Visual Detection and Evaluation of Latent and Lytic Gene Expression during Epstein-Barr Virus Infection Using One-Step Reverse Transcription Loop-Mediated Isothermal Amplification

Abstract
:1. Introduction
2. Results and Discussion
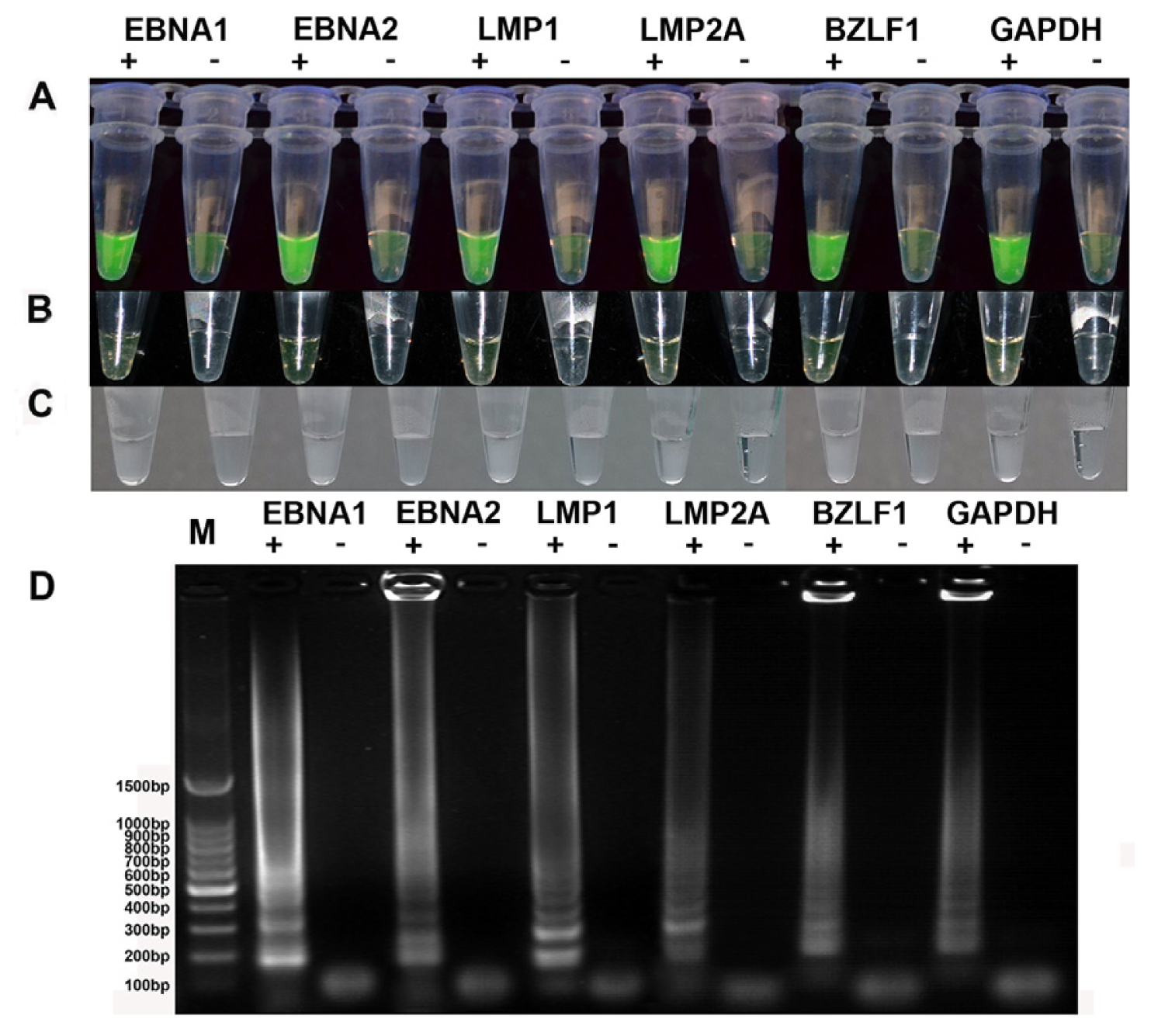
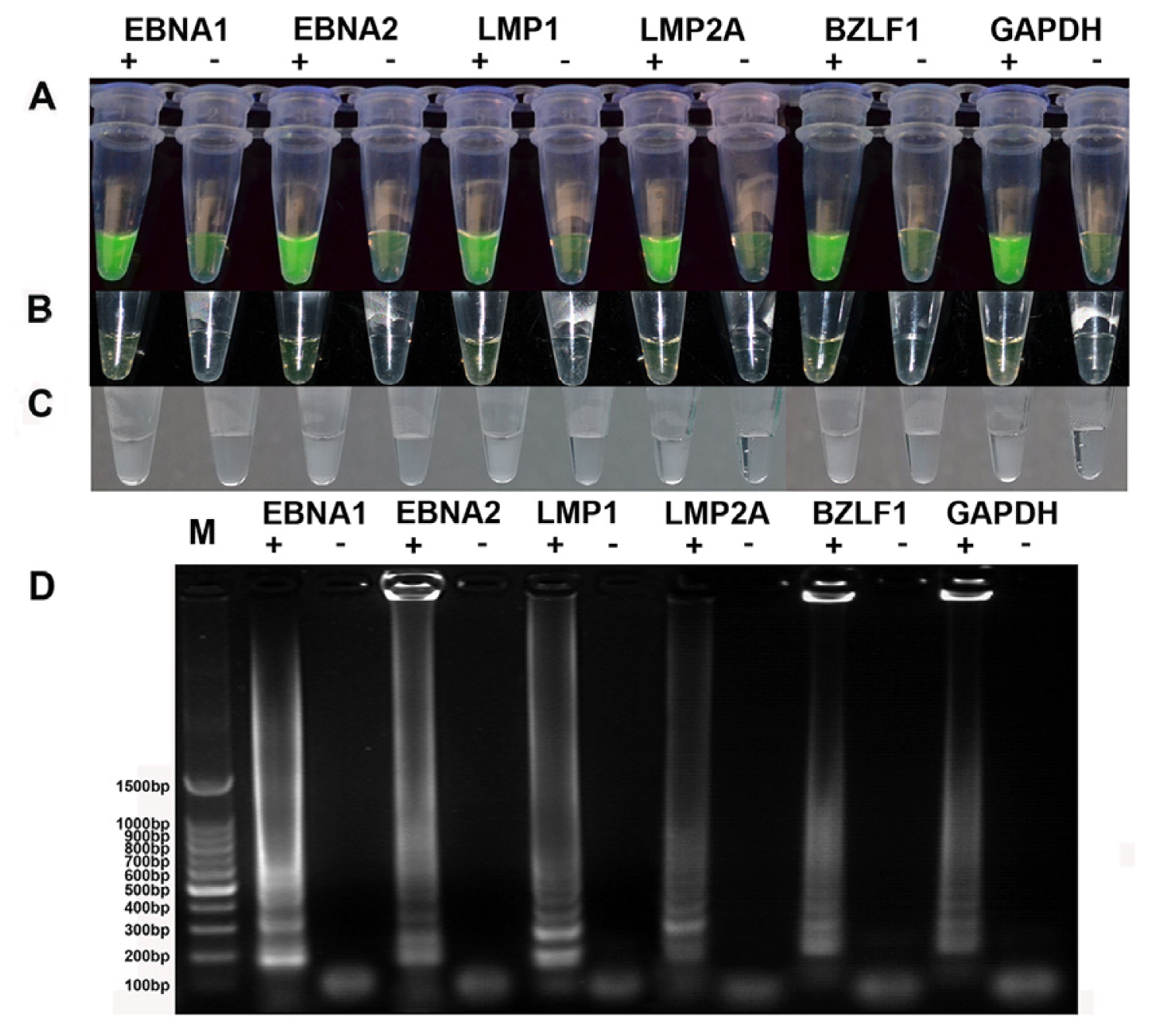
2.1. Establishment of a One-Step RT-LAMP Assay
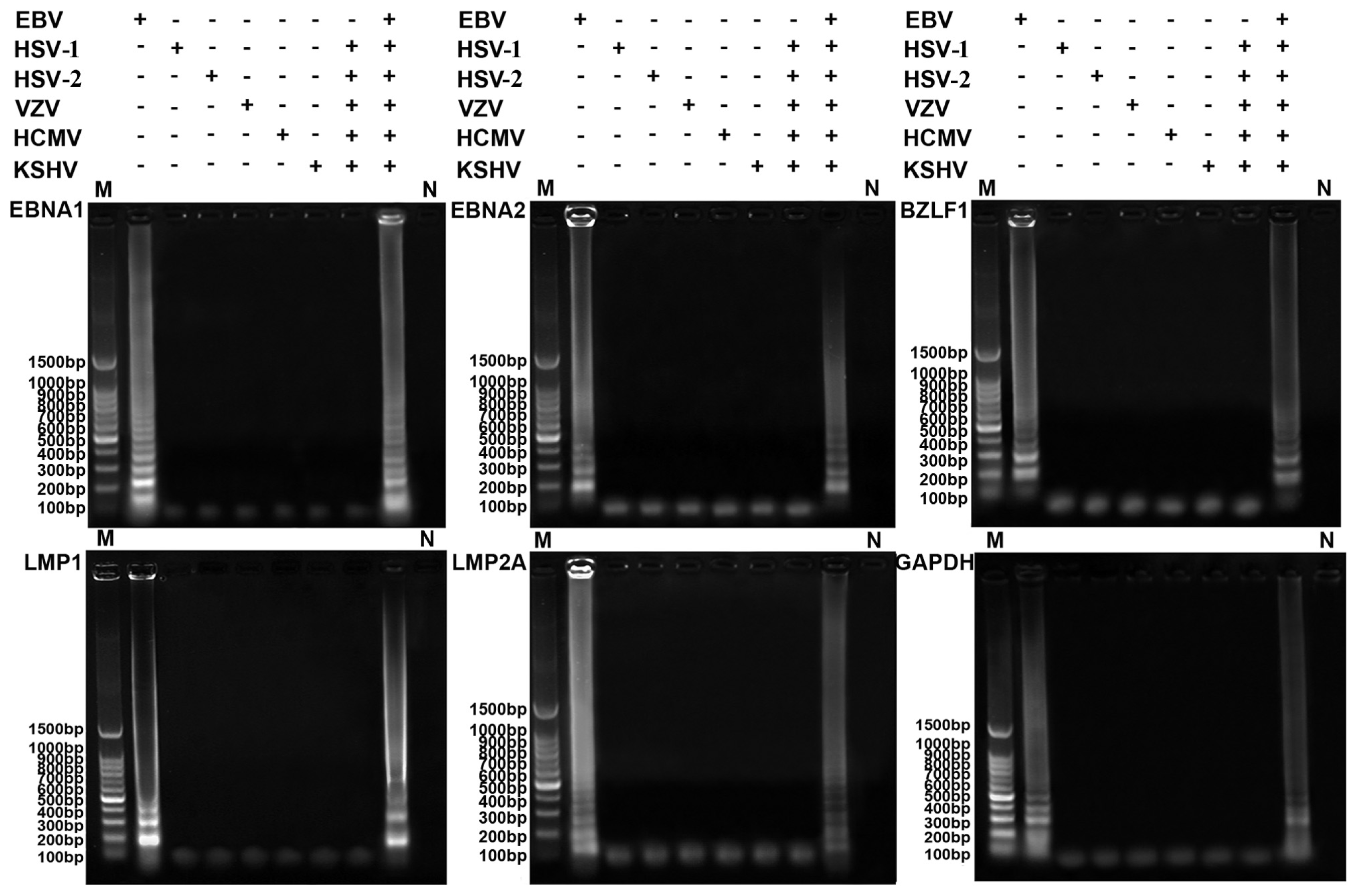
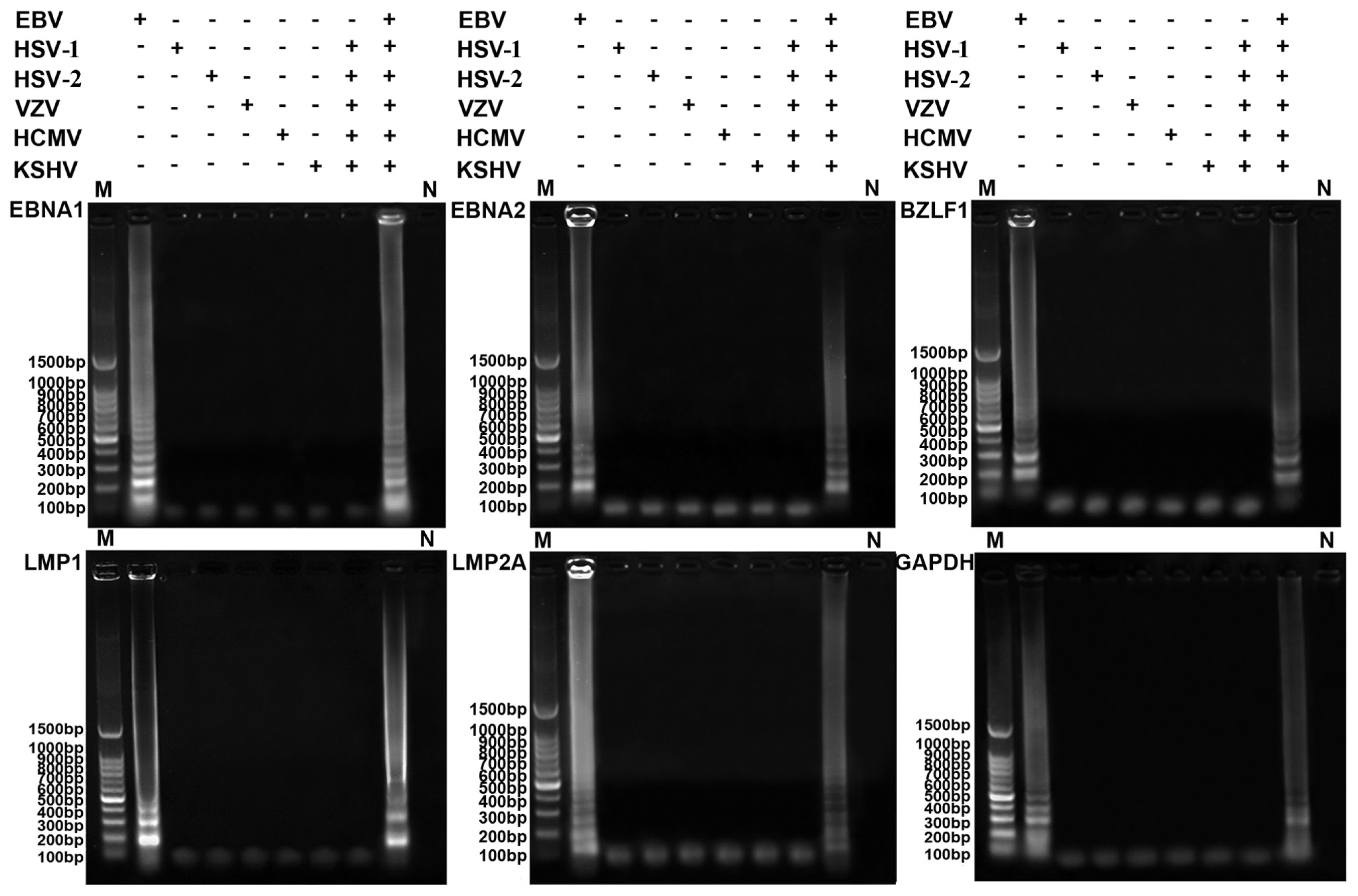
2.2. Specificity of the RT-LAMP Assay
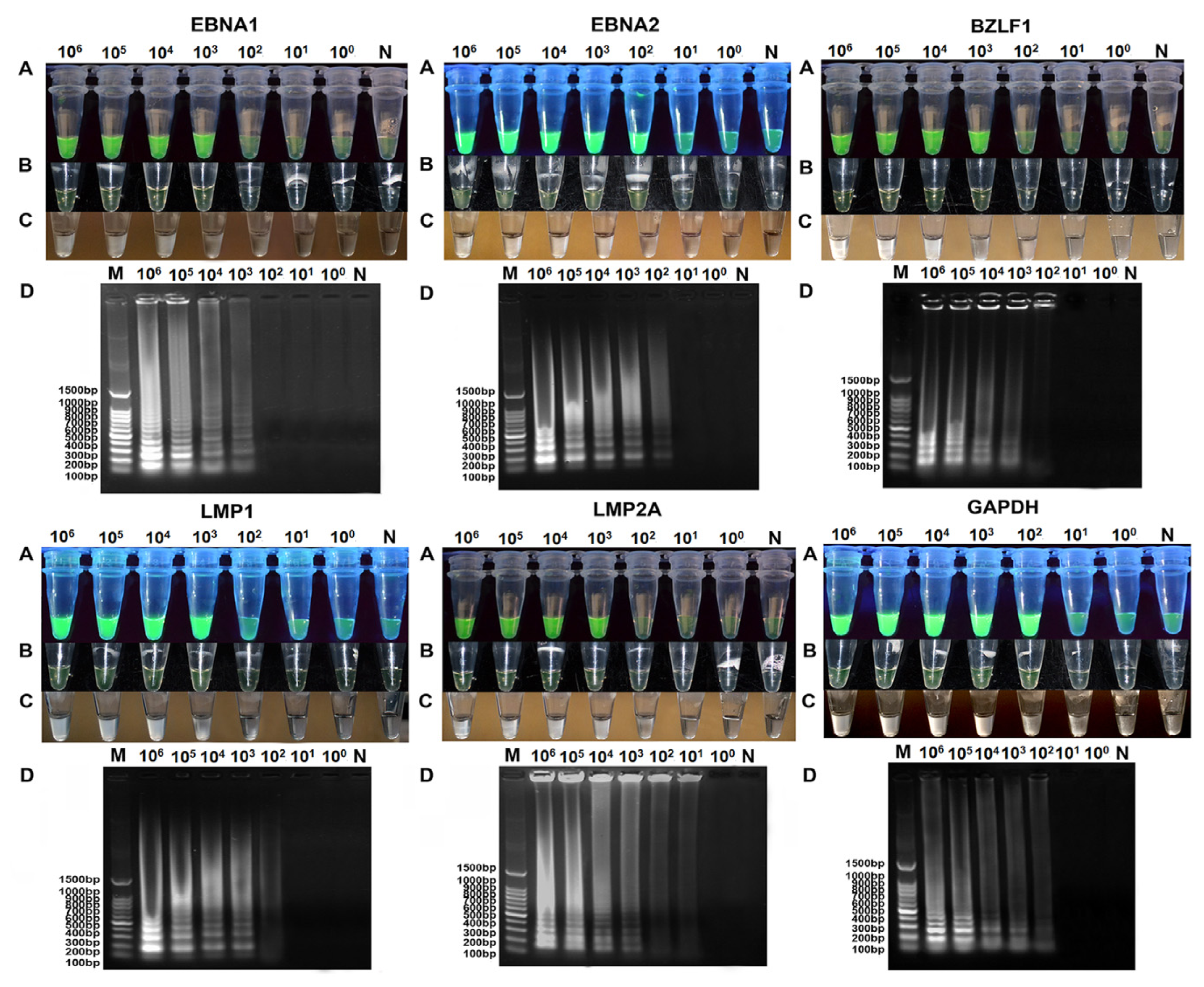
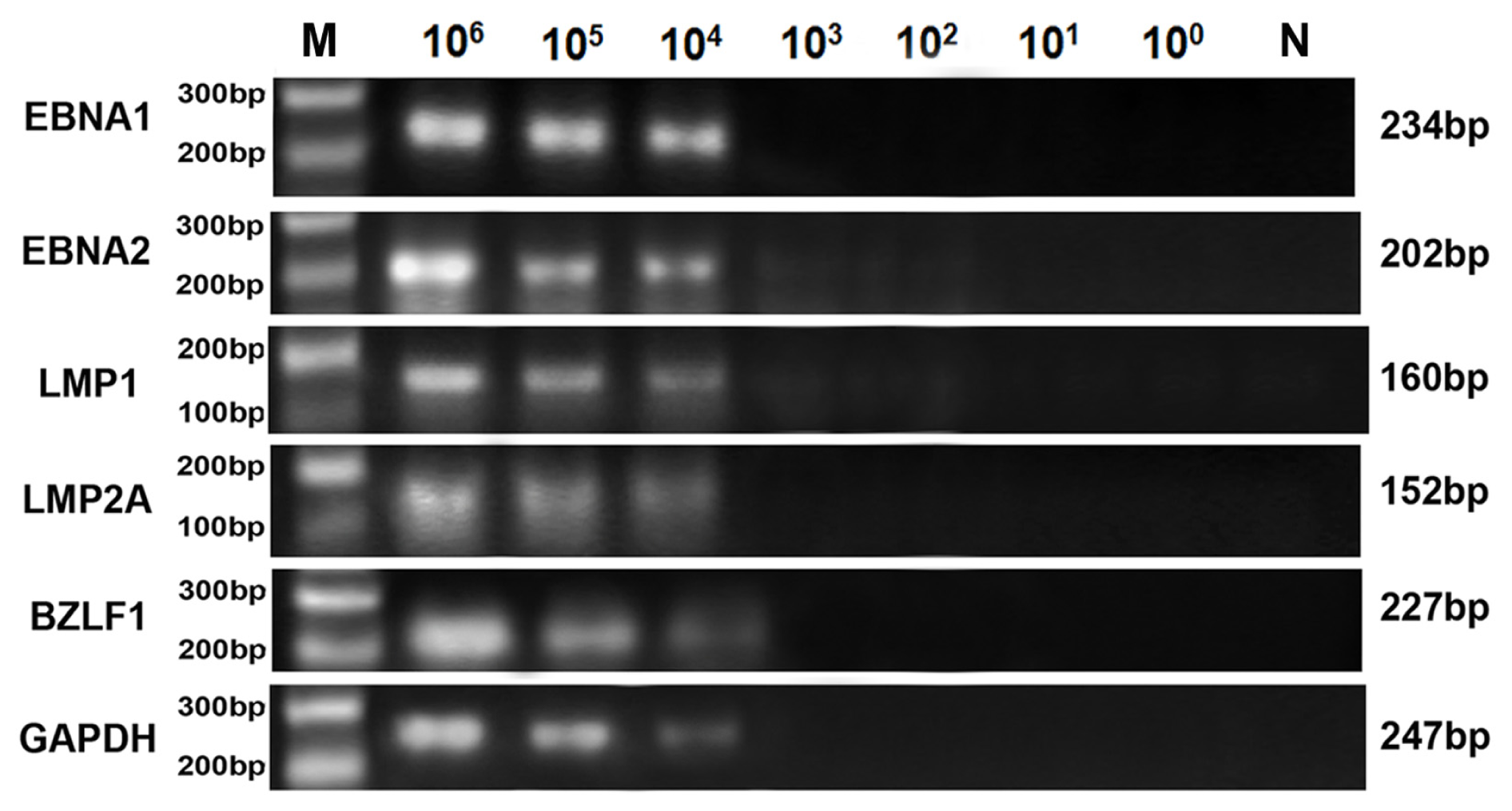
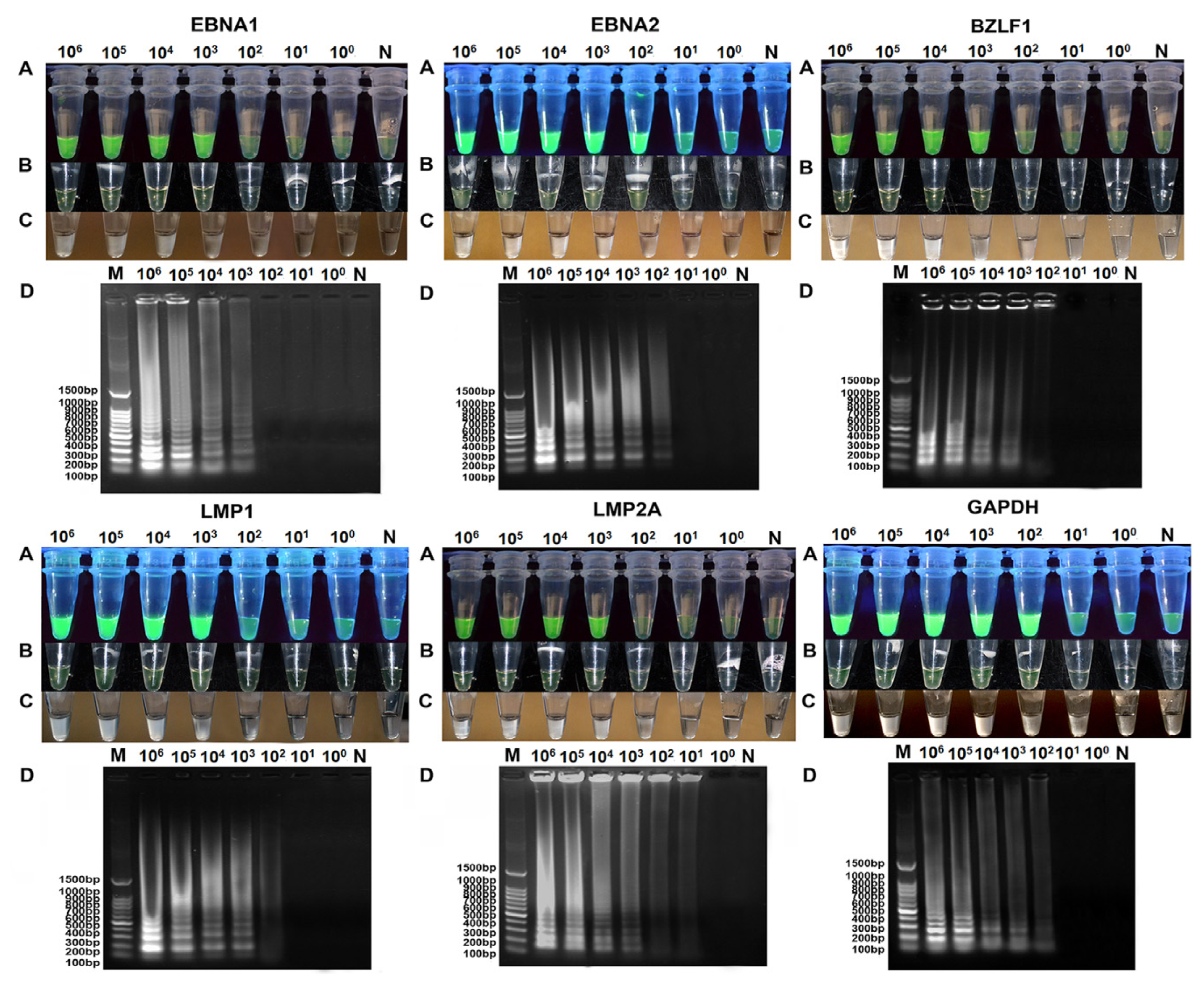
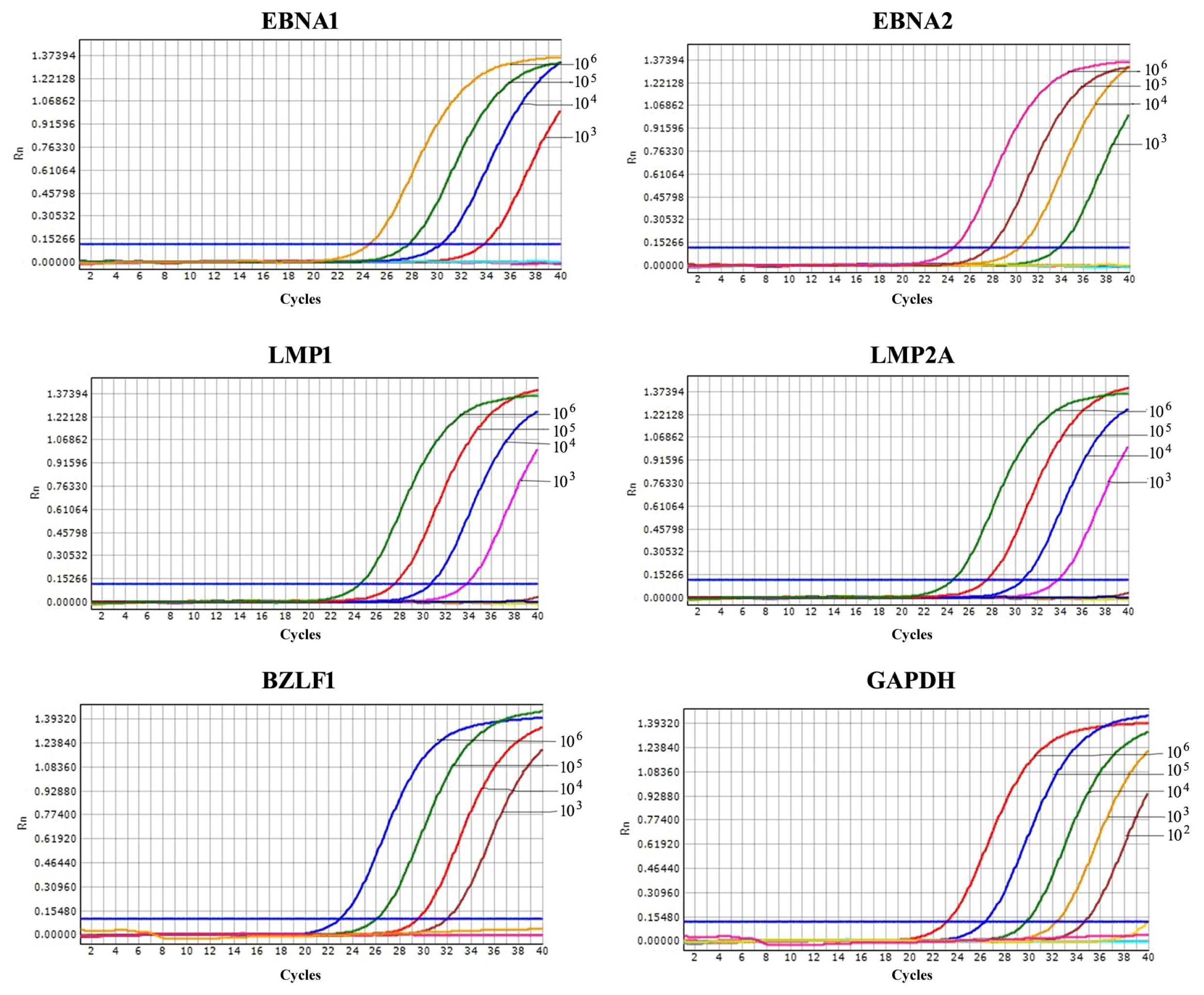
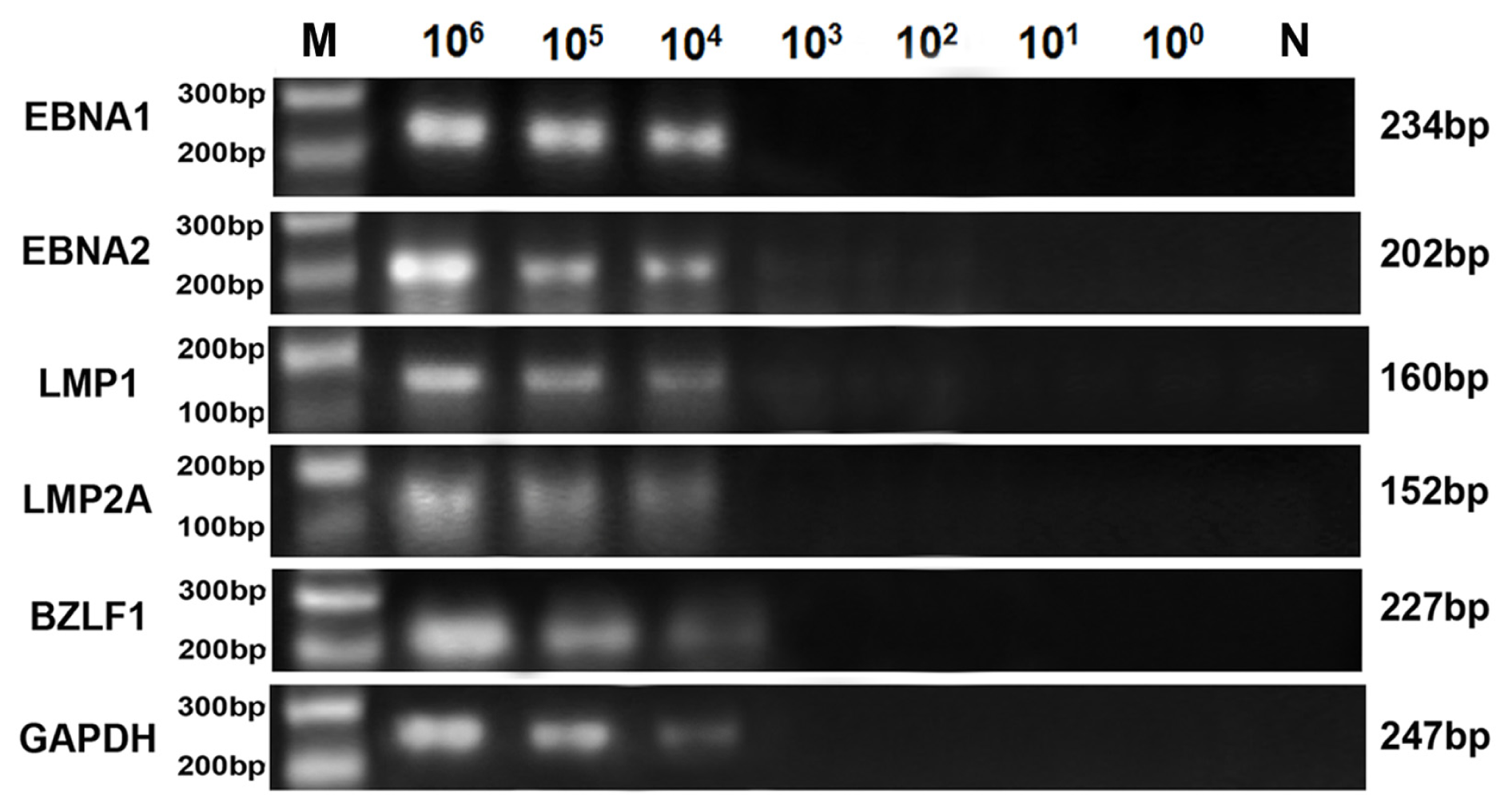
2.3. Comparison of Sensitivity between RT-LAMP, RT-qPCR and Conventional RT-PCR
2.4. Application of RT-LAMP Assays for the Surveillance of EBV Gene Expression
3. Experimental Section
3.1. Clinical Specimens
3.2. Cell Lines
3.3. PBMC Isolation and Total RNA Preparation
3.4. RT-LAMP Reaction
3.5. Conventional RT-PCR and RT-qPCR
3.6. Specificity of the RT-LAMP Assay
3.7. Sensitivity Comparison of the Assays
3.8. Application of RT-LAMP in Clinical Specimens and Comparison with RT-qPCR and Conventional RT-PCR
4. Conclusions
Supplementary Information
ijms-14-23922-s001.pdfAcknowledgments
Conflicts of Interest
References
- Bornkamm, G.W.; Hammerschmidt, W. Molecular virology of Epstein-Barr virus. Philos. Trans. R. Soc. Lond B Biol. Sci 2001, 356, 437–459. [Google Scholar]
- Niedobitek, G.; Agathanggelou, A.; Steven, N.; Young, L.S. Epstein-Barr virus (EBV) in infectious mononucleosis: Detection of the virus in tonsillar B lymphocytes but not in desquamated oropharyngeal epithelial cells. Mol. Pathol 2000, 53, 37–42. [Google Scholar]
