Small Molecule Binding, Docking, and Characterization of the Interaction between Pth1 and Peptidyl-tRNA


Abstract
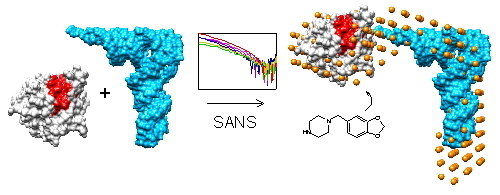
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
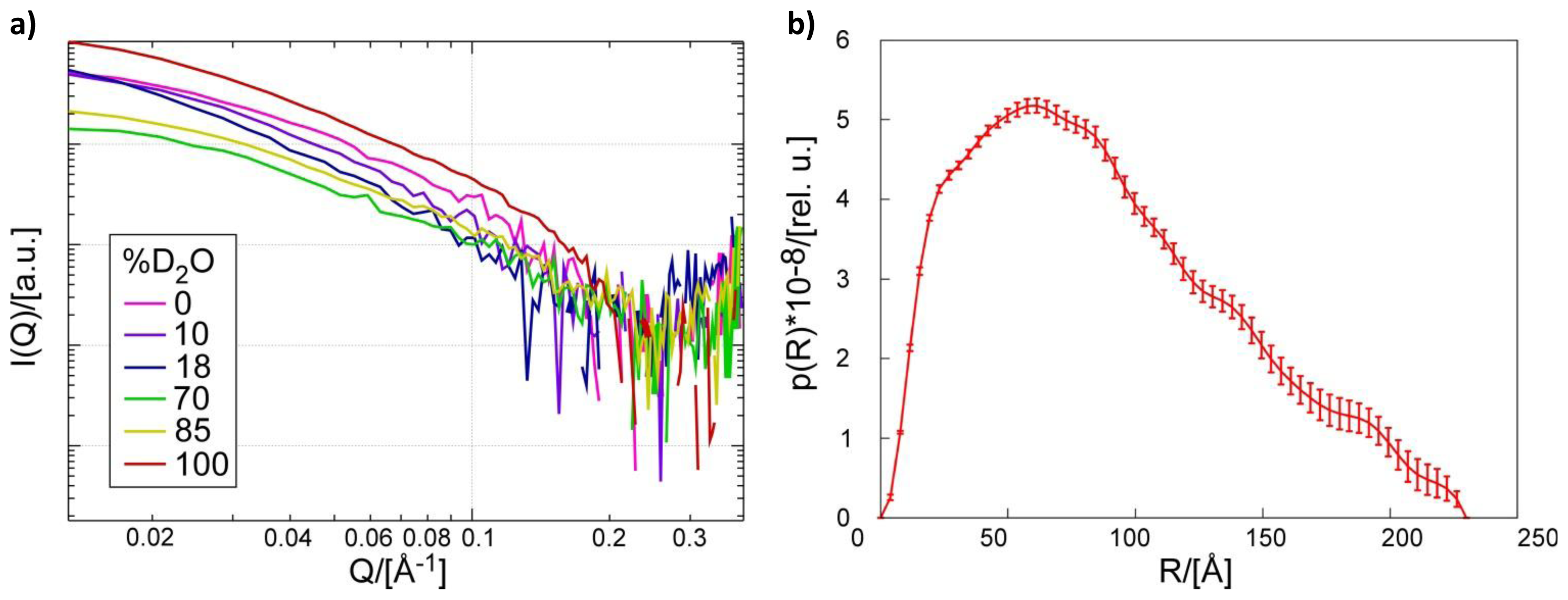
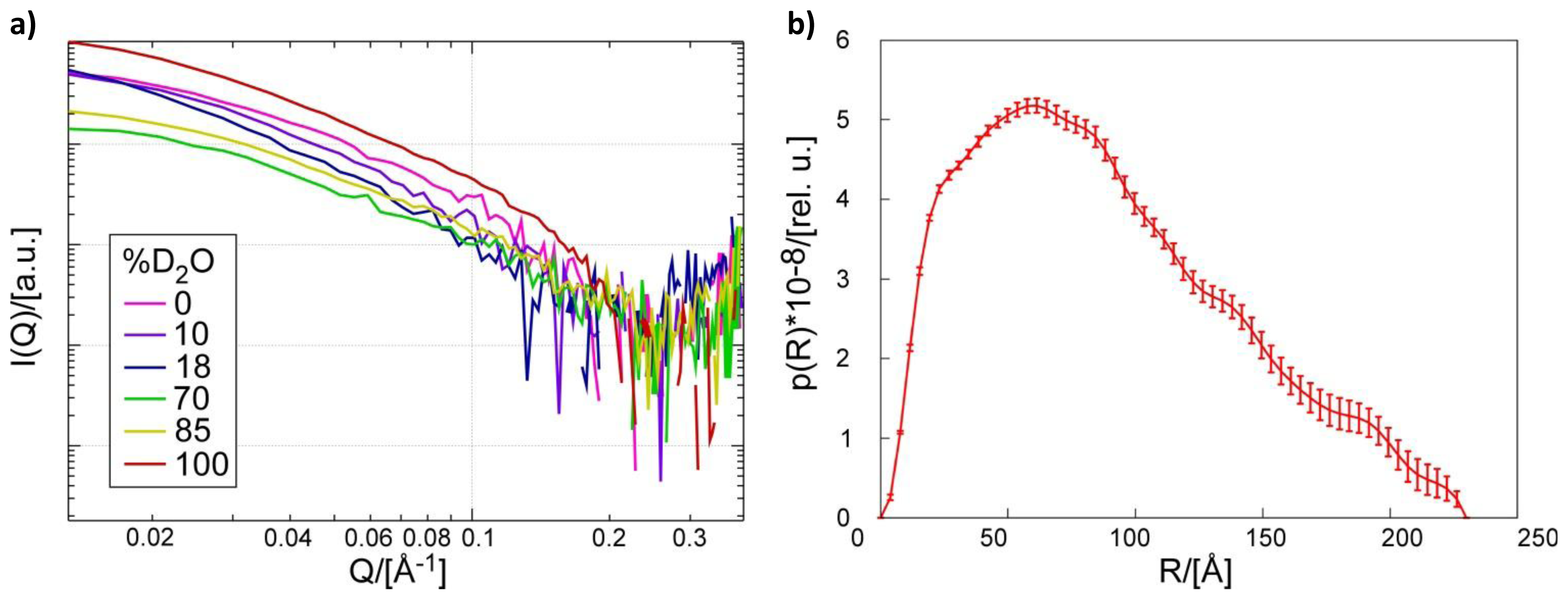
2.1. Small Angle Neutron Scattering
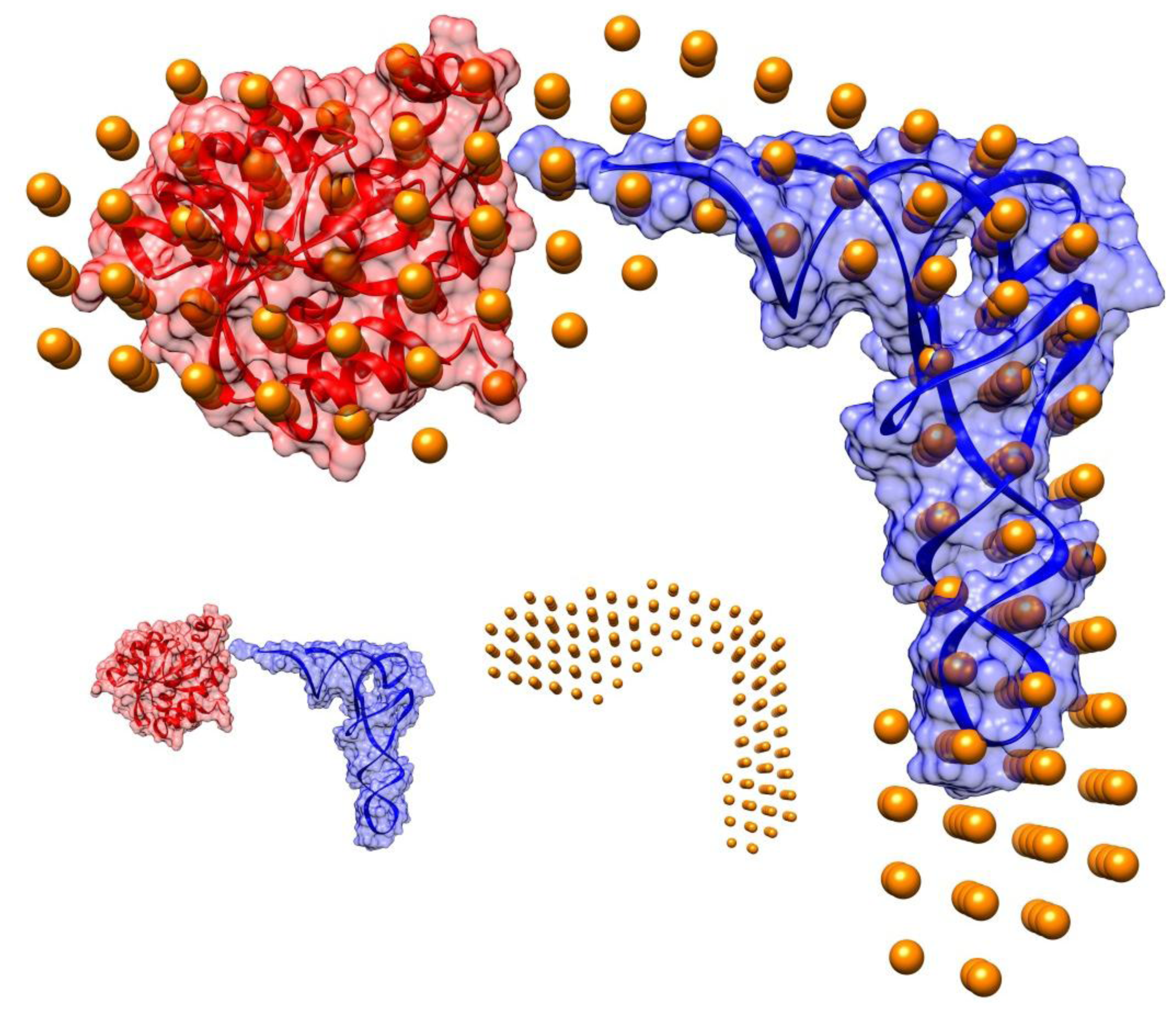
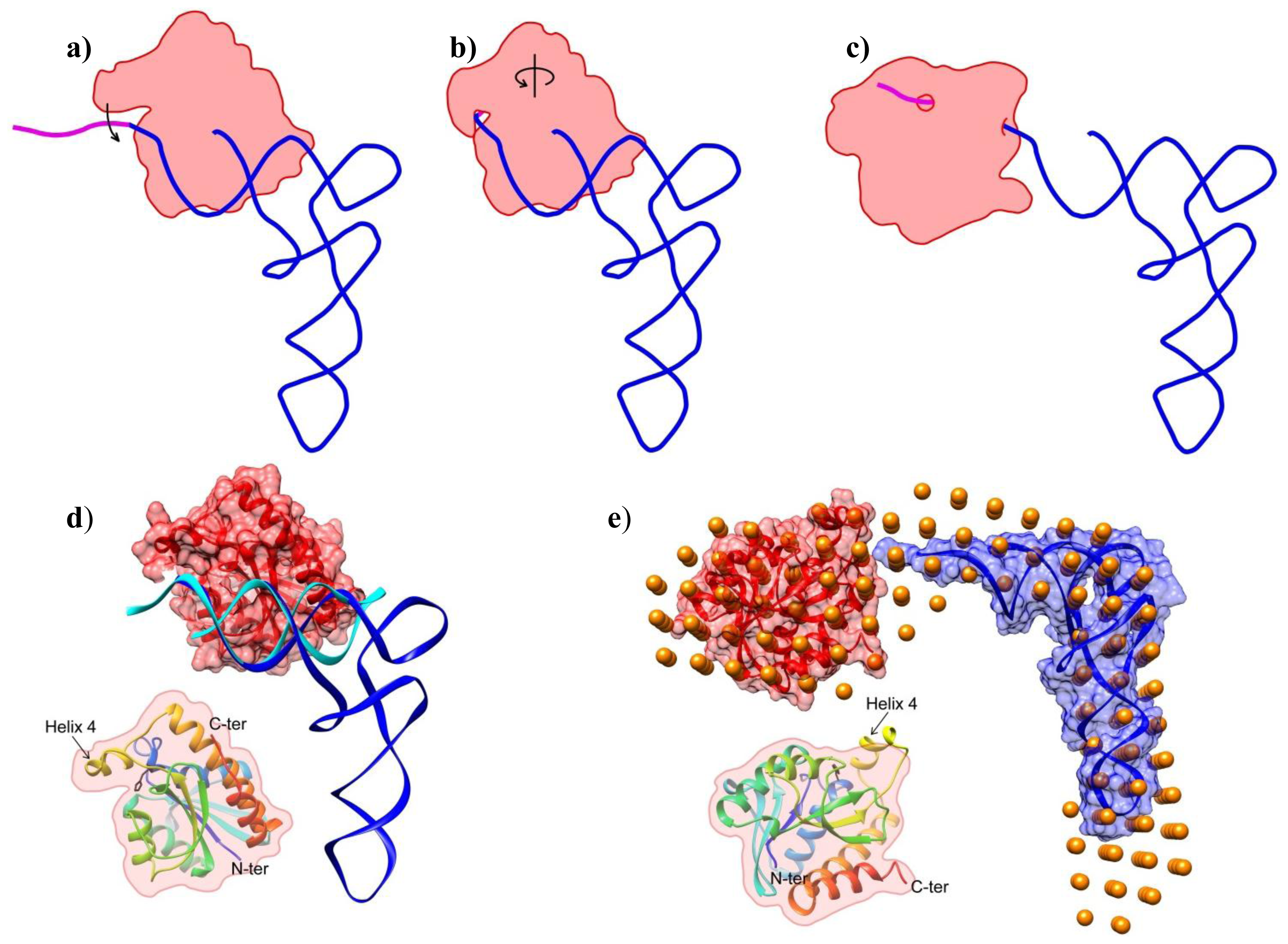
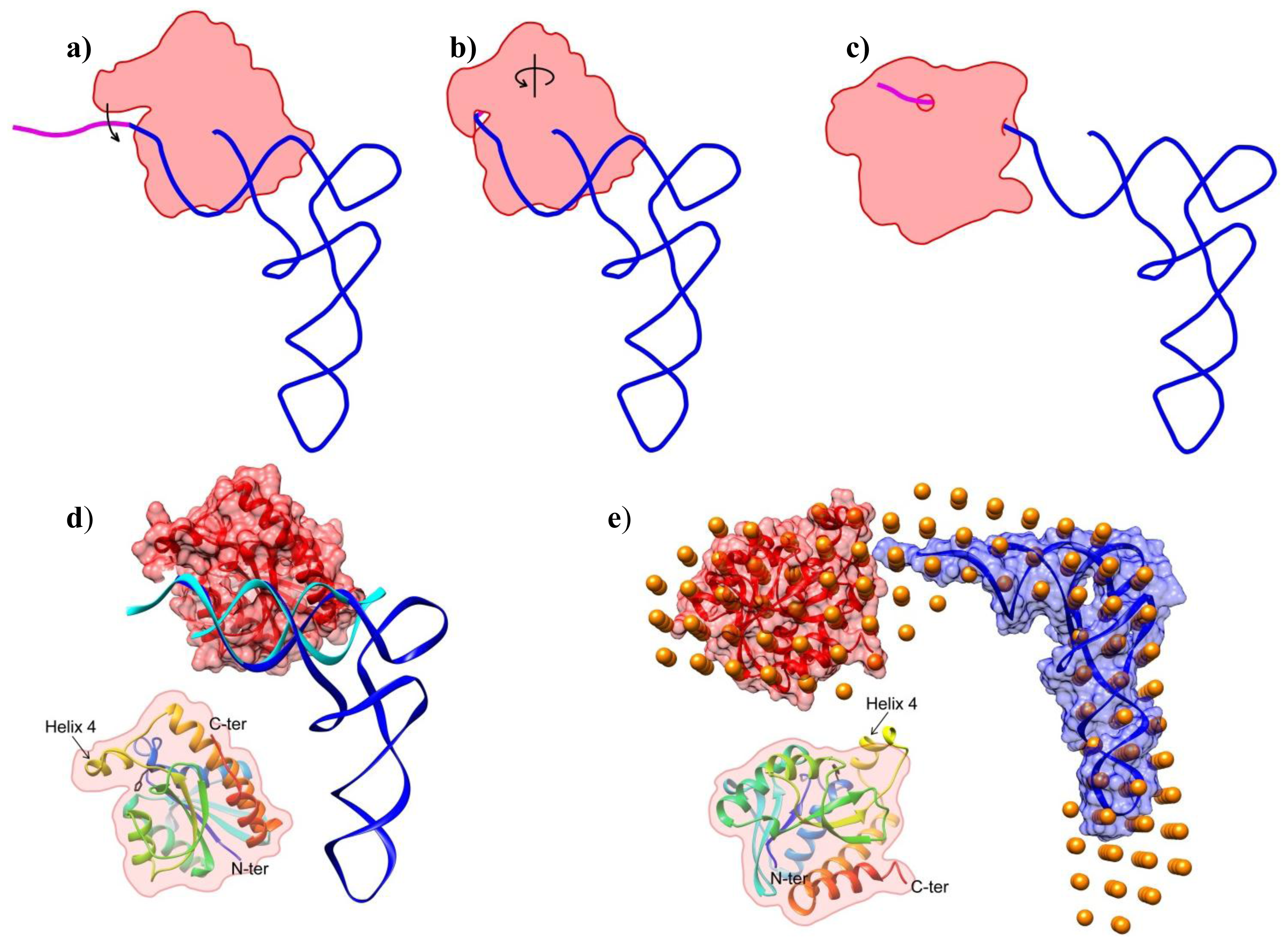
2.2. Shape of the Pth1:peptidyl-tRNA Complex and Their Relative Orientation
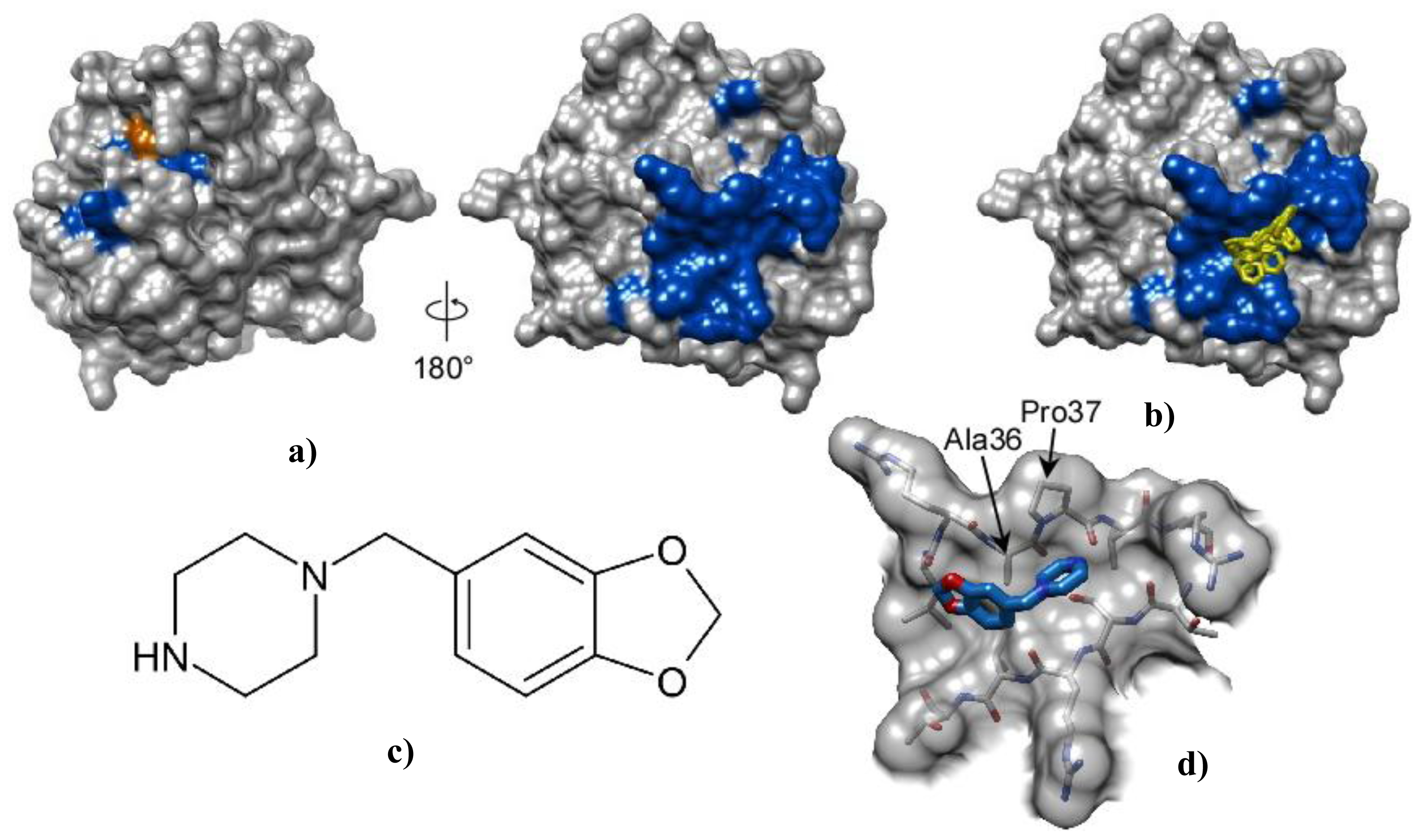
2.3. Piperonylpiperazine Binding and Interaction with Pth1
3. Experimental Section
3.1. Expression and Purification of E. coli Pth1
3.2. Production of Bulk Peptidyl-tRNAs
3.3. Preparation of Pth1:peptidyl-tRNA Complex
3.4. Dynamic Light Scattering
3.5. Small Angle Neutron Scattering of the Pth1:peptidyl-tRNAComplex
3.6. Overall Shape Determination
3.7. Chemical Shift Perturbation Mapping of Piperonylpiperazine Binding to Pth1
3.8. Computational Docking
4. Conclusions


Acknowledgments
Conflicts of Interest
References
- Jorgensen, F.; Kurland, C.G. Processivity errors of gene expression in Escherichia coli. J. Mol. Biol 1990, 215, 511–521. [Google Scholar]
