Phylogenetic Relationships between Four Salix L. Species Based on DArT Markers

 , and
, and
Abstract
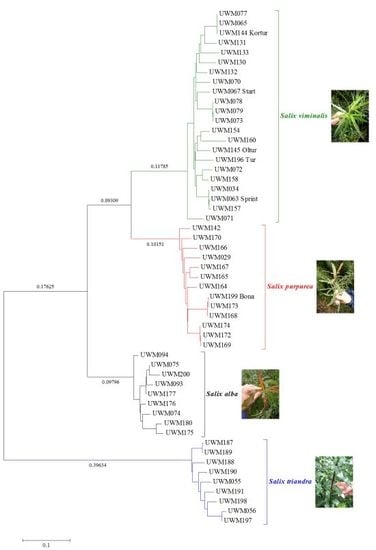
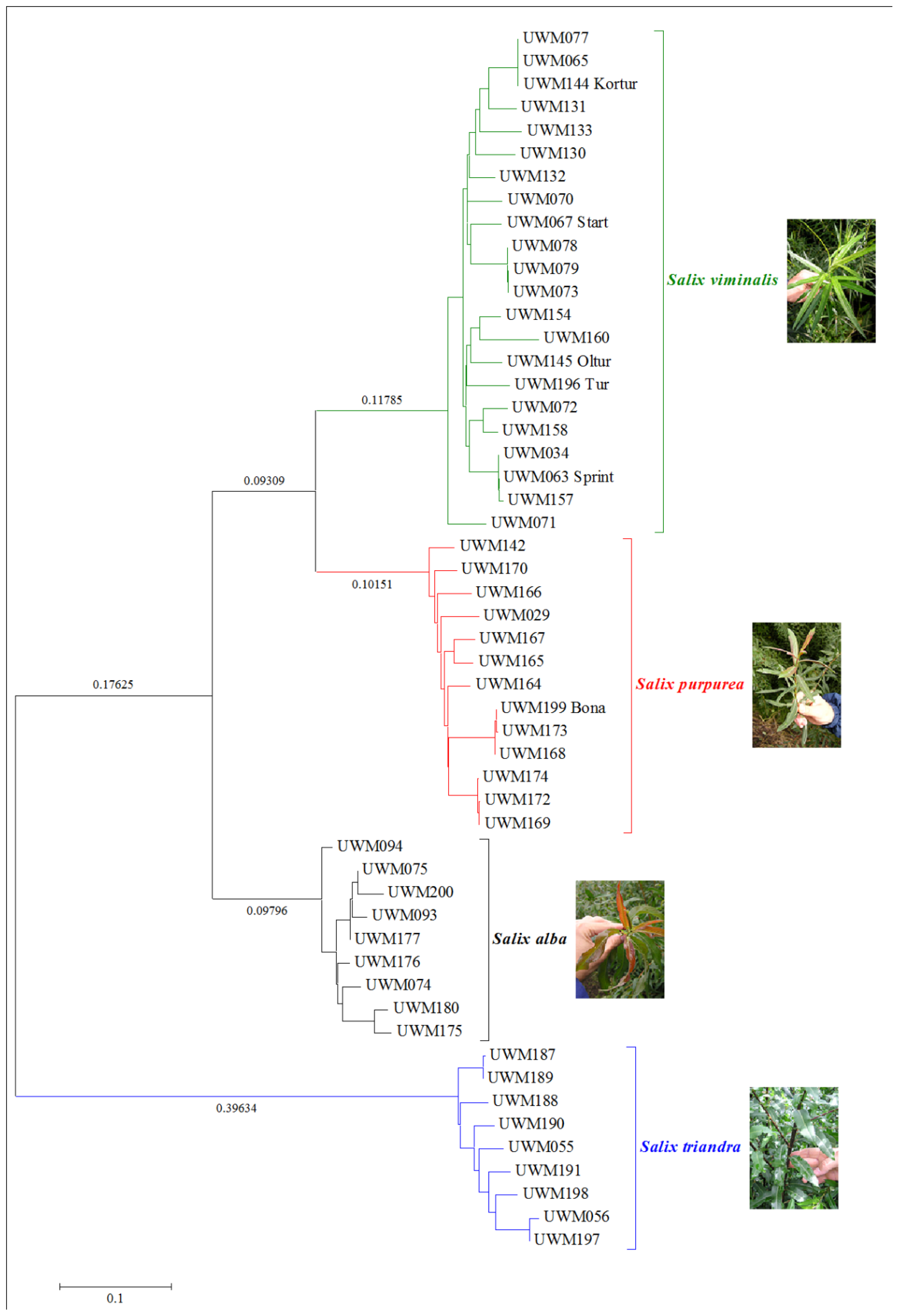
:
1. Introduction
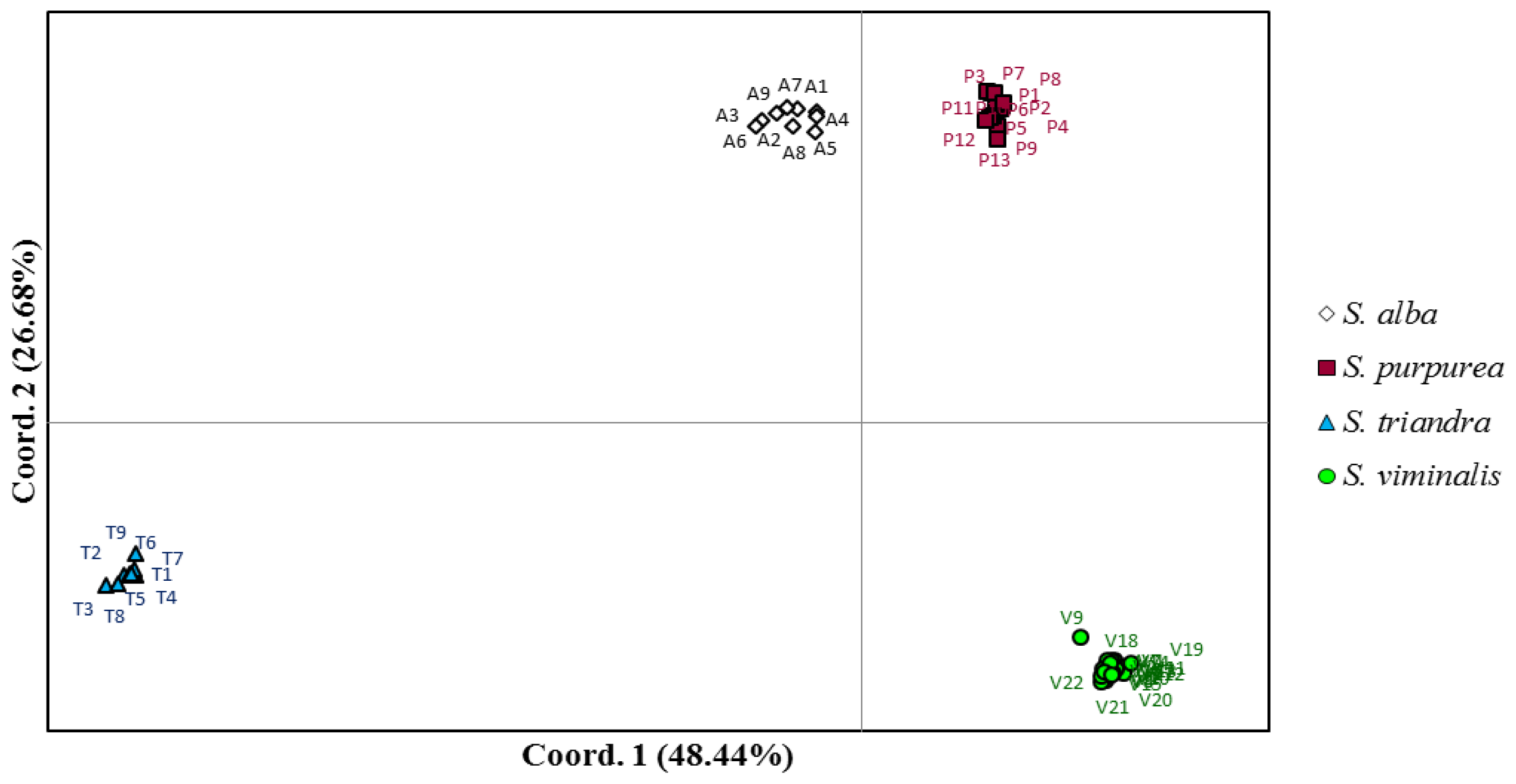
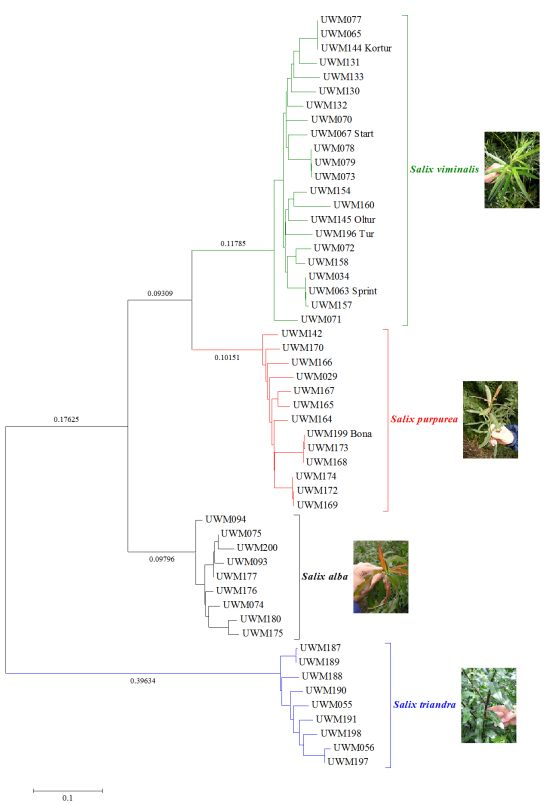
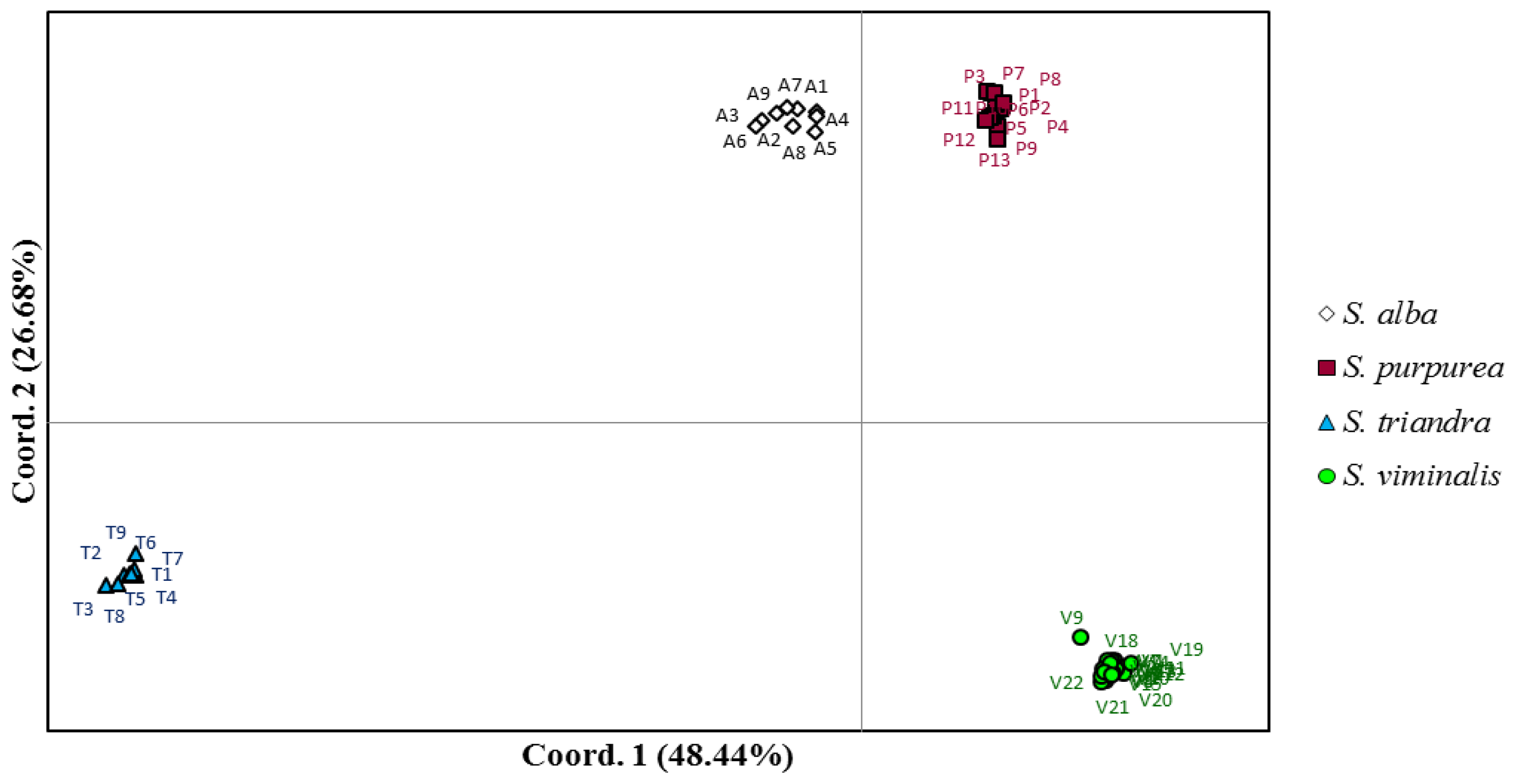
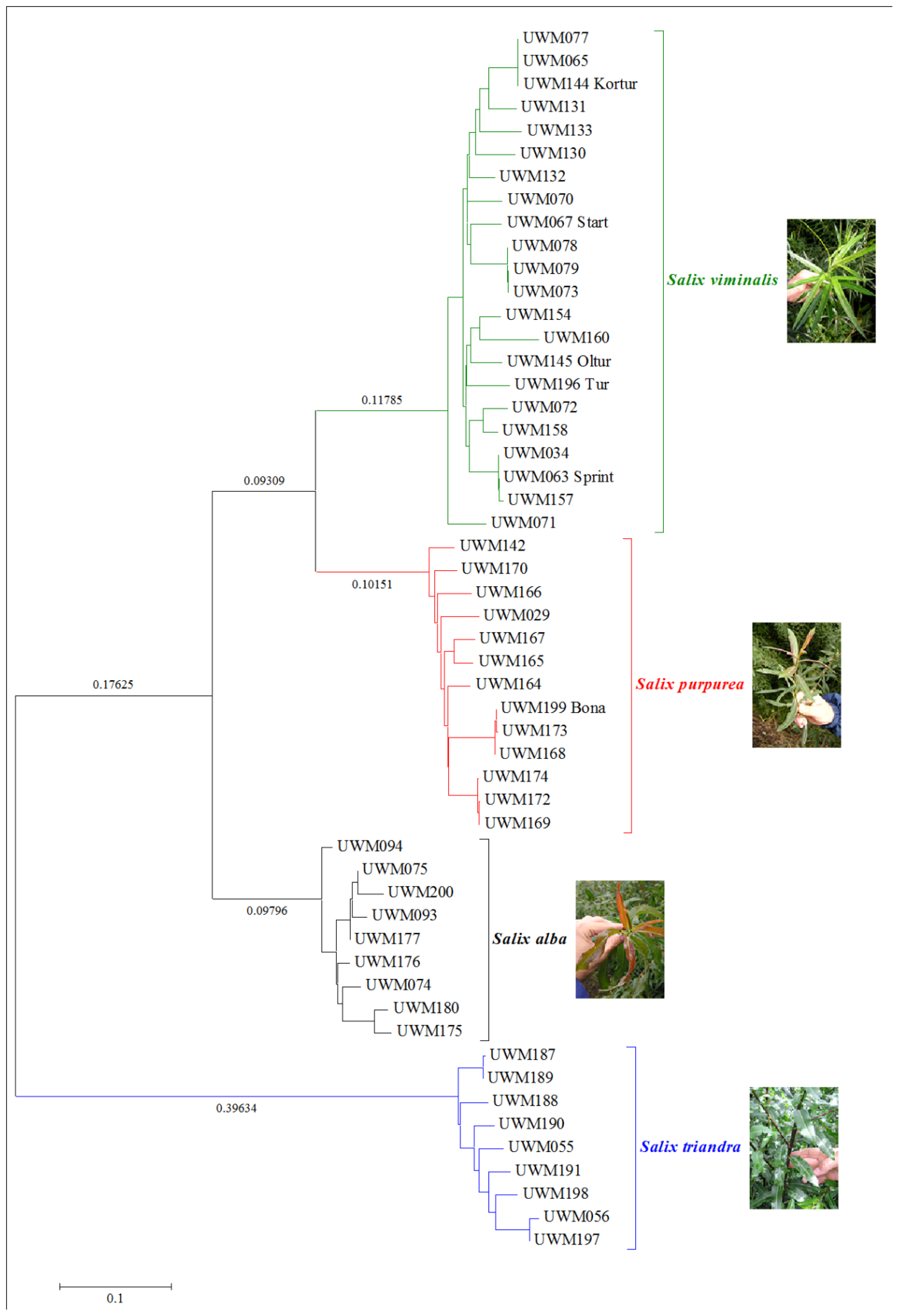
2. Results and Discussion
3. Materials and Methods
3.1. Plant Material
3.2. DNA Isolation and DArT Protocol
3.3. Preparation of Genomic Representations
3.4. Preparation of DArT Libraries and Arrays
3.5. Fingerprinting of DNA Samples
3.6. Image Analysis and Polymorphism Scoring
3.7. Marker Scoring and Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Argus, G.W. Salix (Salicaceae) distribution maps and a synopsis of their classification in North America, north of Mexico. Harv. Pap. Bot 2007, 12, 335–368. [Google Scholar]
- Skvortsov, A.K. Willows of the USSR; Nauka: Moscow, Russia, 1968. [Google Scholar]
- Hörandl, E.; Florineth, F.; Hadacek, F. Weiden in Österreich und angrenzenden Gebieten; Arbeitsbereich Ingenieurbiologie u. Landschaftsbau, Univ. Bodenkultur Wien: Wien, Austria, 2002. [Google Scholar]