- Sitki-Green, D.L.; Edwards, R.H.; Covington, M.M.; Raab-Traub, N. Biology of Epstein-Barr virus during infectious mononucleosis. J. Infect. Dis 2004, 189, 483–92. [Google Scholar]
- Jenson, H.B. Acute complications of Epstein-Barr virus infectious mononucleosis. Curr. Opin. Pediatr 2000, 12, 263–268. [Google Scholar]
- Middeldorp, J.M.; Brink, A.A.; van den Brule, A.J.; Meijer, C.J. Pathogenic roles for Epstein-Barr virus (EBV) gene products in EBV-associated proliferative disorders. Crit. Rev. Oncol. Hematol 2003, 45, 1–36. [Google Scholar]
- Saha, A.; Robertson, E.S. Epstein-Barr virus-associated B-cell lymphomas: Pathogenesis and clinical outcomes. Clin. Cancer Res 2011, 17, 3056–3063. [Google Scholar]
- Yen, C.Y.; Lu, M.C.; Tzeng, C.C.; Huang, J.Y.; Chang, H.W.; Chen, R.S.; Liu, S.Y.; Liu, S.T.; Shieh, B.; Li, C. Detection of EBV infection and gene expression in oral cancer from patients in Taiwan by microarray analysis. J. Biomed. Biotechnol 2009, 2009. [Google Scholar] [CrossRef]
- Kuppers, R. B cells under influence: Transformation of B cells by Epstein-Barr virus. Nat. Rev. Immunol 2003, 3, 801–812. [Google Scholar]
- Gulley, M.L.; Tang, W. Laboratory assays for Epstein-Barr virus-related disease. J. Mol. Diagn 2008, 10, 279–292. [Google Scholar]
- Tsuchiya, S. Diagnosis of Epstein-Barr virus-associated diseases. Crit. Rev. Oncol. Hematol 2002, 44, 227–238. [Google Scholar]
- Rechsteiner, M.P.; Bernasconi, M.; Berger, C.; Nadal, D. Role of latent membrane protein 2 isoforms in Epstein-Barr virus latency. Trends Microbiol 2008, 16, 520–527. [Google Scholar]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol 2007, 81, 1037–1042. [Google Scholar]
