Transcriptional Profiling of Hilar Nodes from Pigs after Experimental Infection with Actinobacillus Pleuropneumoniae

Abstract
:1. Introduction
2. Results
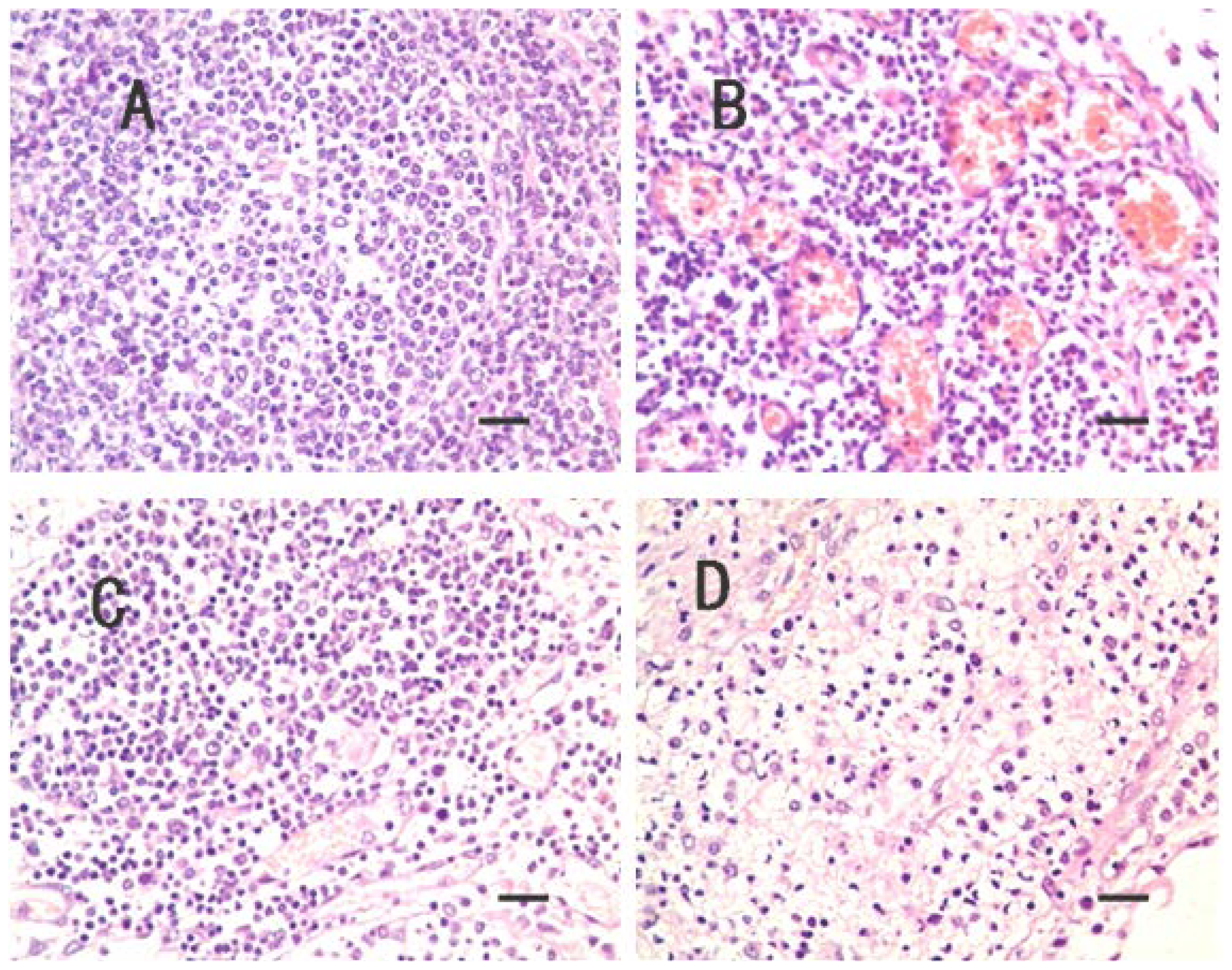
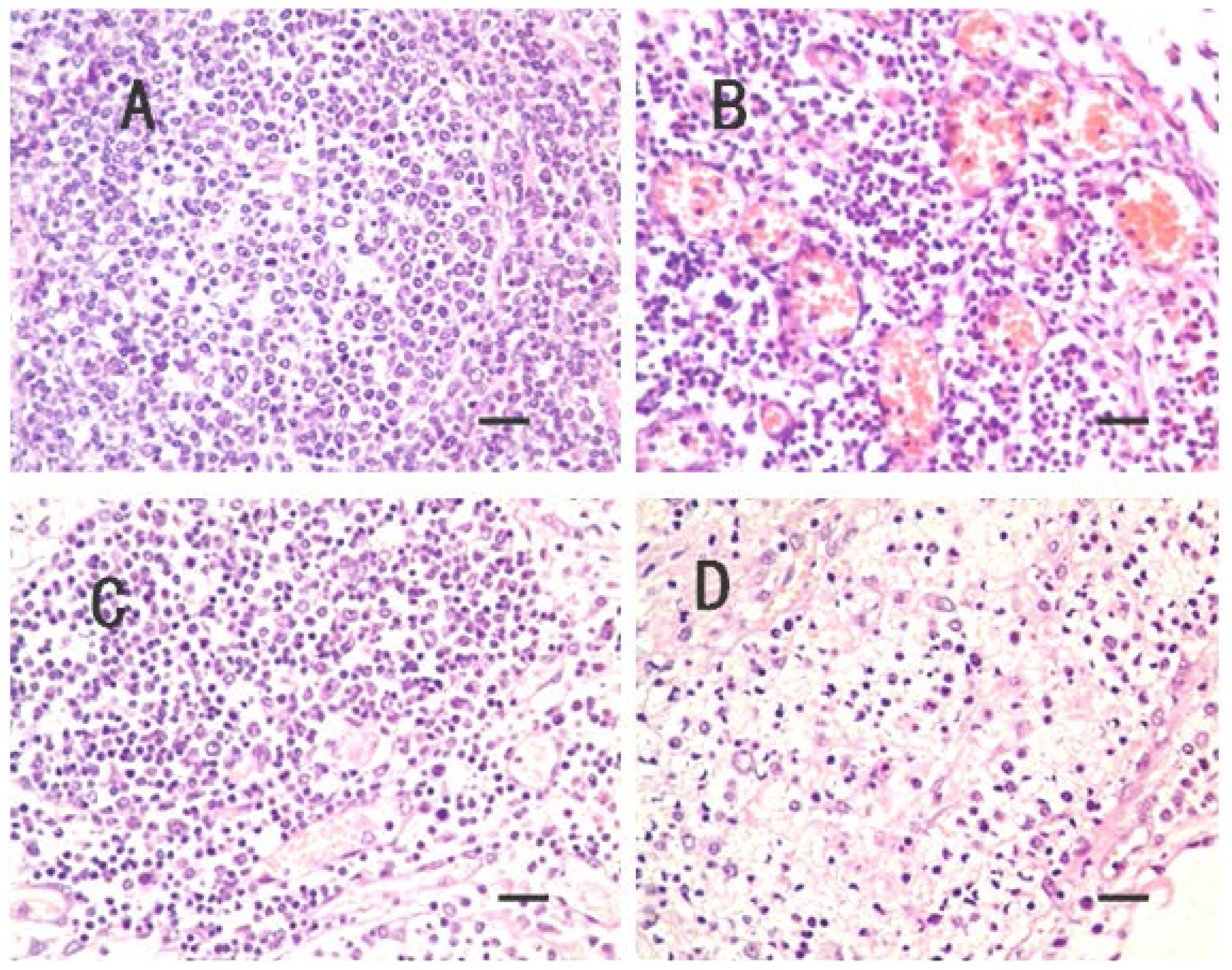
2.1. Clinical Symptoms and Necropsy Findings
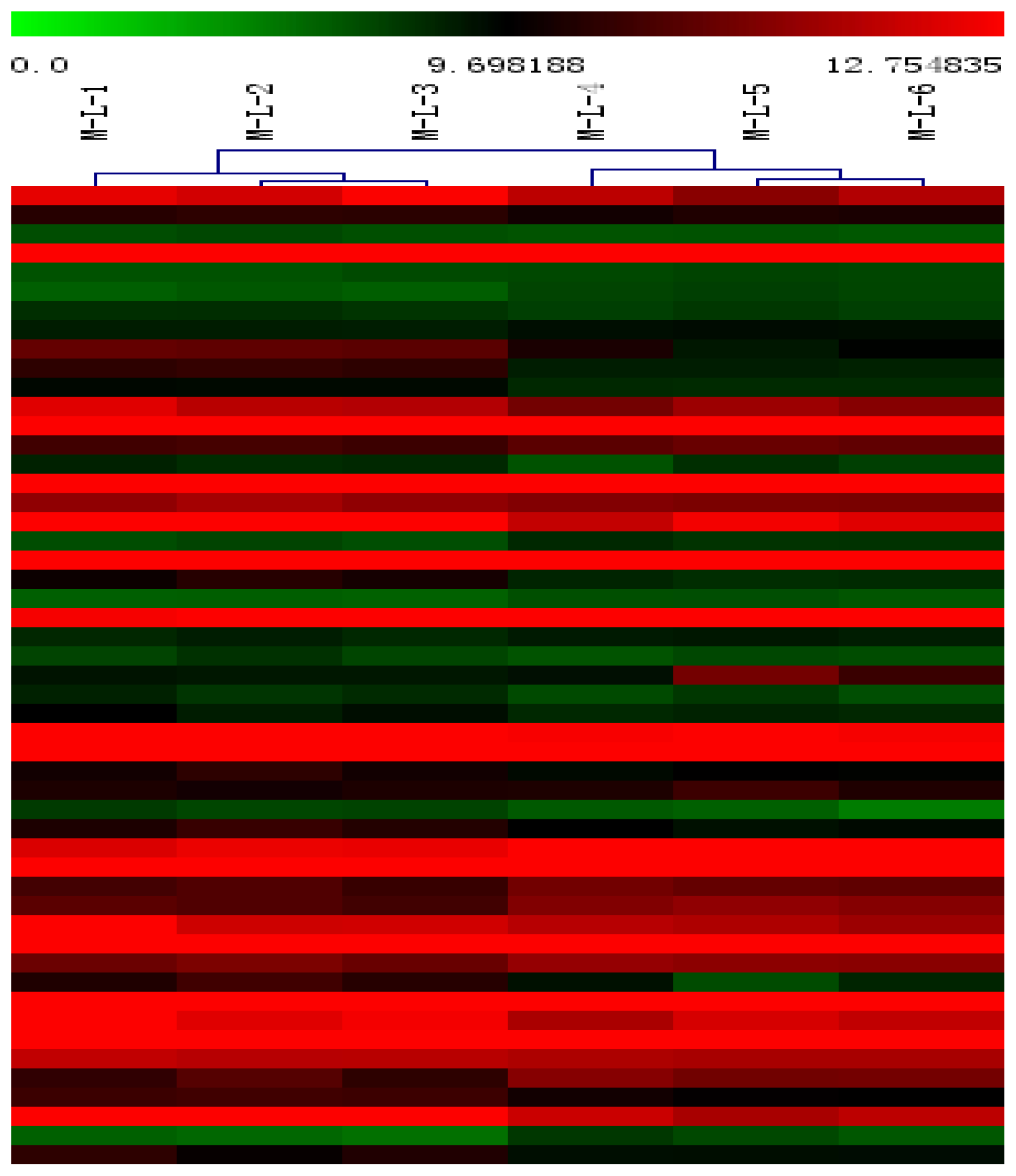
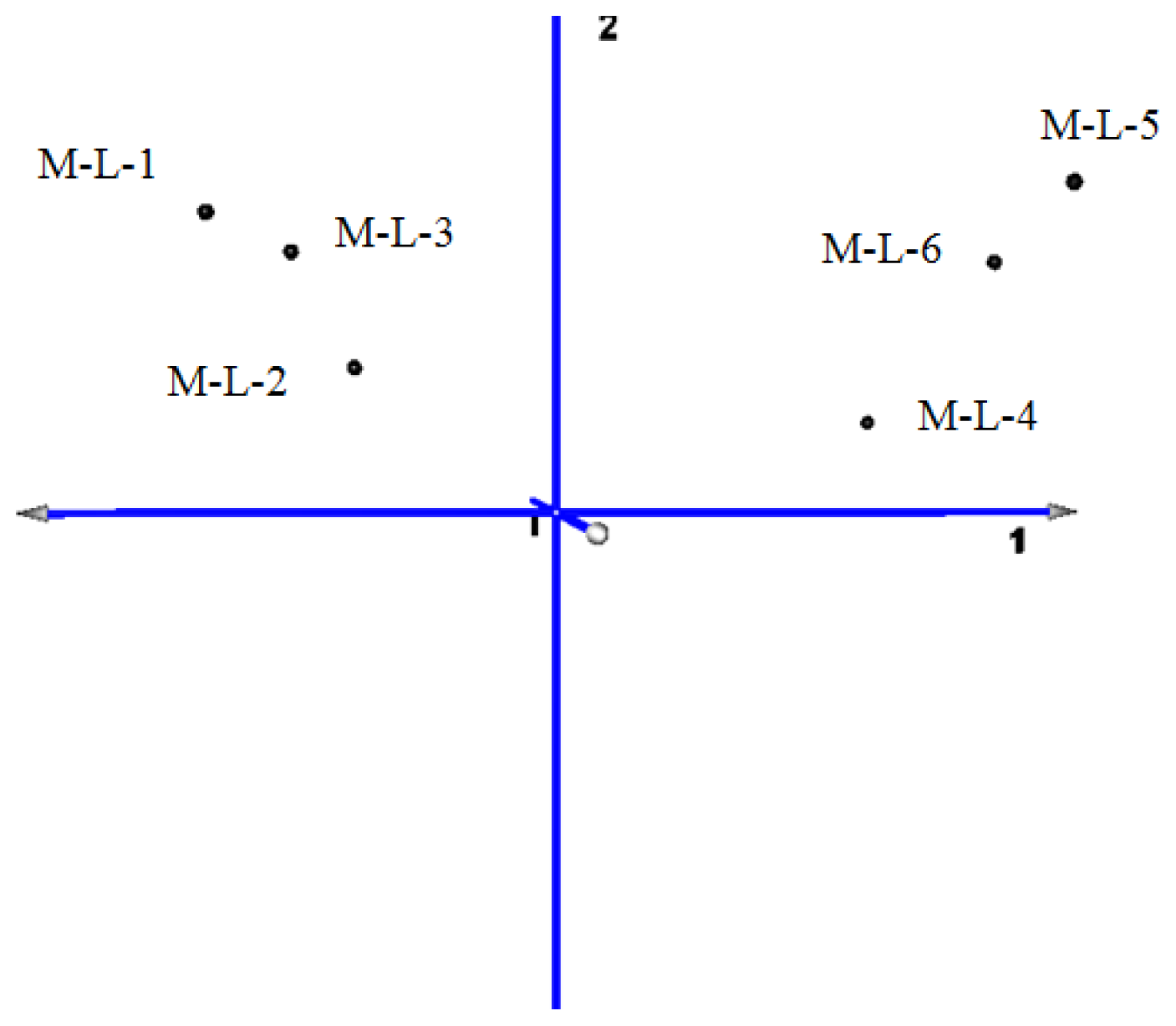
2.2. Microarray Profiling
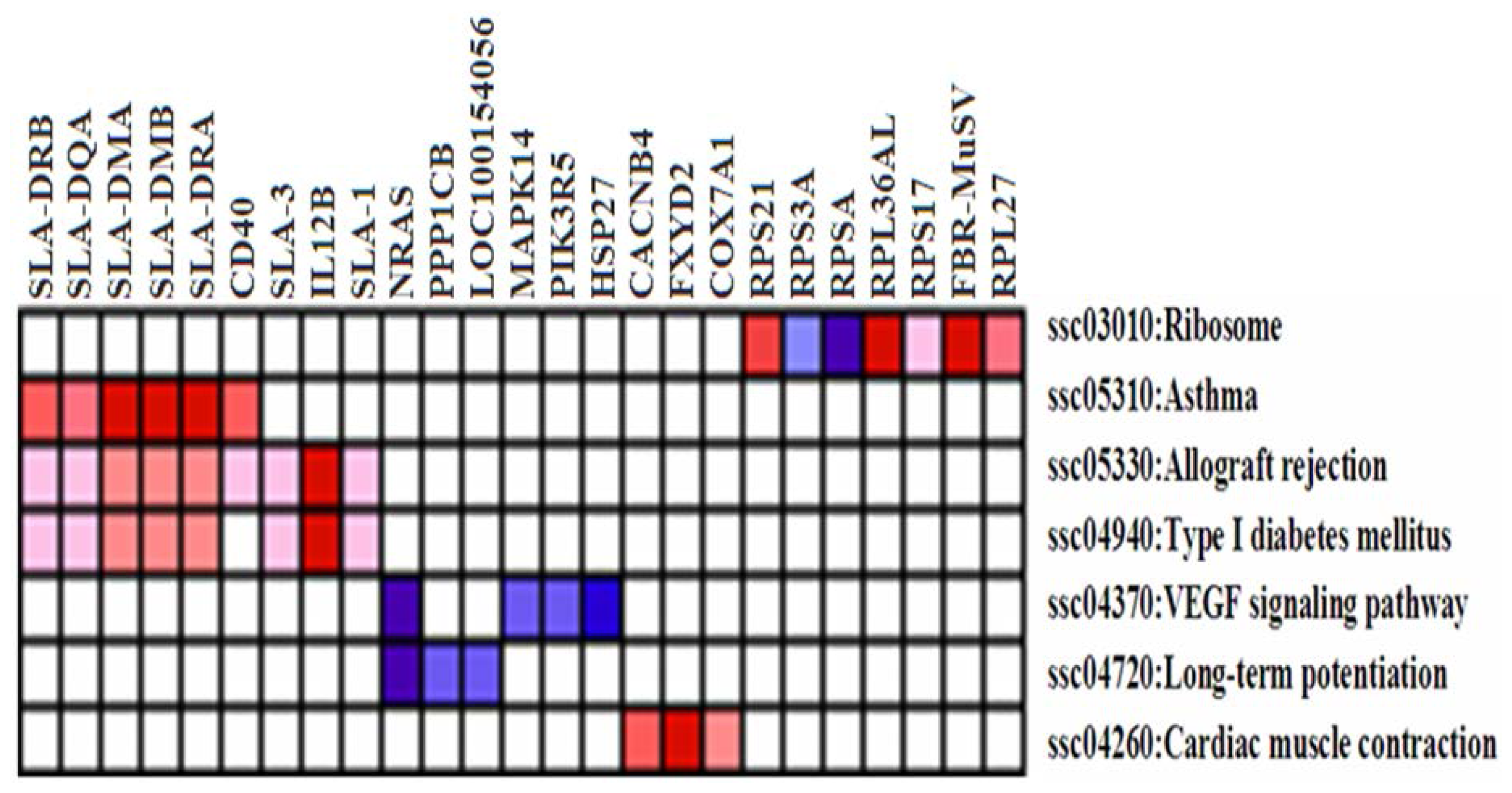
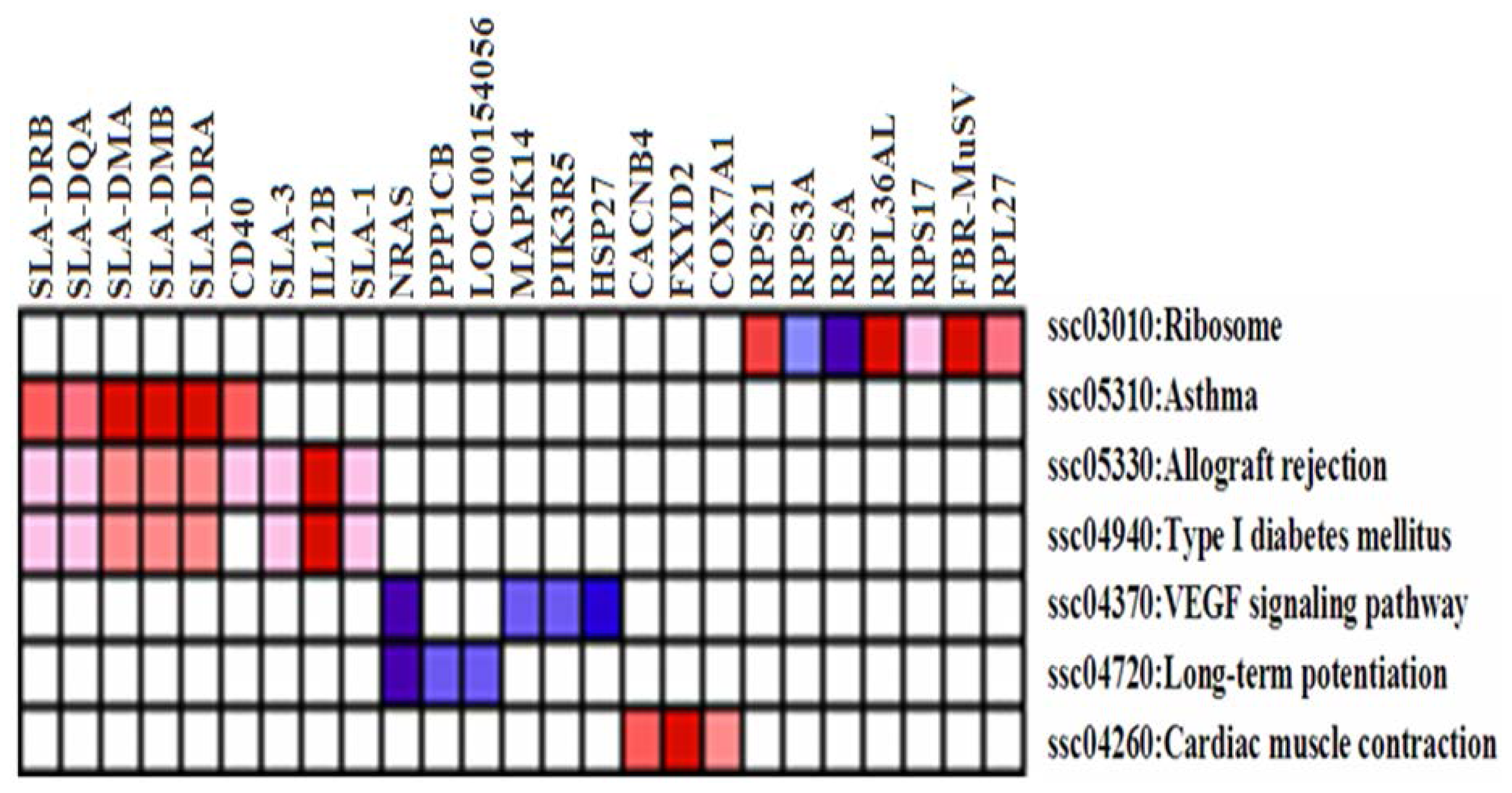
2.3. Differentially Expressed (DE) Gene Analysis
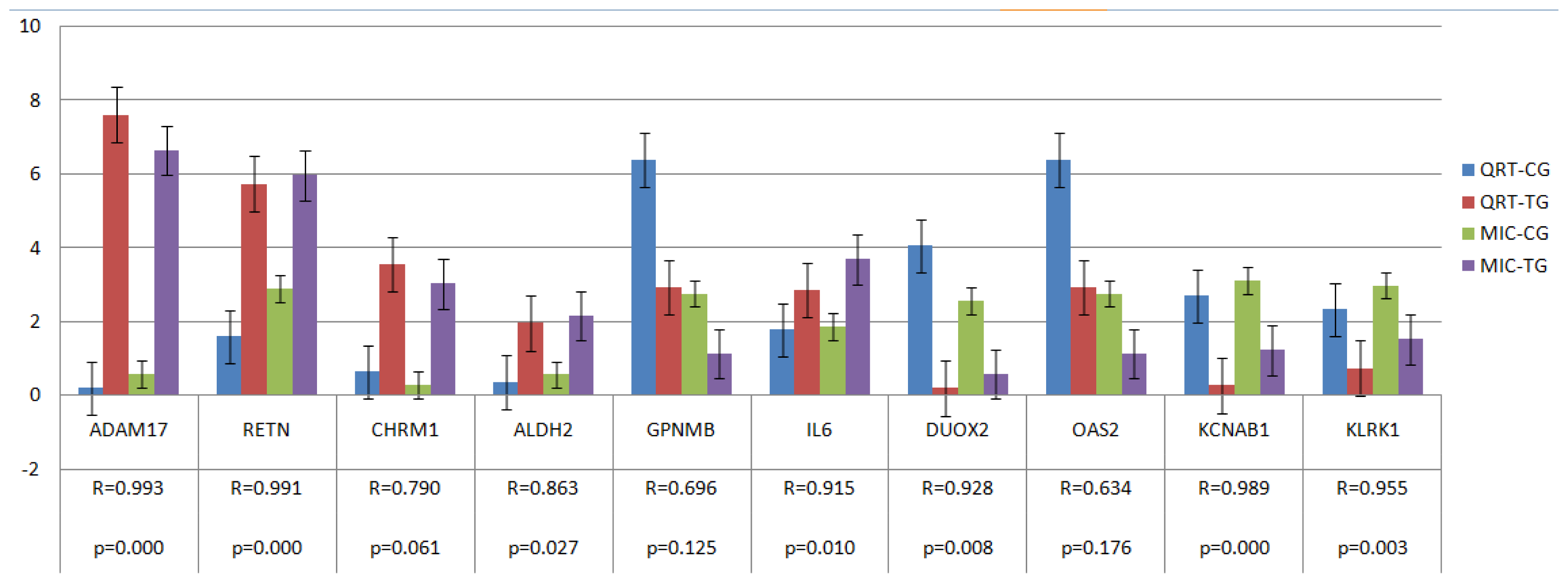
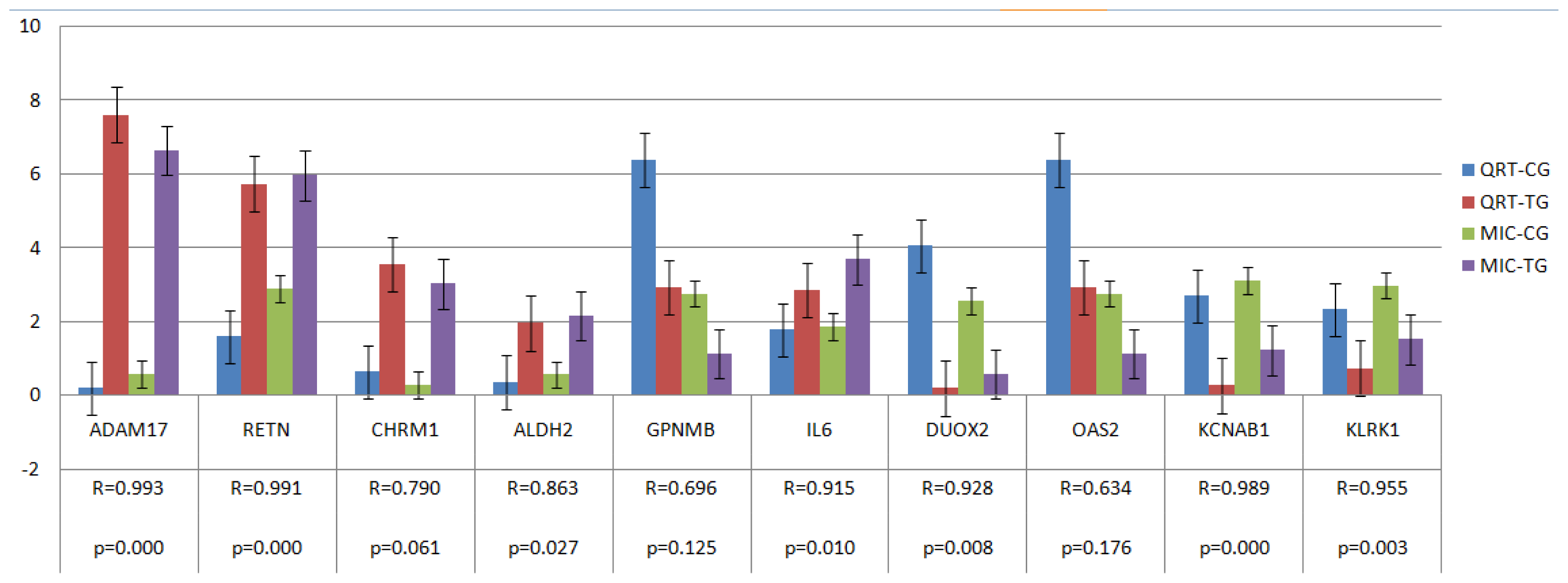
2.4. Verification of Gene Expression Pattern from Microarray Data Using Real-Time QRT-PCR
3. Discussion
4. Experimental Section
4.1. Animals, Bacterial Inoculation and Samples
4.2. Microarray Hybridizations and Data Analysis
4.3. Real-Time QRT-PCR
5. Conclusions
Supplementary Information
ijms-14-23516-s001.pdfAcknowledgments
Conflicts of Interest
References
- Gottschalk, M.; Broes, A.; Mittal, K.R.; Kobisch, M.; Kuhnert, P.; Lebrun, A.; Frey, J. Non-pathogenic Actinobacillus isolates antigenically and biochemically similar to Actinobacillus pleuropneumoniae: A novel species? Vet. Microbiol 2003, 92, 87–101. [Google Scholar]
- Christensen, H.; Bisgaard, M. Revised definition of Actinobacillus sensu strict isolated from animals: A review with special emphasis on diagnosis. Vet. Microbiol 2004, 99, 13–30. [Google Scholar]
