Transcriptional Activity of the FUT1 Gene Promoter Region in Pigs


Abstract
:
1. Introduction
2. Results and Discussion
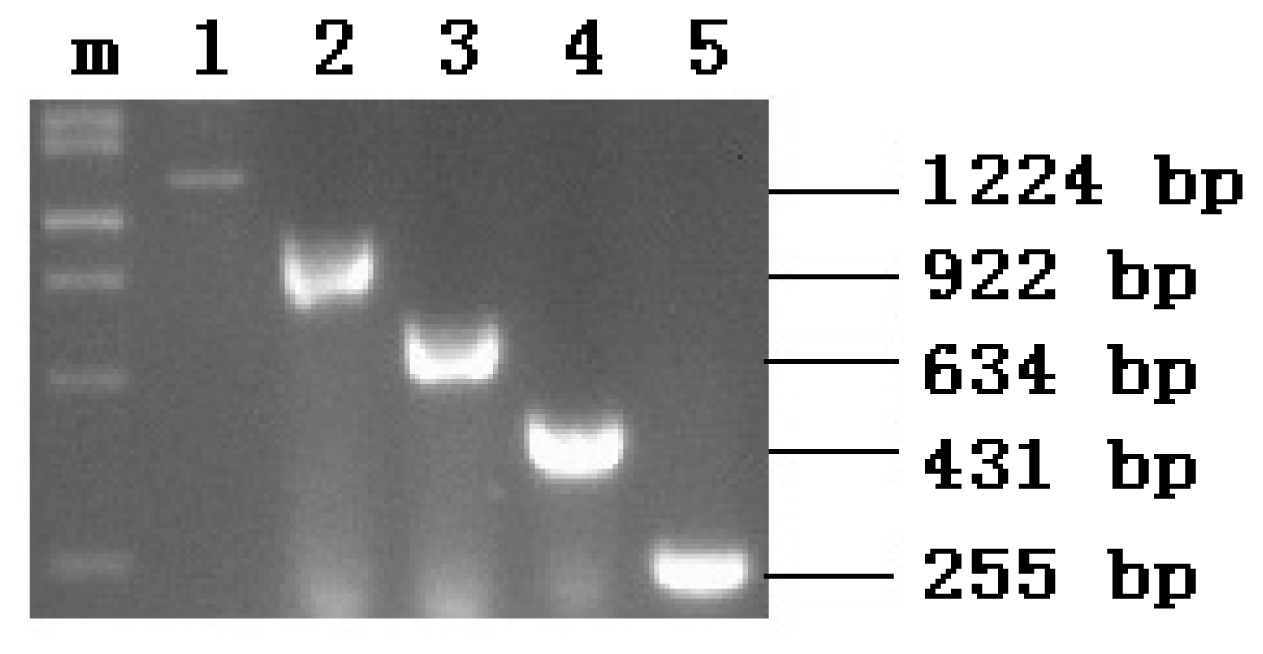
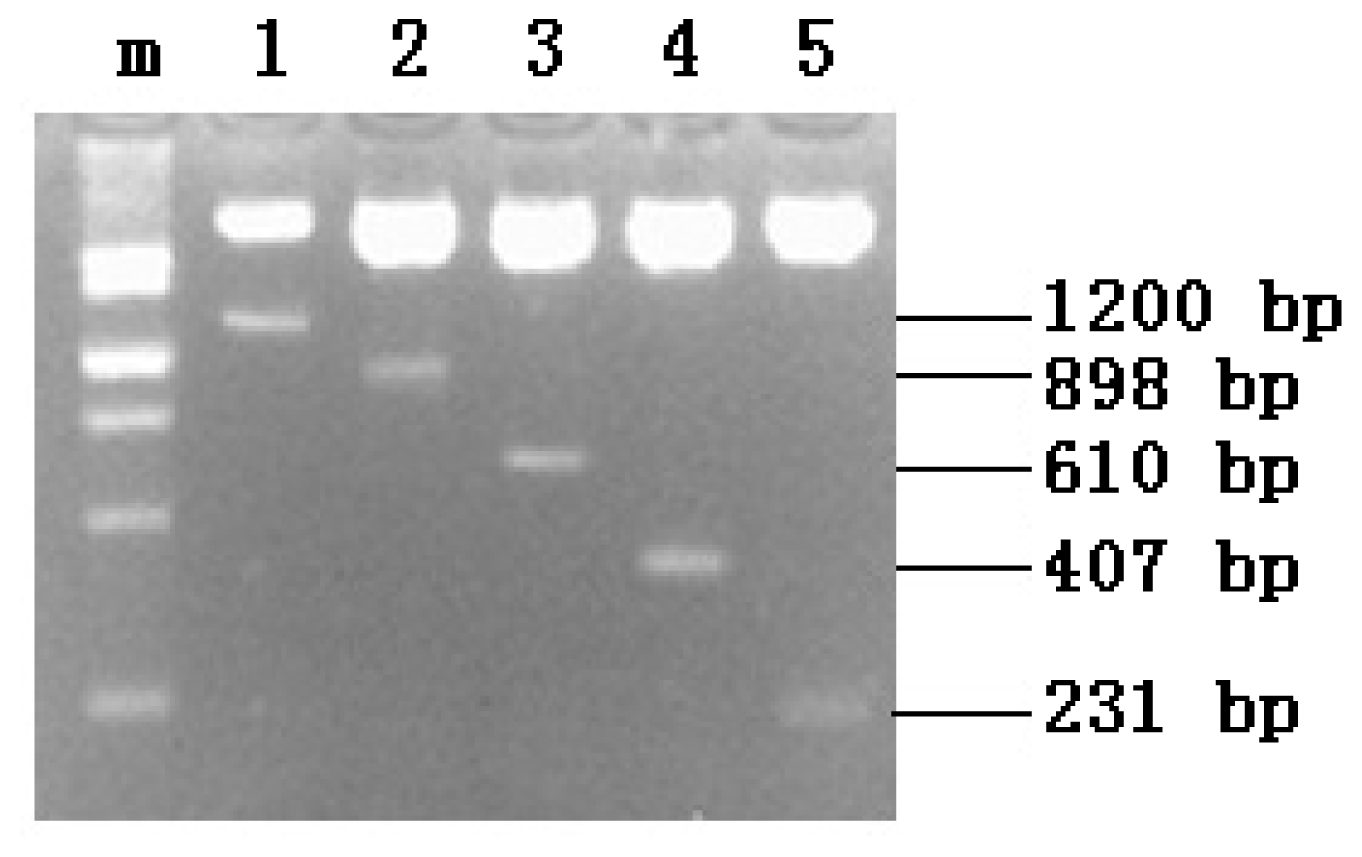
2.1. PCR Amplification
2.2. Recombinant Plasmid Enzyme Identification
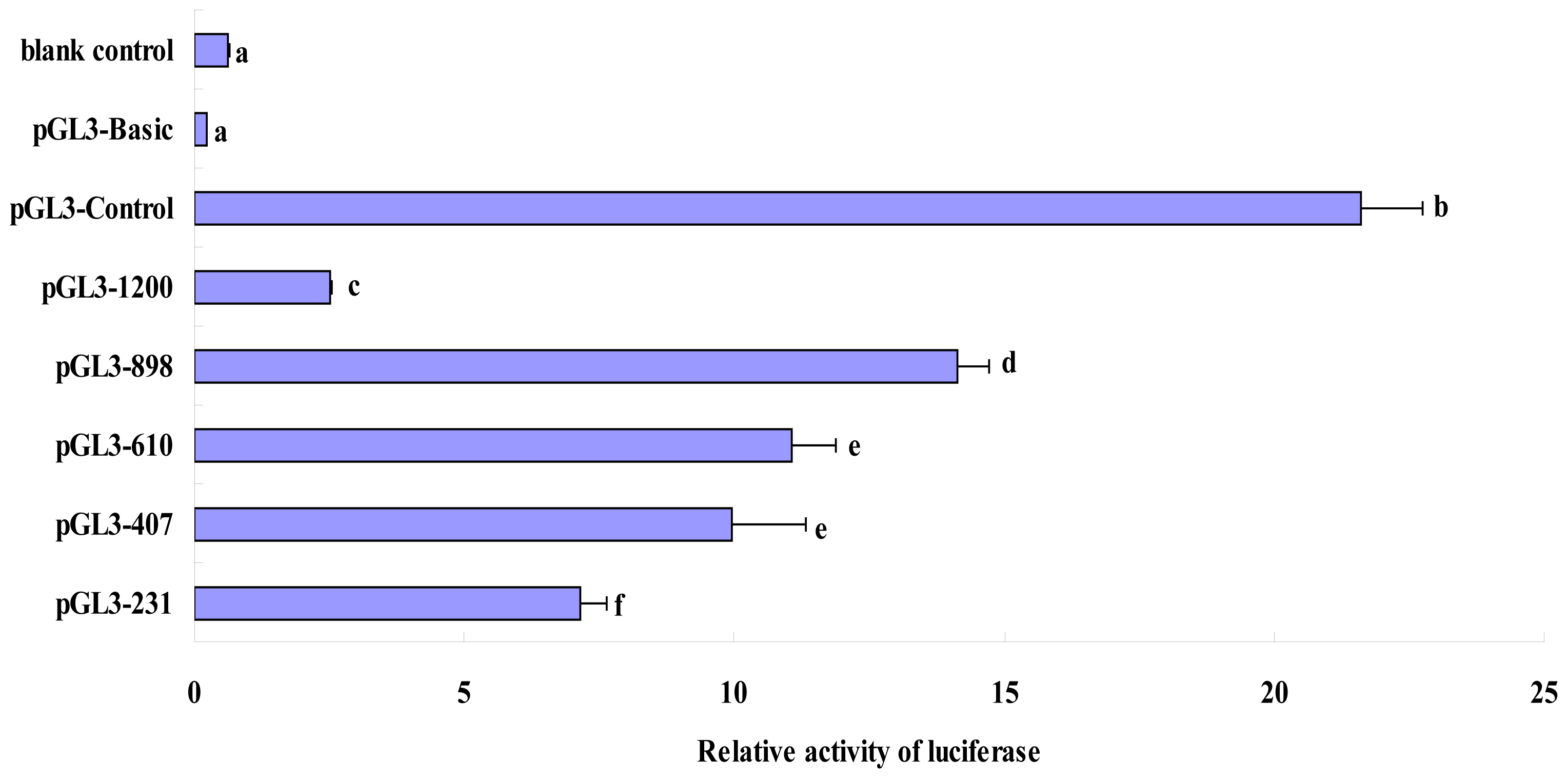
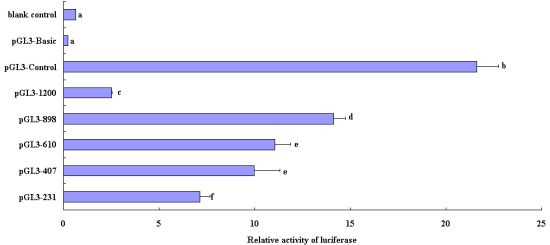
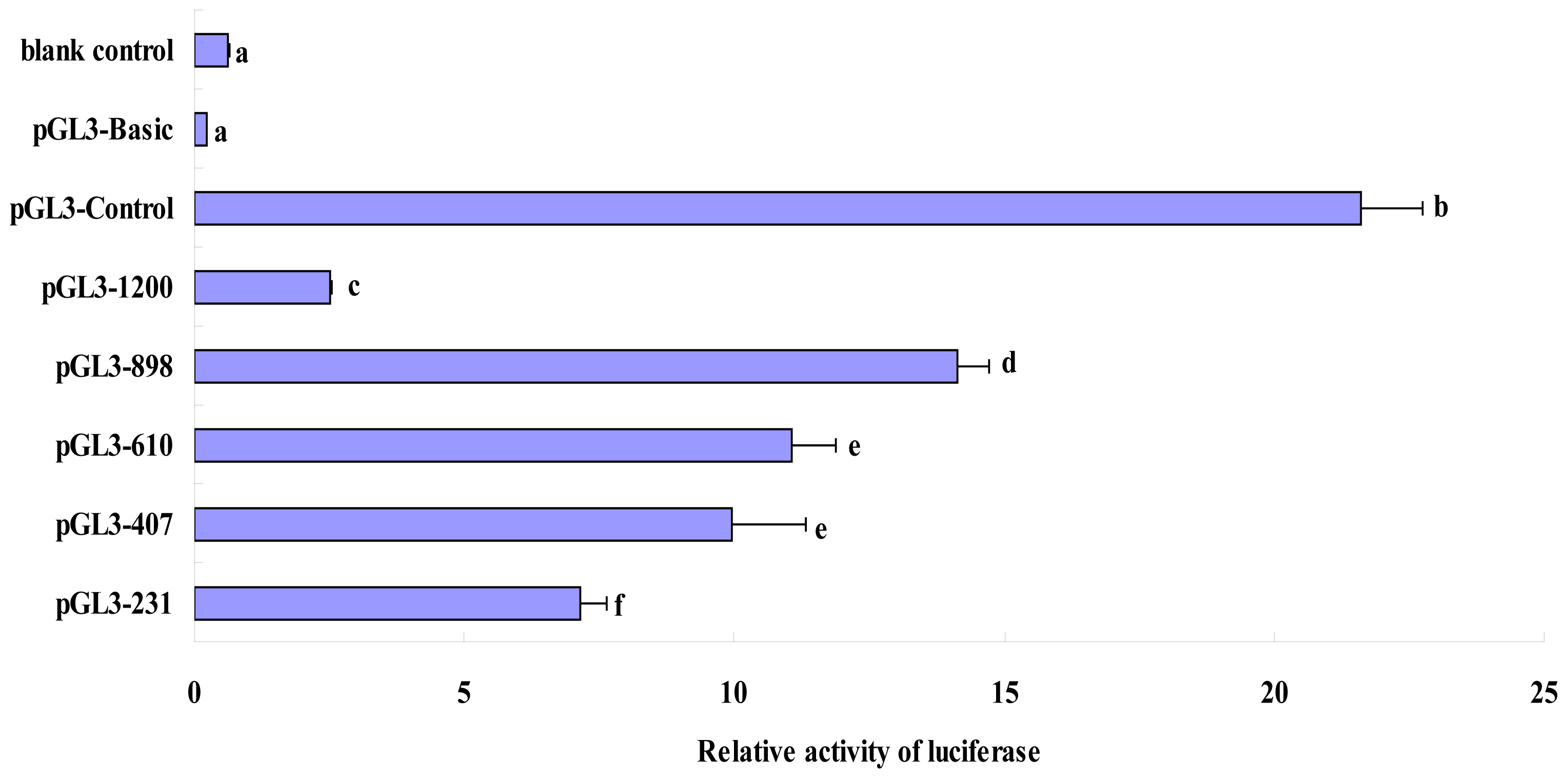
2.3. Luciferase Activity Analysis of Recombinant Plasmids in the Deleted Fragments of Pig FUT1 Gene Promoter
3. Experimental Section
3.1. Experimental Materials
3.2. Primer Design of Pig FUT1 Gene Promoter
3.3. Preparation of Deleted Fragments of Pig FUT1 Gene Promoter
3.4. Vector Construction and Verification
3.5. Luciferase Activity Assay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Boldin, B. Persistence and spread of gastro-intestinal infections: The case of enterotoxigenic Escherichia coli in piglets. Bull. Math. Biol 2008, 70, 2077–2101. [Google Scholar]
