzimichael, E. Non-coding RNAs and EZH2 interactions in cancer: Long and short tales from the transcriptome. Int. J. Cancer 2013, 133, 267–274. [Google Scholar]
- Pogribny, I.P.; Starlard-Davenport, A.; Tryndyak, V.P.; Han, T.; Ross, S.A.; Rusyn, I.; Beland, F.A. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab. Investig 2010, 90, 1437–1446. [Google Scholar]
- Alisi, A.; Da Sacco, L.; Bruscalupi, G.; Piemonte, F.; Panera, N.; de Vito, R.; Leoni, S.; Bottazzo, G.F.; Masotti, A.; Nobili, V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab. Investig 2011, 91, 283–293. [Google Scholar]
- Ciarapica, R.; Annibali, D.; Raimondi, L.; Savino, M.; Nasi, S.; Rota, R. Targeting Id protein interactions by an engineered HLH domain induces human neuroblastoma cell differentiation. Oncogene 2009, 28, 1881–1891. [Google Scholar]
- Ciarapica, R.; Russo, G.; Verginelli, F.; Raimondi, L.; Donfrancesco, A.; Rota, R.; Giordano, A. Deregulated expression of miR-26a and Ezh2 in rhabdomyosarcoma. Cell Cycle 2009, 8, 172–175. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007, 21, 1050–1063. [Google Scholar]
- Ciferri, C.; Lander, G.C.; Maiolica, A.; Herzog, F.; Aebersold, R.; Nogales, E. Molecular architecture of human polycomb repressive complex 2. Elife 2012, 1, e00005. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vella, S.; Gnani, D.; Crudele, A.; Ceccarelli, S.; De Stefanis, C.; Gaspari, S.; Nobili, V.; Locatelli, F.; Marquez, V.E.; Rota, R.; et al. EZH2 Down-Regulation Exacerbates Lipid Accumulation and Inflammation in in Vitro and in Vivo NAFLD. Int. J. Mol. Sci. 2013, 14, 24154-24168. https://doi.org/10.3390/ijms141224154
Vella S, Gnani D, Crudele A, Ceccarelli S, De Stefanis C, Gaspari S, Nobili V, Locatelli F, Marquez VE, Rota R, et al. EZH2 Down-Regulation Exacerbates Lipid Accumulation and Inflammation in in Vitro and in Vivo NAFLD. International Journal of Molecular Sciences. 2013; 14(12):24154-24168. https://doi.org/10.3390/ijms141224154
Chicago/Turabian StyleVella, Serena, Daniela Gnani, Annalisa Crudele, Sara Ceccarelli, Cristiano De Stefanis, Stefania Gaspari, Valerio Nobili, Franco Locatelli, Victor E Marquez, Rossella Rota, and et al. 2013. "EZH2 Down-Regulation Exacerbates Lipid Accumulation and Inflammation in in Vitro and in Vivo NAFLD" International Journal of Molecular Sciences 14, no. 12: 24154-24168. https://doi.org/10.3390/ijms141224154