- Aarestrup, F.M.; Jensen, N.E. Susceptibility testing of Actinobacillus pleuropneumoniae in Denmark: Evaluation of three different media of MIC-determinations and tablet diffusion tests. Vet. Microbiol 1999, 64, 299–305. [Google Scholar]
- Bossé, J.T.; Janson, H.; Sheehan, B.J.; Beddek, A.J.; Rycroft, A.N.; Kroll, J.S.; Langford, P.R. Actinobacillus pleuropneumoniae: Pathobiology and pathogenesis of infection. Microbes Infect 2002, 4, 225–235. [Google Scholar]
- Gutiérrez-Martín, C.B.; del Blanco, N.G.; Blanco, M.; Navas, J.; Rodríguez-Ferri, E.F. Changes in antimicrobial susceptibility of Acti-nobacillus pleuropneumoniae isolated from pigs in Spain during the last decade. Vet. Microbiol 2006, 115, 218–222. [Google Scholar]
- Matter, D.; Rossano, A.; Limat, S.; Vorlet-Fawer, L.; Brodard, I.; Perreten, V. Antimicrobial resistance profile of Actinobacillus pleurop- neumoniae and Actinobacillus porcitonsillarum. Vet. Microbiol 2007, 122, 146–156. [Google Scholar]
- Rycroft, A.N.; Garside, L.H. Actinobacillus species and their role in animal disease. Vet. J 2000, 159, 18–36. [Google Scholar]
- Taylor, D.J. Actinobacillus pleuropneumoniae. In Diseases of Swine, 8th ed.; Straw, B.E., D’Allaire, S., Mengeling, W.L., Taylor, D.J., Eds.; Iowa State University Press: Ames, IA, USA, 1999; Volume 26, pp. 343–354. [Google Scholar]
- Baarsch, M.J.; Foss, D.L.; Murtaugh, M.P. Pathophysiologic correlates of acute porcine pleuropneumonia. Am. J. Vet. Res 2000, 61, 684–690. [Google Scholar]
- Baarsch, M.J.; Scamurra, R.W.; Burger, K.; Foss, D.L.; Maheswaran, S.K.; Murtaugh, M.P. Inflammatory cytokine expression in swine experimentally infected with Actinobacillus pleuropneumoniae. Infect. Immun 1995, 63, 3587–3594. [Google Scholar]