- Katsumura, K.R.; Maruo, S.; Wu, Y.; Kanda, T.; Takada, K. Quantitative evaluation of the role of Epstein-Barr virus immediate-early protein BZLF1 in B-cell transformation. J. Gen. Virol 2009, 90, 2331–2341. [Google Scholar]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H., Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. In Clin. Microbiol. Rev; 2011; Volume 24, pp. 193–209. [Google Scholar]
- Kasahara, Y.; Yachie, A. Cell type specific infection of Epstein-Barr virus (EBV) in EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Crit. Rev. Oncol. Hematol 2002, 44, 283–294. [Google Scholar]
- Bell, A.I.; Groves, K.; Kelly, G.L.; Croom-Carter, D.; Hui, E.; Chan, A.T.; Rickinson, A.B. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt’s lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol 2006, 87, 2885–2890. [Google Scholar]
- Kubota, N.; Wada, K.; Ito, Y.; Shimoyama, Y.; Nakamura, S.; Nishiyama, Y.; Kimura, H. One-step multiplex real-time PCR assay to analyse the latency patterns of Epstein-Barr virus infection. J. Virol. Methods 2008, 147, 26–36. [Google Scholar]
- Iwata, S.; Wada, K.; Tobita, S.; Gotoh, K.; Ito, Y.; Demachi-Okamura, A.; Shimizu, N.; Nishiyama, Y.; Kimura, H. Quantitative analysis of Epstein-Barr virus (EBV)-related gene expression in patients with chronic active EBV infection. J. Gen. Virol 2010, 91, 42–50. [Google Scholar]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 2000, 28, E63. [Google Scholar]
- Wang, C.H.; Lien, K.Y.; Wang, T.Y.; Chen, T.Y.; Lee, G.B. An integrated microfluidic loop-mediated-isothermal-amplification system for rapid sample pre-treatment and detection of viruses. Biosens Bioelectron 2011, 26, 2045–2052. [Google Scholar]
- Parida, M.; Sannarangaiah, S.; Dash, P.K.; Rao, P.V.; Morita, K. Loop mediated isothermal amplification (LAMP): A new generation of innovative gene amplification technique; Perspectives in clinical diagnosis of infectious diseases. Rev. Med. Virol 2008, 18, 407–421. [Google Scholar]