- Manley, J.L. Synthesis and degradation of termination and premature-termination fragments of beta-galactosidase in vitro and in vivo. J. Mol. Biol. 1978, 125, 407–432. [Google Scholar]
- Kurland, C.G.; Ehrenberg, M. Constraints on the accuracy of messenger RNA movement. Q. Rev. Biophys 1985, 18, 423–450. [Google Scholar]
- Heurgue-Hamard, V.; Karimi, R.; Mora, L.; MacDougall, J.; Leboeuf, C.; Grentzmann, G.; Ehrenberg, M.; Buckingham, R.H. Ribosome release factor RF4 and termination factor RF3 are involved in dissociation of peptidyl-tRNA from the ribosome. EMBO J 1998, 17, 808–816. [Google Scholar]
- Karimi, R.; Pavlov, M.Y.; Heurgue-Hamard, V.; Buckingham, R.H.; Ehrenberg, M. Initiation factors IF1 and IF2 synergistically remove peptidyl-tRNAs with short polypeptides from the P-site of translating Escherichia coli ribosomes. J. Mol. Biol 1998, 281, 241–252. [Google Scholar]
- Menninger, J.R. The accumulation as peptidyl-transfer RNA of isoaccepting transfer RNA families in Escherichia coli with temperature-sensitive peptidyl-transfer RNA hydrolase. J. Biol. Chem 1978, 253, 6808–6813. [Google Scholar]
- Cruz-Vera, L.R.; Hernandez-Ramon, E.; Perez-Zamorano, B.; Guarneros, G. The rate of peptidyl-tRNA dissociation from the ribosome during minigene expression depends on the nature of the last decoding interaction. J. Biol. Chem 2003, 278, 26065–26070. [Google Scholar]