- Vögeli, P.; Meijerink, E.; Fries, R.; Neuenschwander, S.; Vorländer, N.; Stranzinger, G.; Bertschinger, H.U. A molecular test for the detection of E. coli F18 receptors: A breakthrough in the struggle against edema disease and post-weaning diarrhea in swine. Schweiz. Arch. Tierheilkd 1997, 139, 479–484. [Google Scholar]
- Meijerink, E.; Neuenschwander, S.; Fries, R.; Dinter, A.; Bertschinger, H.U.; Stranzinger, G.; Vögeli, P. A DNA polymorphism influencing alpha(1,2)-fucosyltransferase activity of the pig FUT1 enzyme determines susceptibility of small intestinal epithelium to Escherichia coli F18 adhesion. Immunogenetics 2000, 52, 129–136. [Google Scholar]
- Bao, W.B.; Wu, S.L.; Musa, H.H.; Zhu, G.Q.; Chen, G.H. Genetic variation at the alpha-1-fucosyltransferase (FUT1) gene in Asian wild boar and Chinese and Western commercial pig breeds. J. Anim. Breed. Genet 2008, 125, 427–430. [Google Scholar]
- Yan, X.M.; Ren, J.; Guo, Y.M.; Ding, N.S.; Chen, K.F.; Gao, J.; Ai, H.S.; Chen, C.Y.; Ma, J.W.; Huang, L.S. Research on the genetic variations of a1-Fucosytransferase(FUT1) gene in 26 pig breeds (in Chinese). Acta Genet. Sin 2003, 30, 830–834. [Google Scholar]