- Nie, G.; Dong, H.; He, G.; Xu, X.; Shi, L.; Cao, Y.; Chen, X. The value of loop-mediated isothermal amplification method for rapid diagnosis of EBV DNA. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2008, 22, 555–557. [Google Scholar]
- Iwata, S.; Shibata, Y.; Kawada, J.; Hara, S.; Nishiyama, Y.; Morishima, T.; Ihira, M.; Yoshikawa, T.; Asano, Y.; Kimura, H. Rapid detection of Epstein-Barr virus DNA by loop-mediated isothermal amplification method. J. Clin. Virol 2006, 37, 128–133. [Google Scholar]
- Abd-Elsalam, K.; Bahkali, A.; Moslem, M.; Amin, O.E.; Niessen, L. An optimized protocol for DNA extraction from wheat seeds and loop-mediated isothermal amplification (LAMP) to detect Fusarium graminearum contamination of wheat grain. Int. J. Mol. Sci 2011, 12, 3459–3472. [Google Scholar]
- Horibe, D.; Ochiai, T.; Shimada, H.; Tomonaga, T.; Nomura, F.; Gun, M.; Tanizawa, T.; Hayashi, H. Rapid detection of metastasis of gastric cancer using reverse transcription loop-mediated isothermal amplification. Int. J. Cancer 2007, 120, 1063–1069. [Google Scholar]
- Maeda, J.; Inoue, M.; Nakabayashi, K.; Otomo, Y.; Shintani, Y.; Ohta, M.; Okumura, M.; Matsuura, N. Rapid diagnosis of lymph node metastasis in lung cancer with loop-mediated isothermal amplification assay using carcinoembryonic antigen-mRNA. Lung Cancer 2009, 65, 324–327. [Google Scholar]
- Countryman, J.K.; Gradoville, L.; Miller, G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol 2008, 82, 4706–4719. [Google Scholar]
- Ohga, S.; Nomura, A.; Takada, H.; Hara, T. Immunological aspects of Epstein-Barr virus infection. Crit. Rev. Oncol. Hematol 2002, 44, 203–215. [Google Scholar]