- Hernandez-Sanchez, J.; Valadez, J.G.; Herrera, J.V.; Ontiveros, C.; Guarneros, G. Lambda bar minigene-mediated inhibition of protein synthesis involves accumulation of peptidyl-tRNA and starvation for tRNA. EMBO J 1998, 17, 3758–3765. [Google Scholar]
- Tenson, T.; Herrera, J.V.; Kloss, P.; Guarneros, G.; Mankin, A.S. Inhibition of translation and cell growth by minigene expression. J. Bacteriol 1999, 181, 1617–1622. [Google Scholar]
- Rosas-Sandoval, G.; Ambrogelly, A.; Rinehart, J.; Wei, D.; Cruz-Vera, L.R.; Graham, D.E.; Stetter, K.O.; Guarneros, G.; Soll, D. Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 16707–16712. [Google Scholar]
- Gross, M.; Crow, P.; White, J. The site of hydrolysis by rabbit reticulocyte peptidyl-tRNA hydrolase is the 3′-AMP terminus of susceptible tRNA substrates. J. Biol. Chem 1992, 267, 2080–2086. [Google Scholar]
- Schulman, L.H.; Pelka, H. The structural basis for the resistance of Escherichia coli formylmethionyl transfer ribonucleic acid to cleavage by Escherichia coli peptidyl transfer ribonucleic acid hydrolase. J. Biol. Chem 1975, 250, 542–547. [Google Scholar]
- Dutka, S.; Meinnel, T.; Lazennec, C.; Mechulam, Y.; Blanquet, S. Role of the 1–72 base pair in tRNAs for the activity of Escherichia coli peptidyl-tRNA hydrolase. Nucleic Acids Res 1993, 21, 4025–4030. [Google Scholar]