- Kuzovkina, Y.A.; Quigley, M.F. Willows beyond wetlands: Uses of Salix L. species for environmental projects. Water Air Soil Pollut 2005, 162, 183–204. [Google Scholar]
- Sulima, P.; Przyborowski, J.A.; Wiwart, M. Willow bark-herbal raw material harvested from plants cultivated on arable lands. Herba Polonica 2006, 52, 18–25. [Google Scholar]
- Smart, L.B.; Cameron, K.D. Genetic Improvement of Willow (Salix spp.) as a Dedicated Bioenergy Crop. In Genetic Improvement of Bioenergy Crops; Springer: New York, NY, USA, 2008; pp. 377–396. [Google Scholar]
- Förster, N.; Ulrichs, C.; Zander, M.; Kätzel, R. Factors influencing the variability of antioxidative phenolic glycosides in Salix species. J. Agricul. Food Chem 2010, 58, 8205–8210. [Google Scholar]
- Stolarski, M.J.; Szczukowski, S.; Tworkowski, J.; Wróblewska, H.; Krzyżaniak, M. Short rotation willow coppice biomass as an industrial and energy feedstock. Industrial Crops Prod 2011, 33, 217–223. [Google Scholar]
- Stolarski, M.J.; Szczukowski, S.; Tworkowski, J.; Krzyżaniak, M. Cost of heat energy generation from willow biomass. Renew. Energy 2013, 59, 100–104. [Google Scholar]