- Williams, H.; Crawford, D.H. Epstein-Barr virus: The impact of scientific advances on clinical practice. Blood 2006, 107, 862–869. [Google Scholar]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar]
- Amon, W.; Farrell, P.J. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol 2005, 15, 149–156. [Google Scholar]
- Swanson-Mungerson, M.; Bultema, R.; Longnecker, R. Epstein-Barr virus LMP2A imposes sensitivity to apoptosis. J. Gen. Virol 2010, 91, 2197–2202. [Google Scholar]
- Sivachandran, N.; Wang, X.; Frappier, L. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J. Virol 2012, 86, 6146–6158. [Google Scholar]
- Lorenzetti, M.A.; de Matteo, E.; Gass, H.; Martinez Vazquez, P.; Lara, J.; Gonzalez, P.; Preciado, M.V.; Chabay, P.A. Characterization of Epstein Barr virus latency pattern in Argentine breast carcinoma. PLoS One 2010, 5, e13603. [Google Scholar]
- Rafailidis, P.I.; Mavros, M.N.; Kapaskelis, A.; Falagas, M.E. Antiviral treatment for severe EBV infections in apparently immunocompetent patients. J. Clin. Virol 2010, 49, 151–157. [Google Scholar]
- Bergallo, M.; Costa, C.; Baro, S.; Musso, T.; Balbo, L.; Merlino, C.; Cavallo, R. Multiplex-nested RT-PCR to evaluate latent and lytic Epstein Barr virus gene expression. J. Biotechnol 2007, 128, 462–476. [Google Scholar]
- McKenna, J.P.; Fairley, D.J.; Shields, M.D.; Cosby, S.L.; Wyatt, D.E.; McCaughey, C.; Coyle, P.V. Development and clinical validation of a loop-mediated isothermal amplification method for the rapid detection of Neisseria meningitidis. Diagn. Microbiol. Infect. Dis 2011, 69, 137–144. [Google Scholar]
- Teoh, B.T.; Sam, S.S.; Tan, K.K.; Johari, J.; Danlami, M.B.; Hooi, P.S.; Md-Esa, R.; Abubakar, S. Detection of dengue viruses using reverse transcription-loop-mediated isothermal amplification. BMC Infect. Dis 2013, 13, 387. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Position a | Primer Sequences (5′→3′) |