- Fromant, M.; Schmitt, E.; Mechulam, Y.; Lazennec, C.; Plateau, P.; Blanquet, S. Crystal structure at 1.8 Å resolution and identification of active site residues of Sulfolobus solfataricus peptidyl-tRNA hydrolase. Biochemistry 2005, 44, 4294–4301. [Google Scholar]
- Pulavarti, S.V.; Jain, A.; Pathak, P.P.; Mahmood, A.; Arora, A. Solution structure and dynamics of peptidyl-tRNA hydrolase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol 2008, 378, 165–177. [Google Scholar]
- Selvaraj, M.; Roy, S.; Singh, N.S.; Sangeetha, R.; Varshney, U.; Vijayan, M. Structural plasticity and enzyme action: Crystal structures of Mycobacterium tuberculosis peptidyl-tRNA hydrolase. J. Mol. Biol 2007, 372, 186–193. [Google Scholar]
- Schmitt, E.; Fromant, M.; Plateau, P.; Mechulam, Y.; Blanquet, S. Crystallization and preliminary X-ray analysis of Escherichia coli peptidyl-tRNA hydrolase. Proteins 1997, 28, 135–136. [Google Scholar]
- Hughes, R.C.; McFeeters, H.; Coates, L.; McFeeters, R.L. Recombinant production, crystallization and X-ray crystallographic structure determination of the peptidyl-tRNA hydrolase of Pseudomonas aeruginosa. Acta Crystallogr. F 2012, 68, 1472–1476. [Google Scholar]