- Serapiglia, M.J.; Cameron, K.D.; Stipanovic, A.J.; Abrahamson, L.P.; Volk, T.A.; Smart, L.B. Yield and woody biomass traits of novel shrub willow hybrids at two contrasting sites. Bioenerg. Res 2012, 6, 1–14. [Google Scholar]
- Rosso, L.; Facciotto, G.; Bergante, S.; Vietto, L.; Nervo, G. Selection and testing of Populus alba and Salix spp. as bioenergy feedstock: Preliminary results. Appl. Energy 2012, 102, 87–92. [Google Scholar]
- Trybush, S.O.; Jahodová, Š.; Čížková, L.; Karp, A.; Hanley, S.J. High levels of genetic diversity in Salix viminalis of the Czech Republic as revealed by microsatellite markers. Bioenerg. Res 2012, 5, 1–9. [Google Scholar]
- Linnaeus, C. Species Plantarum; Laurentius Salvius: Stockholm, Sweden, 1753. [Google Scholar]
- Du Mortier, B.C. Verhandeling over het Geschlacht der Wilgen (Salix) en de Natuurlijke Familie der Amentaceae; bij Joh; van der Hey en zoon: Amsterdam, The Netherlands, 1825. [Google Scholar]
- Nakai, T. Chosenia, a new genus of Salicaceae. Botanical Mag. (Tokyo) 1920, 34, 66–69. [Google Scholar]