- Shi, Q.S.; Huang, S.Q.; Liu, X.C.; He, C.Q.; Jiang, X. Polymorphism of E. coli F18 receptor gene in different pig breeds (in Chinese). Acta Genet. Sin 2003, 30, 221–224. [Google Scholar]
- Zhang, Y.H.; Zhou, Z.X.; Cao, G.Q. FUT1 gene polymorphism and its association with litter size in pigs (in Chinese). Hereditas 2007, 29, 52–56. [Google Scholar]
- Zheng, X.R.; Wang, J.; Zhao, Q.H.; Zhu, S.P.; Wu, Z.C.; Huo, Y.J.; Bao, W.B.; Wu, S.L. Cloning and Polymorphism Analysis of Promoter Region of Porcine α-l,2-fucosyltransferase (FUT1) Gene (in Chinese). China Science Paper Online. Available online: http://www.paper.edu.cn/releasepaper/content/201212-54 (accessed on 11 September 2013).
- Burchard, E.G.; Silverman, E.K.; Rosenwasser, L.J.; Borish, L.; Yandava, C.; Pillari, A.; Weiss, S.T.; Hasday, J.; Lilly, C.M.; Ford, J.G. Association between a sequence variant in the IL-4 gene promoter and FEV(1) in asthma. Am. J. Respir. Crit. Care Med 1999, 160, 919–922. [Google Scholar]