- Clarke, T.E.; Romanov, V.; Lam, R.; Gothe, S.A.; Peddi, S.R.; Razumova, E.B.; Lipman, R.S.; Branstrom, A.A.; Chirgadze, N.Y. Structure of Francisella tularensis peptidyl-tRNA hydrolase. Acta crystallogr. F 2011, 67, 446–449. [Google Scholar]
- Giorgi, L.; Bontems, F.; Fromant, M.; Aubard, C.; Blanquet, S.; Plateau, P. RNA-binding site of Escherichia coli peptidyl-tRNA hydrolase. J. Biol. Chem 2011, 286, 39585–39594. [Google Scholar]
- Giorgi, L.; Plateau, P.; O’Mahony, G.; Aubard, C.; Fromant, M.; Thureau, A.; Grotli, M.; Blanquet, S.; Bontems, F. NMR-based substrate analog docking to Escherichia coli Peptidyl-tRNA hydrolase. J. Mol. Biol 2011, 412, 619–633. [Google Scholar]
- Ito, K.; Murakami, R.; Mochizuki, M.; Qi, H.; Shimizu, Y.; Miura, K.; Ueda, T.; Uchiumi, T. Structural basis for the substrate recognition and catalysis of peptidyl-tRNA hydrolase. Nucleic Acids Res 2012, 40, 10521–10531. [Google Scholar]
- Harris, S.M.; McFeeters, H.; Ogungbe, I.V.; Cruz-Vera, L.R.; Setzer, W.N.; Jackes, B.R.; McFeeters, R.L. Peptidyl-tRNA hydrolase screening combined with molecular docking reveals the antibiotic potential of Syzygium johnsonii bark extract. Nat. Prod. Commun 2011, 6, 1421–1424. [Google Scholar]