- Fang, Z.; Zhao, S.; Skvortsov, A. Salicaceae. In Flora of China; Wu, Z., Raven, P., Eds.; Science Press and Missouri Botanical Garden Press: Beijing, China and St. Louis, MO, USA, 1999; Volume 4, pp. 139–274. [Google Scholar]
- Chen, J.; Sun, H.; Wen, J.; Yang, Y. Molecular phylogeny of Salix L.(Salicaceae) inferred from three chloroplast datasets and its systematic implications. Taxon 2010, 59, 29–37. [Google Scholar]
- Bachmann, K. Evolution and the genetic analysis of populations: 1950–2000. Taxon 2001, 50, 7–45. [Google Scholar]
- Ford, C.S.; Ayres, K.L.; Toomey, N.; Haider, N.; Stahl, J.V.; Kelly, L.J.; Wikstrom, N.; Hollingsworth, P.M.; Duff, R.J.; Hoot, S.B.; et al. Selection of candidate coding DNA barcoding regions for use on land plants. Botanical J. Linn. Soc 2009, 159, 1–11. [Google Scholar]
- Stuessy, T.F. Plant Taxonomy: The Systematic Evaluation of Comparative Data; Columbia University Press: New York, NY, USA, 2009. [Google Scholar]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS One 2011, 6, e19254. [Google Scholar]
- Zanke, C.; Hemleben, V. A new Solanum satellite DNA containing species-specific sequences which can be used for identification of genome parts in somatic hybrids of potato. Plant Sci 1997, 126, 185–191. [Google Scholar]