|---|---|---|
| LMP1b F3 | 944–961 | ACCCTAGCGACTCTGCTG |
| LMP1 B3 | 1100–1118 | TGGGGGTCGTCATCATCTC |
| LMP1 FIP | F1c: 1022–1041 | GTCTGTCATCGAAGGCGGGCTTTT c |
| F2: 971–989 | GAGGCCCTCCAGAATTGAC | |
| LMP1 BIP | B1c: 1055–1076 | ATCCACACCTTCCTACCCTGCTTTTT c |
| B2: 1081–1099 | CACCGGAACCAGAAGTACC | |
| LMP2Ab F3 | 1084–1104 | GGCAGTATTTTGCAAACAAAC |
| LMP2A B3 | 1250–1267 | ATGAAGAGTATGCCAGCG |
| LMP2A FIP | F1c: 1158–1182 | GCTTGTTTTCTTCAACTAAACAGGATTTT c |
| F2: 1106–1125 | TCAAGAGTTTAAGCAGCACT | |
| LMP2A BIP | B1c: 1185–1209 | TTGGATTGTAACACACATTTTACGCTTTT c |
| B2: 1226–1249 | ACAATCAGTAATAACATGCAGAAC | |
| EBNA1b F3 | 407–426 | GAGTAGTCTCAGGGCATCCT |
| EBNA1 B3 | 927–946 | ATGTGTCTCCCTTCTCTCCT |
| EBNA1 FIP | F1c: 468–489 | CGGTGAATCTGCTCCCAGGTCTTTTT c |
| F2: 427–444 | CTGGAGCCTGACCTGTGA | |
| EBNA1 BIP | B1c: 492–512 | GCGGCCGTCTCCTTTAAGTGTTTTT c |
| B2: 904–923 | CCATTTCCAGGTCCTGTACC | |
| EBNA2b F3 | 711–730 | AATCAACCTGATTCCCCCTG |
| EBNA2 B3 | 919–936 | CGCTGGGTGGTTACTGTG |
| EBNA2 FIP | F1c: 777–796 | AGCACTTACCCGAGCGGGAGTTTT c |
| F2: 737–756 | CCTCCACTTACAACCAAGCC | |
| EBNA2 BIP | B1c: 835–856 | AGGCAAACCTCCAATCACCAGCTTTT c |
| B2: 880–899 | TAATGCGTAGCAGCCACTCT | |
| BZLF1b F3 | 6–25 | GGACCCAAACTCGACTTCTG |
| BZLF1 B3 | 210–228 | AGGAAACCACGACCCAGTT |
| BZLF1 FIP | F1c: 94–115 | GCCCTCCCAGGTCCTGATAGACTTTT c |
| F2: 45–62 | TGACCCATACCAGGTGCC | |
| BZLF1 BIP | B1c: 130–149 | TTGCCTTGTGTGCTGTGGCCTTTT c |
| B2: 192–209 | GGTGCGGCTGAAACATGA | |
| GAPDHb F3 | 248–267 | CCATCTTCCAGGAGCGAGAT |
| GAPDH B3 | 449–468 | GCTGATGATCTTGAGGCTGT |
| GAPDH FIP | F1c: 313–333 | GGTGAAGACGCCAGTGGACTCTTTT c |
| F2: 273–292 | CAAAATCAAGTGGGGCGATG | |