- McFeeters, H.; Gilbert, M.J.; Thompson, R.M.; Setzer, W.N.; Cruz-Vera, L.R.; McFeeters, R.L. Inhibition of essential bacterial peptidyl-tRNA hydrolase activity by tropical plant extracts. Nat. Prod. Commun 2012, 7, 1107–1110. [Google Scholar]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr 1992, 25, 495–503. [Google Scholar]
- Goodall, J.J.; Chen, G.J.; Page, M.G. Essential role of histidine 20 in the catalytic mechanism of Escherichia coli peptidyl-tRNA hydrolase. Biochemistry 2004, 43, 4583–4591. [Google Scholar]
- Schmitt, E.; Mechulam, Y.; Fromant, M.; Plateau, P.; Blanquet, S. Crystal structure at 1.2 Å resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J 1997, 16, 4760–4769. [Google Scholar]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst 2001, 34, 33–41. [Google Scholar]
- Giege, R. Toward a more complete view of tRNA biology. Nat. Struct. Mol. Biol 2008, 15, 1007–1014. [Google Scholar]
- Alexander, R.W.; Eargle, J.; Luthey-Schulten, Z. Experimental and computational determination of tRNA dynamics. FEBS Lett 2010, 584, 376–386. [Google Scholar]
- Atherly, A.G.; Menninger, J.R. Mutant E. coli strain with temperature sensitive peptidyl-transfer RNA hydrolase. Nat. New Biol 1972, 240, 245–246. [Google Scholar]
- Cruz-Vera, L.R.; Toledo, I.; Hernandez-Sanchez, J.; Guarneros, G. Molecular basis for the temperature sensitivity of Escherichia coli pth(Ts). J. Bacteriol 2000, 182, 1523–1528. [Google Scholar]