- King, R.A.; Harris, S.L.; Karp, A.; Barker, J.H. Characterisation and inheritance of nuclear microsatellite loci for use in population studies of the allotetraploid Salix alba–Salix fragilis complex. Tree Genet. Gen 2010, 6, 247–258. [Google Scholar]
- Rafalski, J.A. Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci 2002, 162, 329–333. [Google Scholar]
- Semagn, K.; Bjørnstad, Å.; Ndjiondjop, M. An overview of molecular marker methods for plants. Afr. J. Biotechnol 2010, 5, 2540–2568. [Google Scholar]
- Sá, O.; Pereira, J.A.; Baptista, P. Optimization of DNA extraction for RAPD and ISSR analysis of Arbutus unedo L. Leaves. Int. J. Mol. Sci 2011, 12, 4156–4164. [Google Scholar]
- Jaccoud, D.; Peng, K.; Feinstein, D.; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucl. Acids Res 2001, 29, e25. [Google Scholar]
- Kilian, A.; Huttner, E.; Wenzl, P.; Jaccoud, D.; Carling, J.; Caig, V.; Evers, M.; Heller-Uszynska, K.; Cayla, C.; Patarapuwadol, S. The Fast and the Cheap: SNP and DArT-based Whole Genome Profiling for Crop Improvement; Proceedings of International Congress “In the Wake of the Double Helix: From the Green Revolution to the Gene Revolution”, Bologna, Italy, 27–31 May 2003, Tuberosa, R., Phillips, R.L., Gale, M., Eds.; Avenue Media: Bologna, Italy, 2005; pp. 443–461. [Google Scholar]
- Steane, D.A.; Nicolle, D.; Sansaloni, C.P.; Petroli, C.D.; Carling, J.; Kilian, A.; Myburg, A.A.; Grattapaglia, D.; Vaillancourt, R.E. Population genetic analysis and phylogeny reconstruction in Eucalyptus (Myrtaceae) using high-throughput, genome-wide genotyping. Mol. Phylogenet. Evolut 2011, 59, 206–224. [Google Scholar]
- Hudson, C.J.; Kullan, A.R.; Freeman, J.S.; Faria, D.A.; Grattapaglia, D.; Kilian, A.; Myburg, A.A.; Potts, B.M.; Vaillancourt, R.E. High synteny and colinearity among Eucalyptus genomes revealed by high-density comparative genetic mapping. Tree Genet. Genomes 2012, 8, 339–352. [Google Scholar]
- Belaj, A.; del Carmen Dominguez-García, M.; Atienza, S.G.; Martín Urdíroz, N.; De la Rosa, R.; Satovic, Z.; Martín, A.; Kilian, A.; Trujillo, I.; Valpuesta, V. Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genomes 2012, 8, 365–378. [Google Scholar]
- Diversity Arrays Technology Homepage. Available online: http://www.diversityarrays.com (accessed on 20 October 2013).
