Synthesis and Characterization of Ferroelectric Liquid Crystalline Organosiloxanes Containing 4-(4-undecanyloxy bi-phenyl-1-carboxyloxy)phenyl (2S,3S)-2-chloro-3-methylvalerate and 4-(4-undecanyloxybenzoyloxy)biphenyl (2S,3S)-2-chloro-3-methylvalerate

Abstract
:1. Introduction
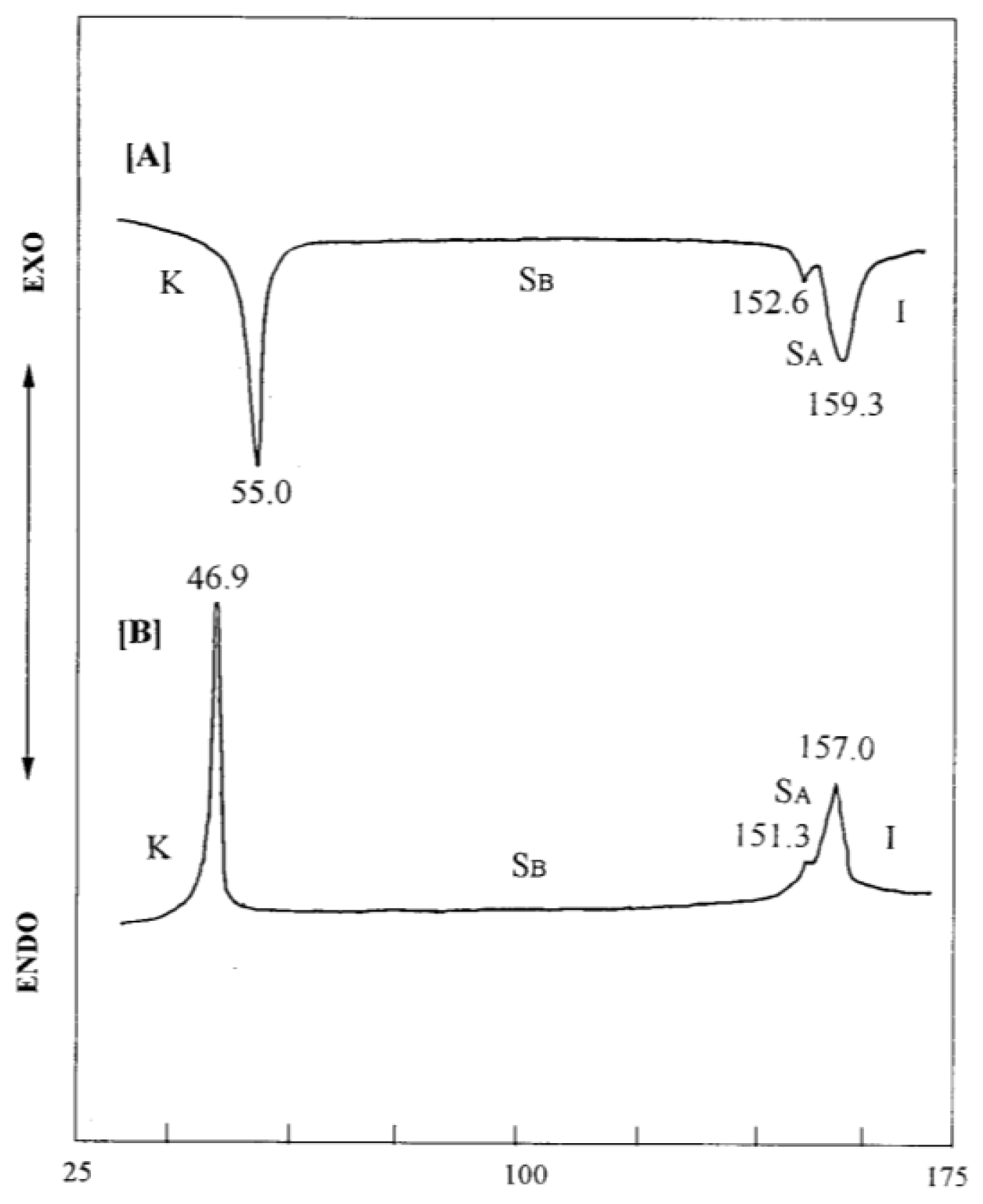
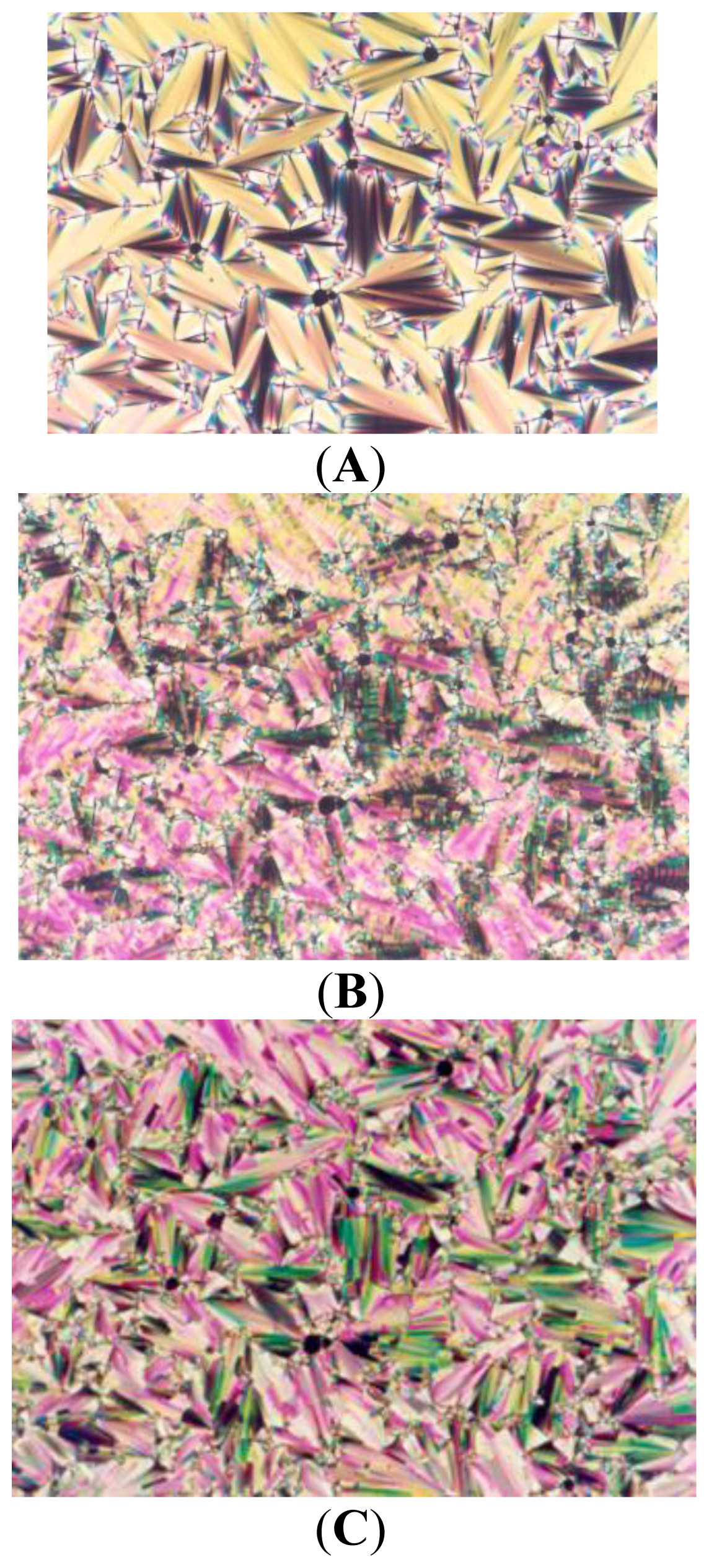