- Choi, C.; Kwon, D.; Min, K.; Chae, C. In-situ hybridization for the detection of inflammatory cytokines (IL-1, TNF-α and IL-6) in pigs naturally infected with Actinobacillus pleuropneumoniae. J. Comp. Pathol 1999, 121, 349–356. [Google Scholar]
- Huang, H.; Potter, A.A.; Campos, M.; Campos, M.; Leighton, F.A.; Willson, P.J.; Haines, D.M.; Yates, W.D. Pathogenesis of porcine Actinobacillus pleuropneumonia, part II: Roles of proinflammatory cytokines. Can. J. Vet. Res 1999, 63, 69–78. [Google Scholar]
- Wattrang, E.; Wallgren, P.; Fossum, C. Actinobacillus pleuropneumonia serotype 2-effects on the interferon-alpha production of porcine leukocytes in vivo and in vitro. Comp. Immunol. Microbiol. Infect. Dis. 1998, 21, 135–154. [Google Scholar]
- Cho, W.S.; Jung, K.; Kim, J.; Ha, Y.; Chae, C. Expression of mRNA encoding interleukin (IL)-10, IL-12p35 and IL-12p40 in lungs from pigs experimentally infected with Actinobacillus pleuropneumoniae. Vet. Res. Commun 2005, 29, 111–122. [Google Scholar]
- Cho, W.S.; Chae, C. Expression of nitric oxide synthase 2 and tumor necrosis factor alpha in swine naturally infected with Actinobacillus pleuropneumoniae. Vet. Pathol 2002, 39, 27–32. [Google Scholar]
- Moser, R.J.; Reverter, A.; Kerr, C.A.; Beh, K.J.; Lehnert, S.A. A mixedmodel approach for the analysis of cDNA microarray gene expression data from extreme-performing pigs after infection with Actinobacillus pleuropneumoniae. J. Anim. Sci 2004, 82, 1261–1271. [Google Scholar]