- Kulozik, A.E.; Bellan-Koch, A.; Bail, S.; Kohne, E.; Kleihauer, E. Thalassemia intermedia: Moderate reduction of beta globin gene transcriptional activity by a novel mutation of the proximal CACCC promoter element. Blood 1991, 77, 2054–2058. [Google Scholar]
- Jha, A.K.; Nikbakht, M.; Jain, V.; Capalash, N.; Kaur, J. p16(INK4a) and p15(INK4b) gene promoter methylation in cervical cancer patients. Oncol. Lett 2012, 3, 1331–1335. [Google Scholar]
- Zhao, R.X.; Zhao, W.M.; Xu, Q.; Duan, X.J.; Dong, B.; Sun, G.B.; Bi, Y.L.; Li, X.; Zhang, Y.; Huang, Z.Y. Analysis of transcription regulation in the promoter region of PIT-1 gene in goose (in Chinese). Xumu Shouyi Xuebao Acta Veteri 2012, 43, 220–225. [Google Scholar]
- Chen, H.; Chen, H.Q.; Zhou, Q.Q.; Zhang, Y.P.; Wei, H.Q.; Li, C.M.; Zhang, X.R.; Peng, Y.L. Identification of transcription regulation activity in 5′ flanking region of pig THRSP gene (in Chinese). Acta Vet. Zootechn. Sin 2011, 42, 329–334. [Google Scholar]
- Ren, Z.Q.; Liu, W.H.; Zheng, R.; Zuo, B.; Xu, D.Q.; Lei, M.G.; Li, F.E.; Li, J.L.; Ni, D.B.; Xiong, Y.Z. A 304 bp insertion/deletion mutation in promoter region induces the increase of porcine IDH3β gene expression. Mol. Biol. Rep 2012, 39, 1419–1426. [Google Scholar]