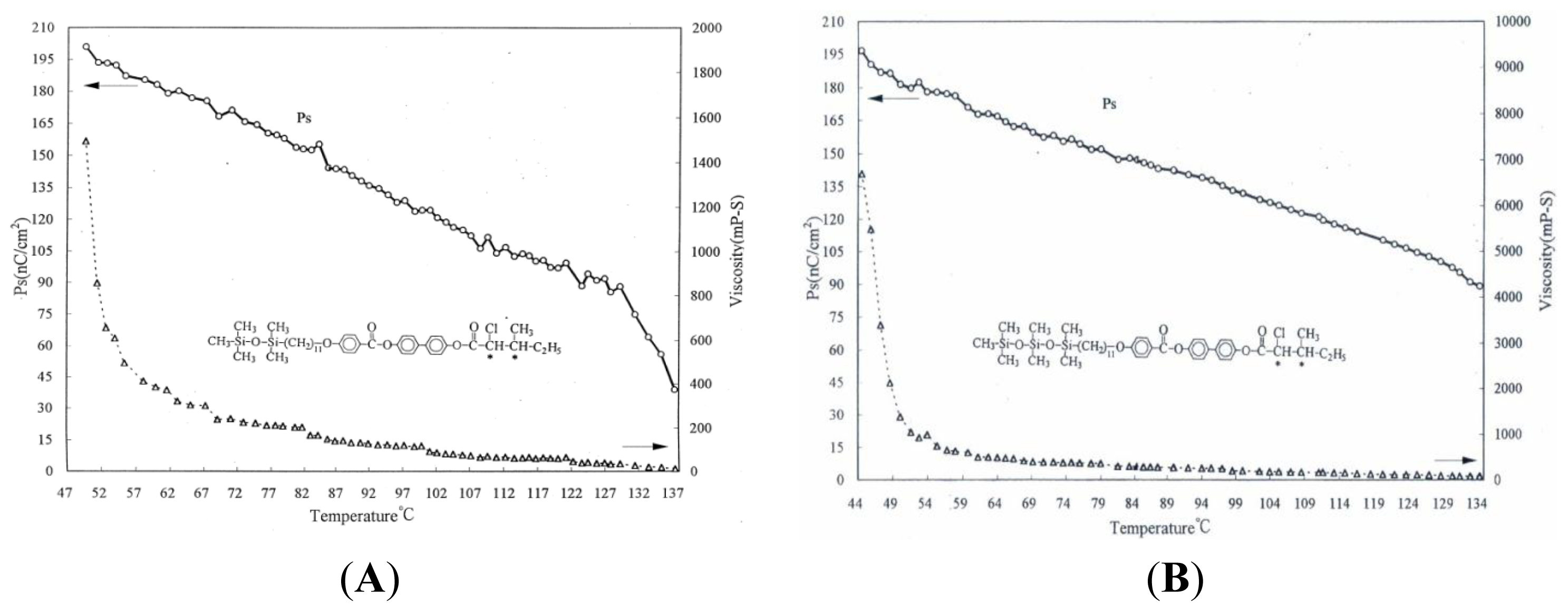
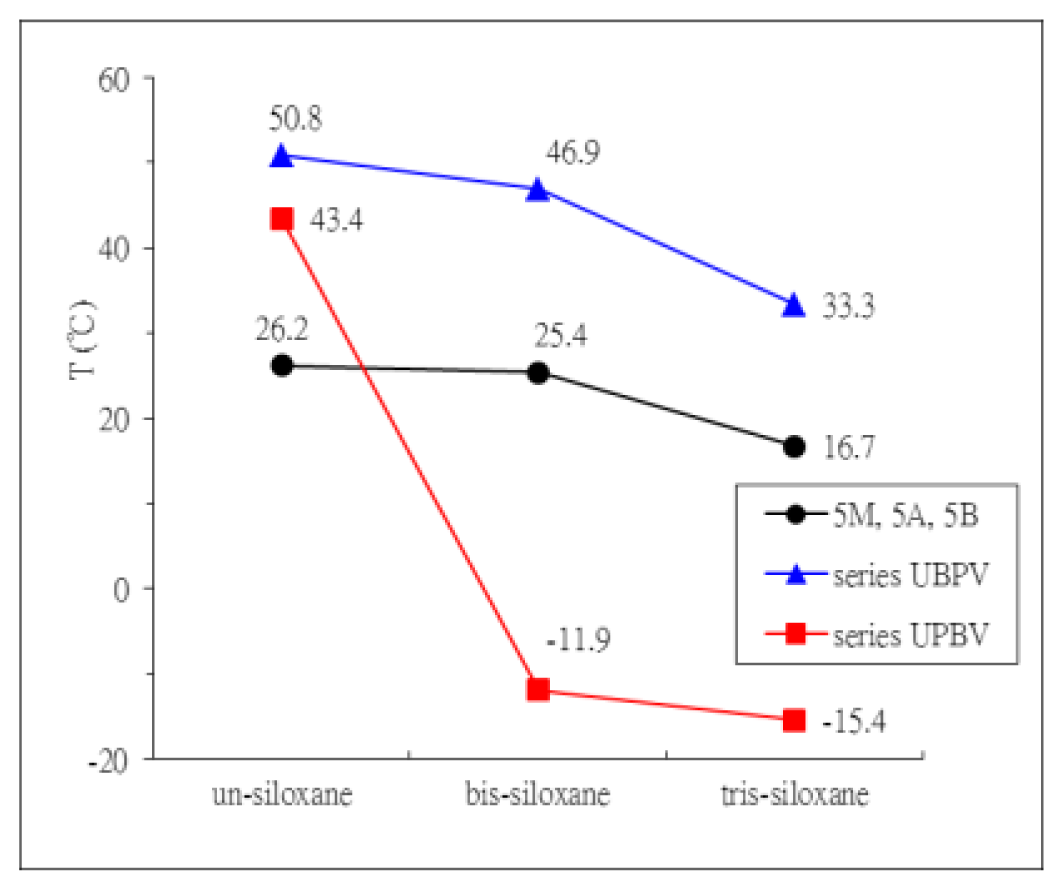