- Hedegaard, J.; Skovgaard, K.; Mortensen, S.; Sorensen, P.; Jensen, T.; Hornshoj, H.; Bendixen, C.; Heegaard, P.M.H. Molecular characterisation of the early response in swines to experimental infection with Actinobacillus pleuropneumoniae using cDNA microarrays. Acta Venterinaria Scand 2007, 49, 11. [Google Scholar]
- Mortensen, S.; Skovgaard, K.; Hedegaard, J.; Bendixen, C.; Heegaard, P.M. Transcriptional profiling at different sites in lungs of pigs during acute bacterial respiratory infection. Innate Immun 2011, 17, 41–53. [Google Scholar]
- Zuo, Z.; Cui, H.; Li, M.; Peng, X.; Zhu, L.; Zhang, M.; Ma, J.; Xu, Z.; Gan, M.; Deng, J.; et al. Transcriptional profiling of swine lung tissue after experimental infection with Actinobacillus pleuropneumoniae. Int. J. Mol. Sci 2013, 14, 10626–10660. [Google Scholar]
- Kelly, P.T.; Mackinnon, R.L.; Dietz, R.V.; Maher, B.J.; Wang, J.; et al. Postsynaptic IP3 receptor- mediated Ca2+ release modulates synaptic transmission in hippocampal neurons. Brain Res. Mol. Brain Res 2005, 135, 232–248. [Google Scholar]
- Munoz-Chapuli, R.; Quesada, A.R.; Angel Medina, M. Angiogenesis and signal transduction in endothelial cells. Cell. Mol. Life Sci 2004, 61, 2224–2243. [Google Scholar]
- Zachary, I. VEGF signalling: Integration and multi-tasking in endothelial cell biology. Biochem. Soc. Trans 2003, 31, 1171–1177. [Google Scholar]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. (Lond.) 2005, 109, 227–241. [Google Scholar]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev 2004, 56, 549–580. [Google Scholar]