| GAPDH BIP | B1c: 351–370 | GGCTCATTTGCAGGGGGGAGTTTT c |
| B2: 411–430 | TCACACCCATGACGAACATG |
| Assay | PBMC/1 × 106 BJAB cells | |||||||
|---|---|---|---|---|---|---|---|---|
| 106 | 105 | 104 | 103 | 102 | 101 | 100 | ||
| RT-LAMP | EBNA1 | + | + | + | + | − | − | − |
| EBNA2 | + | + | + | + | ±(2/3) | − | − | |
| LMP1 | + | + | + | + | − | − | − | |
| LMP2A | + | + | + | + | − | − | − | |
| BZLF1 | + | + | + | − | − | − | − | |
| RT-qPCR | EBNA1 | + | + | + | + | − | − | − |
| EBNA2 | + | + | + | + | − | − | − | |
| LMP1 | + | + | + | + | − | − | − | |
| LMP2A | + | + | + | + | − | − | − | |
| BZLF1 | + | + | + | − | − | − | − | |
| Conventional RT-PCR | EBNA1 | + | + | + | − | − | − | − |
| EBNA2 | + | + | − | − | − | − | − | |
| LMP1 | + | + | + | − | − | − | − | |
| LMP2A | + | + | − | − | − | − | − | |
| BZLF1 | + | + | − | − | − | − | − | |
| RT-qPCR a | RT-LAMP | Conventional RT-PCR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Transcript | No. positive patients (%) | No. positive patients (%) | Clinical sensitivity b | Clinical specificity b | Absolute agreement b | κ value c | No. positive patients (%) | Clinical sensitivity d | Clinical specificity d | Absolute agreement d | κ value e |
| Group 1 (n = 51) | EBNA1 | 2 (3.9%) | 2 (3.9%) | 100% | 100% | 100% | 1.00 | 2 (3.9%) | 100% | 100% | 100% | 1.00 |
| EBNA2 | 2 (3.9%) | 2 (3.9%) | 100% | 100% | 100% | 1.00 | 2 (3.9%) | 100% | 100% | 100% | 1.00 | |
| LMP1 | 9 (17.6%) | 8 (15.7%) | 88.9% | 100% | 98% | 0.93 | 6 (11.8%) | 66.7% | 100% | 94.1% | 0.77 | |
| LMP2A | 12 (23.5%) | 11 (21.6%) | 91.7% | 100% | 98% | 0.94 | 9 (17.6%) | 75% | 100% | 94.1% | 0.82 | |
| BZLF1 | 23 (45.1%) | 21 (41.2%) | 91.3% | 100% | 98% | 0.92 | 19 (37.3%) | 73.9% | 92.9% | 84.3% | 0.68 | |