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Data Production and Analysis in Population Genomics; Pompanon, F., Bonin, A., Eds.; Humana Press: New York, NY, USA, 2012; Volume 888, pp. 67–89. [Google Scholar]
- Trybush, S.; Jahodová, Š.; Macalpine, W.; Karp, A. A genetic study of a Salix germplasm resource reveals new insights into relationships among subgenera, sections and species. Bioenergy Res 2008, 1, 67–79. [Google Scholar]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol 2004, 13, 1143–1155. [Google Scholar]
- Thibault, J. Nuclear DNA amount in pure species and hybrid willows (Salix): A flow cytometric investigation. Can. J. Bot 1998, 76, 157–165. [Google Scholar]
- Pei, M.; Hunter, T.; Royle, D. Host–pathogen relationship between Salix and Melampsora sheds light on the parentage of some biomass willows. New Phytol 2002, 141, 155–160. [Google Scholar]
- Hearnden, P.; Eckermann, P.J.; McMichael, G.; Hayden, M.; Eglinton, J.K.; Chalmers, K.J. A genetic map of 1000 SSR and DArT markers in a wide barley cross. TAG Theor.Appl. Genet 2007, 115, 383–391. [Google Scholar]
- Kopecký, D.; Bartoš, J.; Lukaszewski, A.; Baird, J.; Černoch, V.; Kölliker, R.; Rognli, O.; Blois, H.; Caig, V.; Lübberstedt, T. Development and mapping of DArT markers within the Festuca-Lolium complex. BMC Genomics 2009, 10, 473. [Google Scholar]
- Leskinen, E.; Alström-Rapaport, C. Molecular phylogeny of Salicaceae and closely related Flacourtiaceae: Evidence from 5.8 S, ITS 1 and ITS 2 of the rDNA. Plant Syst. Evolut 1999, 215, 209–227. [Google Scholar]
- Azuma, T.; Kajita, T.; Yokoyama, J.; Ohashi, H. Phylogenetic relationships of Salix (Salicaceae) based on rbcL sequence data. Am. J. Bot 2000, 87, 67–75. [Google Scholar]
- Neumann, A. Die Mitteleuropäischen Salix-Arten; Mitt Forstlichen Bundes-versuchsanstalt: Wien, Austria, 1981. [Google Scholar]
- Plant DNA Extraction Protocol for DArT. Available online: http://www.diversityarrays.com/sites/default/files/pub/DArT_DNA_isolation.pdf (accessed on 20 October 2013).
- DArTsoft. Available online: http://www.diversityarrays.com/software.html (accessed on 20 October 2013).
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar]
- Vekemans, X.; Beauwens, T.; Lemaire, M.; Roldán–Ruiz, I. Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol. Ecol 2002, 11, 139–151. [Google Scholar]
- Yeh, F.C.; Yang, R.C.; Boyle, T.B.J.; Ye, Z.H.; Mao, J.X. POPGENE, the User-Friendly Shareware for Population Genetic Analysis. In Molecular Biology and Biotechnology Centre; University of Alberta: Edmonton, AB, Canada, 1997. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihoevolutionary distance and maximum parsimony methods. Mol. Biol. Evolut 2011, 28, 2731–2739. [Google Scholar]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics; Sinauer: Sunderland, UK, 2007. [Google Scholar]
- Nei, M. (1973) analysis of gene diversity in subdivided populations. Proc. National Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL, USA, 1949. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Nei, M. Genetic distance between populations. Am. Nat 1972, 106, 283–292. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evolut 1987, 4, 406–425. [Google Scholar]
- Sulima, P.; Przyborowski, J.A.; Załuski, D. RAPD markers reveal genetic diversity in Salix purpurea L. Crop Sci 2009, 49, 857–863. [Google Scholar]
- Przyborowski, J.P.; Sulima, P. The analysis of genetic diversity of Salix viminalis genotypes as a potential source of biomass by RAPD markers. Industrial Crops Product 2010, 31, 395–400. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Name in Collection | Symbol PCoA |