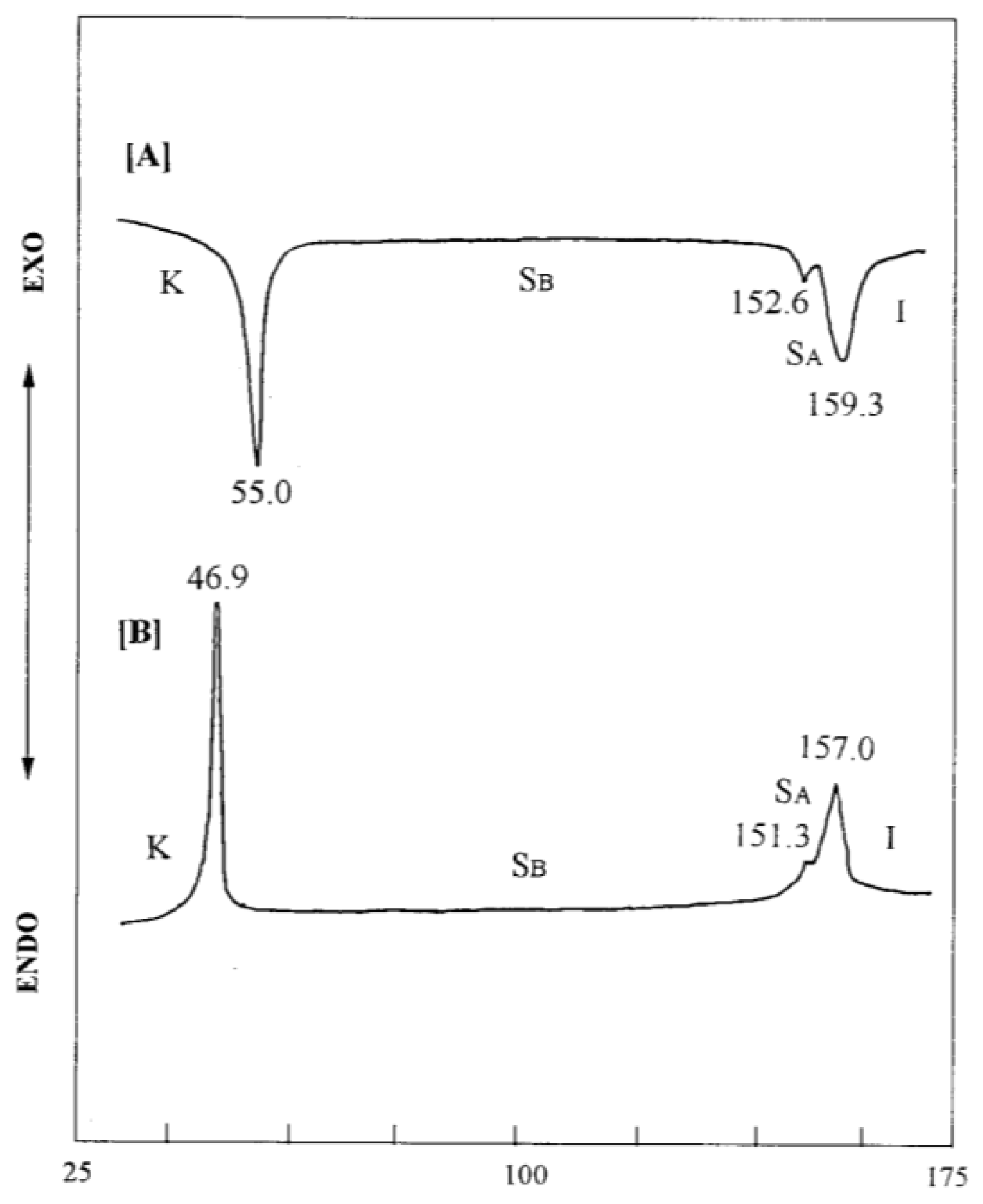
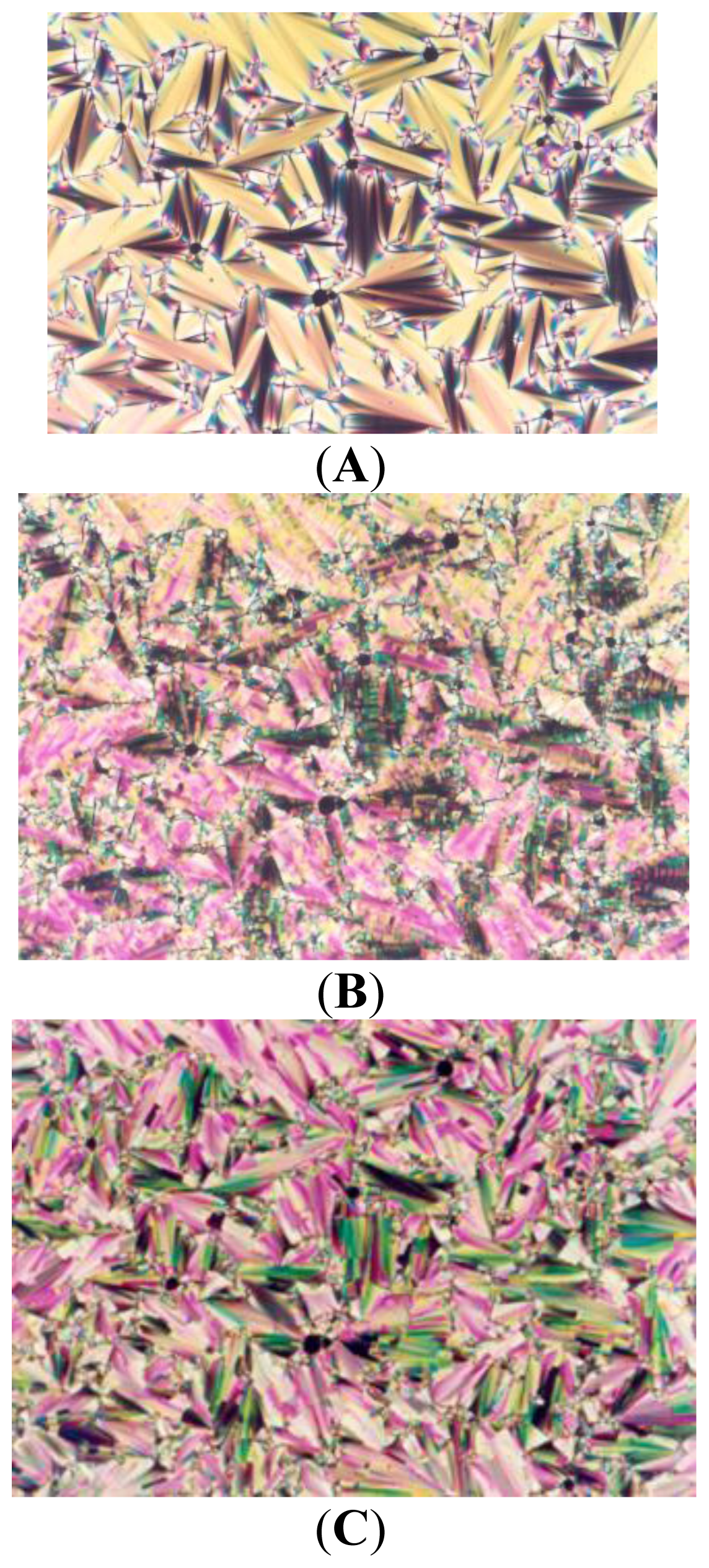