- Varshney, U.; Lee, C.P.; RajBhandary, U.L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem 1991, 266, 24712–24718. [Google Scholar]
- Wignall, G.D.; Bates, F.S. Absolute calibration of small-angle neutron scattering data. J. Appl. Crystallogr 1987, 20, 28–40. [Google Scholar]
- Guinier, A. La diffraction des rayons X aux tres petits angles: Applications a l’etude de phenomenes ultramicroscopiques. Annales de Physique 1939, 12.1939, 161–237. [Google Scholar]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J 1999, 76, 2879–2886. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model 1999, 17, 57–61. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar]
- Falb, M.; Amata, I.; Gabel, F.; Simon, B.; Carlomagno, T. Structure of the K-turn U4 RNA: A combined NMR and SANS study. Nucleic Acids Res 2010, 38, 6274–6285. [Google Scholar]
- Grishaev, A.; Ying, J.; Canny, M.D.; Pardi, A.; Bax, A. Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data. J. Biomol. NMR 2008, 42, 99–109. [Google Scholar]
- Moroder, H.; Steger, J.; Graber, D.; Fauster, K.; Trappl, K.; Marquez, V.; Polacek, N.; Wilson, D.N.; Micura, R. Non-hydrolyzable RNA-peptide conjugates: A powerful advance in the synthesis of mimics for 3′-peptidyl tRNA termini. Angewandte Chemie 2009, 48, 4056–4060. [Google Scholar]