- Uematsu, S.; Akira, S. Toll-like receptors and innate immunity. J. Mol. Med 2006, 84, 712–725. [Google Scholar]
- Arbibe, L.; Mira, J.P.; Teusch, N.; Kline, L.; Guha, M.; Mackman, N.; Godowski, P.J.; Ulevitch, R.J.; Knaus, U.G. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol 2000, 1, 533–540. [Google Scholar]
- Uematsu, S.; Akira, S. Toll-like receptors and Type I interferons. J. Biol. Chem 2007, 282, 15319–15323. [Google Scholar]
- Shaw, M.H.; Reimer, T.; Kim, Y.G.; Nunez, G. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Curr. Opin. Immunol 2008, 20, 377–382. [Google Scholar]
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar]
- Saito, T.; Gale, M., Jr. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med 2008, 205, 1523–1527. [Google Scholar]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev 2009, 227, 54–65. [Google Scholar]
- Ulkü, A.S.; Der, C.J. Ras signaling, deregulation of gene expression and oncogenesis. Cancer Treat. Res 2003, 115, 189–208. [Google Scholar]
- Reuther, G.W.; Der, C.J. The Ras branch of small GTPases: Ras family members don’t fall far from the tree. Curr. Opin. Cell Biol 2000, 12, 157–165. [Google Scholar]
- Brock, C.; Schaefer, M.; Reusch, H.P.; Reusch, H.P.; Czupalla, C.; Michalke, M.; Spicher, K.; Schultz, G.; Nürnberg, B. Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J. Cell Biol 2003, 160, 89–99. [Google Scholar]