| Group 2 (n = 37) | EBNA1 | 22 (59.5%) | 21 (56.8%) | 95.5% | 100% | 97.3% | 0.95 | 16 (43.2%) | 72.7% | 100% | 83.8% | 0.68 |
| EBNA2 | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
| LMP1 | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
| LMP2A | 24 (64.9%) | 23 (62.2%) | 95.8% | 100% | 97.3% | 0.94 | 17 (45.9%) | 70.8% | 100% | 81.1% | 0.63 | |
| BZLF1 | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
| Group 3 (n = 58) | EBNA1 | 0 | 0 | − | − | − | − | 0 | − | − | − | − |
| EBNA2 | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
| LMP1 | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
| LMP2A | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
| BZLF1 | 0 | 0 | − | − | − | − | 0 | − | − | − | − | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, X.; Tang, J.; Wang, M.; Ma, Q.; Wang, Y. Visual Detection and Evaluation of Latent and Lytic Gene Expression during Epstein-Barr Virus Infection Using One-Step Reverse Transcription Loop-Mediated Isothermal Amplification. Int. J. Mol. Sci. 2013, 14, 23922-23940. https://doi.org/10.3390/ijms141223922
Liu X, Tang J, Wang M, Ma Q, Wang Y. Visual Detection and Evaluation of Latent and Lytic Gene Expression during Epstein-Barr Virus Infection Using One-Step Reverse Transcription Loop-Mediated Isothermal Amplification. International Journal of Molecular Sciences. 2013; 14(12):23922-23940. https://doi.org/10.3390/ijms141223922
Chicago/Turabian StyleLiu, Xiaoying, Jingfeng Tang, Man Wang, Qiang Ma, and Yefu Wang. 2013. "Visual Detection and Evaluation of Latent and Lytic Gene Expression during Epstein-Barr Virus Infection Using One-Step Reverse Transcription Loop-Mediated Isothermal Amplification" International Journal of Molecular Sciences 14, no. 12: 23922-23940. https://doi.org/10.3390/ijms141223922