- Smale, S.T.; Kadonaga, J.T. The RNA polymerase II core promoter. Annu. Rev. Biochem 2003, 72, 449–479. [Google Scholar]
- Arkhipova, I.R. Promoter elements in Drosophila melanogaster revealed by sequence analysis. Genetics 1995, 139, 1359–1369. [Google Scholar]
- Judde, J.G.; Max, E.E. Characterization of the human immunoglobulin kappa gene 3′ enhancer: Functional importance of three motifs that demonstrate B-cell-specific in vivo footprints. Mol. Cell. Biol 1992, 12, 5206–5216. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | 5′-Sequences-3′ | Amplification Region (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| A (KpnI) P (HindIII) | ATCATCggtaccCTGGCCTGGCTCAGTGGGTCAGGGACCCAGC ATCATCaagcttCTGAGCTCCTGGCGGGTGGCAGATTCCCAG | (−1150)–50 | 57 |
| B (KpnI) P (HindIII) | ATCATCggtaccCGGCCTTTGTTCCAGAAGCCGGCTGCAGCCCGGGCCT ATCATCaagcttCTGAGCTCCTGGCGGGTGGCAGATTCCCAG | (−848)–50 | 58 |
| C (KpnI) P (HindIII) | ATCATCggtaccTGCTGCCCCCGGGGAGGACTCGGCAGGGGGGCGGGGGG ATCATCaagcttCTGAGCTCCTGGCGGGTGGCAGATTCCCAG | (−560)–50 | 58 |