- Voigt, P.; Brock, C.; Nürnberg, B.; Schaefer, M. Assigning functional domains within the p101 regulatory subunit of phosphoinositide 3-kinase gamma. J. Biol. Chem 2005, 280, 5121–5127. [Google Scholar]
- Stephens, L.; Ellson, C.; Hawkins, P. Roles of PI3Ks in leukocytes chemotaxis and phagocytosis. Curr. Opin. Cell Biol 2002, 14, 203–213. [Google Scholar]
- Wymann, M.P.; Zvelebil, M.; Laffargue, M. Phosphoinositide 3-kinase signalling-which way to target? Trends Pharmacol. Sci 2003, 24, 366–376. [Google Scholar]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; et al. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science 2000, 287, 1049–1053. [Google Scholar]
- Crackower, M.A.; Oudit, G.Y.; Kozieradzki, I.; Sarao, R.; Sun, H.; Sasaki, T.; Hirsch, E.; Suzuki, A.; Shioi, T.; Irie-Sasaki, J.; et al. Regulation of yocardial contractibility and cell size by distinct PI3K-PTEN signaling pathways. Cell 2002, 110, 737–749. [Google Scholar]
- Gesslein, B.; Hakansson, G.; Carpio, R.; Gustafsson, L.; Perez, M.T.; Malmsjö, M. Mitogen-activated protein kinases in the porcine retinal arteries and neuroretina following retinal ischemia-reperfusion. Mol. Vis 2010, 16, 392–407. [Google Scholar]
- Abbas, A.K.; Lichtman, A.H. Antigen Capture and Presentation to Lymphocytes. In Basic Immunology: Functions and Disorders of the Immune System, 3rd ed.; Saunders Elsevier: Philadelphia, PA, USA, 2009; pp. 49–70. [Google Scholar]
- Cheng, G.; Schoenberger, S.P. CD40 signaling and autoimmunity. Curr. Dir. Autoimmun 2002, 5, 51–61. [Google Scholar]