- Geiermann, A.S.; Micura, R. Selective desulfurization significantly expands sequence variety of 3′-peptidyl-tRNA mimics obtained by native chemical ligation. Chembiochem Eur. J. Chem. Biol 2012, 13, 1742–1745. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hames, M.C.; McFeeters, H.; Holloway, W.B.; Stanley, C.B.; Urban, V.S.; McFeeters, R.L. Small Molecule Binding, Docking, and Characterization of the Interaction between Pth1 and Peptidyl-tRNA. Int. J. Mol. Sci. 2013, 14, 22741-22752. https://doi.org/10.3390/ijms141122741
Hames MC, McFeeters H, Holloway WB, Stanley CB, Urban VS, McFeeters RL. Small Molecule Binding, Docking, and Characterization of the Interaction between Pth1 and Peptidyl-tRNA. International Journal of Molecular Sciences. 2013; 14(11):22741-22752. https://doi.org/10.3390/ijms141122741
Chicago/Turabian StyleHames, Mary C., Hana McFeeters, W. Blake Holloway, Christopher B. Stanley, Volker S. Urban, and Robert L. McFeeters. 2013. "Small Molecule Binding, Docking, and Characterization of the Interaction between Pth1 and Peptidyl-tRNA" International Journal of Molecular Sciences 14, no. 11: 22741-22752. https://doi.org/10.3390/ijms141122741