| D (KpnI) P (HindIII) | ATCATCggtaccGCTGCATCTGGCCGCTGGATCTCCGCGGCCG ATCATCaagcttCTGAGCTCCTGGCGGGTGGCAGATTCCCAG | (−357)–50 | 59 |
| E (KpnI) P (HindIII) | ATCATCggtaccGCGCCAGGGAAGGGGTGGGGCTCCGCCTCCG ATCATCaagcttCTGAGCTCCTGGCGGGTGGCAGATTCCCAG | (−181)–50 | 59 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zi, C.; Wu, Z.; Wang, J.; Huo, Y.; Zhu, G.; Wu, S.; Bao, W. Transcriptional Activity of the FUT1 Gene Promoter Region in Pigs. Int. J. Mol. Sci. 2013, 14, 24126-24134. https://doi.org/10.3390/ijms141224126
Zi C, Wu Z, Wang J, Huo Y, Zhu G, Wu S, Bao W. Transcriptional Activity of the FUT1 Gene Promoter Region in Pigs. International Journal of Molecular Sciences. 2013; 14(12):24126-24134. https://doi.org/10.3390/ijms141224126
Chicago/Turabian StyleZi, Chen, Zhengchang Wu, Jing Wang, Yongjiu Huo, Guoqiang Zhu, Shenglong Wu, and Wenbin Bao. 2013. "Transcriptional Activity of the FUT1 Gene Promoter Region in Pigs" International Journal of Molecular Sciences 14, no. 12: 24126-24134. https://doi.org/10.3390/ijms141224126