- Dallman, C.; Johnson, P.W.; Packham, G. Differential regulation of cell survival by CD40. Apoptosis 2003, 8, 45–53. [Google Scholar]
- O’Sullivan, B.; Thomas, R. Recent advances on the role of CD40 and dendritic cells in immunity and tolerance. Curr. Opin. Hematol 2004, 10, 272–278. [Google Scholar]
- Van de Vosse, E.; Ottenhoff, H.M. Human host genetic factors in mycobacterial and Salmonella infection: Lessons from single gene disorders in IL-12/IL-23-dependent signaling that affect innate and adaptive immunity. Microbes Infect 2006, 8, 1167–1173. [Google Scholar]
- De Jong, R.; Altare, F.; Haagen, I.A.; Elferink, D.G.; Boer, T.; van Breda Vriesman, P.J.C.; Kabel, P.J.; Draaisma, J.M.; van Dissel, J.T.; Kroon, F.P.; et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 1998, 280, 1435–1438. [Google Scholar]
- Picard, C.; Fieschi, C.; Altare, F.; Al-Jumaah, S.; Al-Hajjar, S.; Feinberg, J.; Dupuis, S.; Soudais, C.; Al-Mohsen, I.Z.; Génin, E.; et al. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am. J. Hum. Genet 2002, 70, 336–348. [Google Scholar]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar]
- Palma, M.; DeLuca, D.; Worgall, S.; Quadri, L.E.N. Transcriptome analysis of the response of Pseudomonas aeruginosa to hydrogen peroxide. J. Bacteriol 2004, 186, 248–252. [Google Scholar]
- Transcriptional profiling of swine lung and hilar node after experimental infection with Actinobacillus pleuropneumoniae. Available online: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE42317 (accessed on 15 November 2012).