|---|---|---|
| S. alba | UWM075 | A1 |
| S. alba | UWM200 | A2 |
| S. alba | UWM180 | A3 |
| S. alba | UWM177 | A4 |
| S. alba | UWM176 | A5 |
| S. alba | UWM175 | A6 |
| S. alba | UWM074 | A7 |
| S. alba | UWM094 | A8 |
| S. alba | UWM093 | A9 |
| S. purpurea | UWM199 | P1 |
| S. purpurea | UWM174 | P2 |
| S. purpurea | UWM173 | P3 |
| S. purpurea | UWM172 | P4 |
| S. purpurea | UWM170 | P5 |
| S. purpurea | UWM169 | P6 |
| S. purpurea | UWM168 | P7 |
| S. purpurea | UWM167 | P8 |
| S. purpurea | UWM166 | P9 |
| S. purpurea | UWM165 | P10 |
| S. purpurea | UWM164 | P11 |
| S. purpurea | UWM029 | P12 |
| S. purpurea | UWM142 | P13 |
| S. triandra | UWM187 | T1 |
| S. triandra | UWM198 | T2 |
| S. triandra | UWM056 | T3 |
| S. triandra | UWM055 | T4 |
| S. triandra | UWM197 | T5 |
| S. triandra | UWM191 | T6 |
| S. triandra | UWM190 | T7 |
| S. triandra | UWM189 | T8 |
| S. triandra | UWM188 | T9 |
| S. viminalis | UWM079 | V1 |
| S. viminalis | UWM078 | V2 |
| S. viminalis | UWM077 | V3 |
| S. viminalis | UWM145 | V4 |
| S. viminalis | UWM154 | V5 |
| S. viminalis | UWM070 | V6 |
| S. viminalis | UWM073 | V7 |
| S. viminalis | UWM072 | V8 |
| S. viminalis | UWM071 | V9 |
| S. viminalis | UWM067 | V10 |
| S. viminalis | UWM065 | V11 |
| S. viminalis | UWM063 | V12 |
| S. viminalis | UWM034 | V13 |
| S. viminalis | UWM196 | V14 |
| S. viminalis | UWM160 | V15 |
| S. viminalis | UWM158 | V16 |
| S. viminalis | UWM157 | V17 |
| S. viminalis | UWM130 | V18 |
| S. viminalis | UWM144 | V19 |
| S. viminalis | UWM133 | V20 |
| S. viminalis | UWM132 | V21 |
| S. viminalis | UWM131 | V22 |
| S. alba | S. purpurea | S. triandra | S. viminalis | |
|---|---|---|---|---|
| S. alba | – | 0.320 | 0.687 | 0.371 |
| S. purpurea | 0.687 | – | 0.802 | 0.264 |
| S. triandra | 0.782 | 0.828 | – | 0.808 |
| S. viminalis | 0.707 | 0.665 | 0.822 | – |
| Source of Variation | d.f. | Sum of Squares | Mean Squares | Estimated Variation | Total Variance | ΦST (p < 0.01) |
|---|---|---|---|---|---|---|
| Among Species | 3 | 8715.160 | 2905.053 | 225.787 | 75% | 0.754 |
| Within Species | 49 | 3600.991 | 73.490 | 73.490 | 25% | |
| Total | 52 | 12,316.151 | 299.276 | 100% |
| Species | N | Na | Ne | I | He | uHe | PrB |
|---|---|---|---|---|---|---|---|
| S. alba | 9 | 0.866 | 1.095 | 0.079 | 0.053 | 0.057 | 77 |
| S. purpurea | 13 | 0.829 | 1.114 | 0.097 | 0.065 | 0.068 | 37 |
| S. triandra | 9 | 0.647 | 1.081 | 0.071 | 0.047 | 0.050 | 135 |
| S. viminalis | 22 | 0.984 | 1.145 | 0.126 | 0.084 | 0.086 | 83 |
| Mean | 12.860 | 0.831 | 1.109 | 0.093 | 0.062 | 0.065 | 83 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Przyborowski, J.A.; Sulima, P.; Kuszewska, A.; Załuski, D.; Kilian, A. Phylogenetic Relationships between Four Salix L. Species Based on DArT Markers. Int. J. Mol. Sci. 2013, 14, 24113-24125. https://doi.org/10.3390/ijms141224113
Przyborowski JA, Sulima P, Kuszewska A, Załuski D, Kilian A. Phylogenetic Relationships between Four Salix L. Species Based on DArT Markers. International Journal of Molecular Sciences. 2013; 14(12):24113-24125. https://doi.org/10.3390/ijms141224113
Chicago/Turabian StylePrzyborowski, Jerzy A., Paweł Sulima, Anna Kuszewska, Dariusz Załuski, and Andrzej Kilian. 2013. "Phylogenetic Relationships between Four Salix L. Species Based on DArT Markers" International Journal of Molecular Sciences 14, no. 12: 24113-24125. https://doi.org/10.3390/ijms141224113