- Hu, J.; He, X. Enhanced quantile normalization of microarray data to reduce loss of information in gene expression profiles. Biometrics 2007, 63, 50–59. [Google Scholar]
- Xia, X.; McClelland, M.; Wang, Y. WebArray: An online platform for microarray data analysis. BMC Bioinforma 2005, 6, 306. [Google Scholar]
- Romualdi, C.; Vitulo, N.; Favero, M.D.; Lanfranchi, G. MIDAW: A web tool for statistical analysis of microarray data. Nucleic Acids Res 2005, 33, 644–649. [Google Scholar]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PCG-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 2003, 34, 267–273. [Google Scholar]
- Bild, A.; Febbo, P.G. Application of a priori established gene sets to discover biologically important differential expression in microarray data. Proc. Natl. Acad. Sci. USA 2005, 102, 15278–15279. [Google Scholar]
- Aoki-Kinoshita, K.F.; Kanehisa, M. Gene annotation and pathway mapping in KEGG. Methods Mol. Biol 2007, 396, 71–91. [Google Scholar]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol 2003, 4, P3. [Google Scholar]
- Lee, H.K.; Braynen, W.; Keshav, K.; Pavlidis, P. ErmineJ: Tool for functional analysis of gene expression data sets. BMC Bioinforma 2005, 6, 269. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Dopazo, J. Functional interpretation of microarray experiments. OMICS 2006, 10, 398–410. [Google Scholar]
- Gene Set Enrichment Analysis. Available online: http://www.broadinstitute.org/gsea/index.jsp (accessed on 15 October 2012).
- Gene Expression Omnibus. Available online: http://www.ncbi.nih.gov/geo/ (accessed on 7 July 2007).
- Barrett, T.; Suzek, T.O.; Troup, D.B.; Wilhite, S.E.; Ngau, W.C.; Ledoux, P.; Rudnev, D.; Lash, A.E.; Fujibuchi, W.; Edgar, R. NCBI GEO: Mining millionsof expression profiles-database and tools. Nucleic Acids Res 2005, 33, 562–566. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002, 30, 207–210. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Principal Component | Eigenvalues | Contribution ratio |
|---|---|---|
| 1 | 42.273 | 97.16% |
| 2 | 0.978 | 2.25% |
| 3 | 0.147 | 0.34% |
| 4 | 0.072 | 0.17% |
| 5 | 0.025 | 0.06% |
| 6 | 0.014 | 0.03% |
| Confirmation objects | Gene symbol | Primer sequence (5′→3′) | Amplicon length (bp) | Ta (°C) | GenBank No. |
|---|---|---|---|---|---|
| Reference gene | ACTB | TCTGGCACCACACCTTCT | 114 | 60 | DQ178122 |
| TGATCTGGGTCATCTTCTCAC | |||||
| TBP | GATGGACGTTCGGTTTAGG | 124 | 60 | DQ178129 | |
| AGCAGCACAGTACGAGCAA | |||||
| TOP2B | AACTGGATGATGCTAATGATGCT | 137 | 60 | AF222921 | |
| TGGAAAAACTCCGTATCTGTCTC | |||||
| Up gene | RETN | AGTGCGCTGGCATAGACTGG | 197 | 60 | NM_213783 |
| CATCCTCTTCTCAAGGTTTATTTCC | |||||
| ADAM17 | TTGAGGAAGGGGAAGCC | 158 | 56 | NM_001099926 | |
| ACGGAGCCCACGATGTT | |||||
| GPNMB | GAGACCCAGCCTTCCTT | 130 | 51.2 | NM_001098584 | |
| TTGCTTTCTATCGCTTTGTA | |||||
| CHRM1 | CGCTGGTCAAGGAGAAGAA | 185 | 56 | NM_214034 | |
| GCACATGGGGTTGATGGT | |||||
| ALDH2 | AAACTGCTCTGCGGTGGA | 181 | 56 | NM_001044611 | |
| CGTACTTGGAATTGTTGGCTC | |||||
| IL6 | GTCGAGGCTGTGCAGATTAG | 101 | 56 | NM_214399 | |
| GCATTTGTGGTGGGGTTAG | |||||
| Down gene | KLRK1 | TGATGTGATAAACCGTGGTG | 107 | 56 | NM_213813 |
| TGGATCGGGCAAGGAAA | |||||
| DUOX2 | CCCTTCTTCAACTCCCTG | 158 | 51.2 | NM_213999 | |
| CAAAAGTTCTCATAGTGGTGC | |||||
| OAS2 | GACACGGCTGAAGGTTT | 291 | 51.2 | NM_001031796 | |
| TGGCACGTCCCAAGACT | |||||
| KCNAB1 | AAGGGAGAAAACAGCAAAAC | 176 | 56 | NM_001105294 | |
| AACCTGAATGGCACCGA | |||||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, S.; Zuo, Z.; Cui, H.; Li, M.; Peng, X.; Zhu, L.; Zhang, M.; Li, X.; Xu, Z.; Gan, M.; et al. Transcriptional Profiling of Hilar Nodes from Pigs after Experimental Infection with Actinobacillus Pleuropneumoniae. Int. J. Mol. Sci. 2013, 14, 23516-23532. https://doi.org/10.3390/ijms141223516
Yu S, Zuo Z, Cui H, Li M, Peng X, Zhu L, Zhang M, Li X, Xu Z, Gan M, et al. Transcriptional Profiling of Hilar Nodes from Pigs after Experimental Infection with Actinobacillus Pleuropneumoniae. International Journal of Molecular Sciences. 2013; 14(12):23516-23532. https://doi.org/10.3390/ijms141223516
Chicago/Turabian StyleYu, Shumin, Zhicai Zuo, Hengmin Cui, Mingzhou Li, Xi Peng, Ling Zhu, Ming Zhang, Xuewei Li, Zhiwen Xu, Meng Gan, and et al. 2013. "Transcriptional Profiling of Hilar Nodes from Pigs after Experimental Infection with Actinobacillus Pleuropneumoniae" International Journal of Molecular Sciences 14, no. 12: 23516-23532. https://doi.org/10.3390/ijms141223516