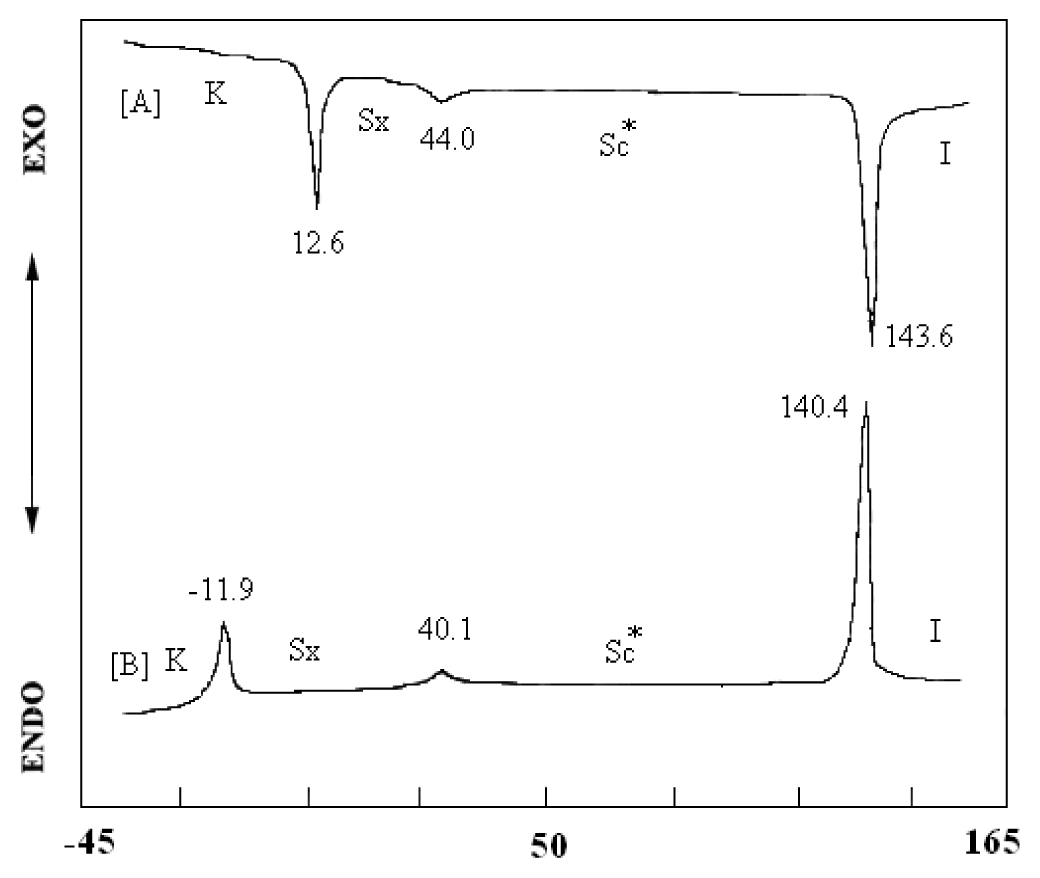
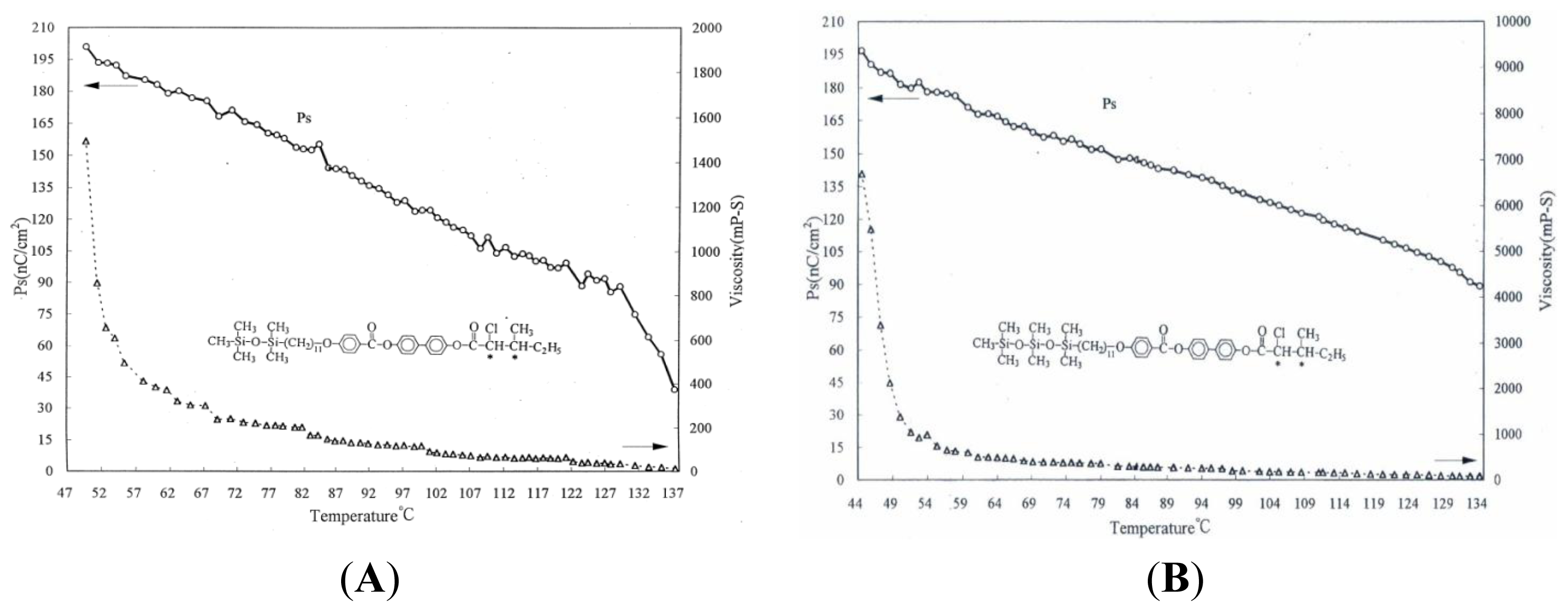
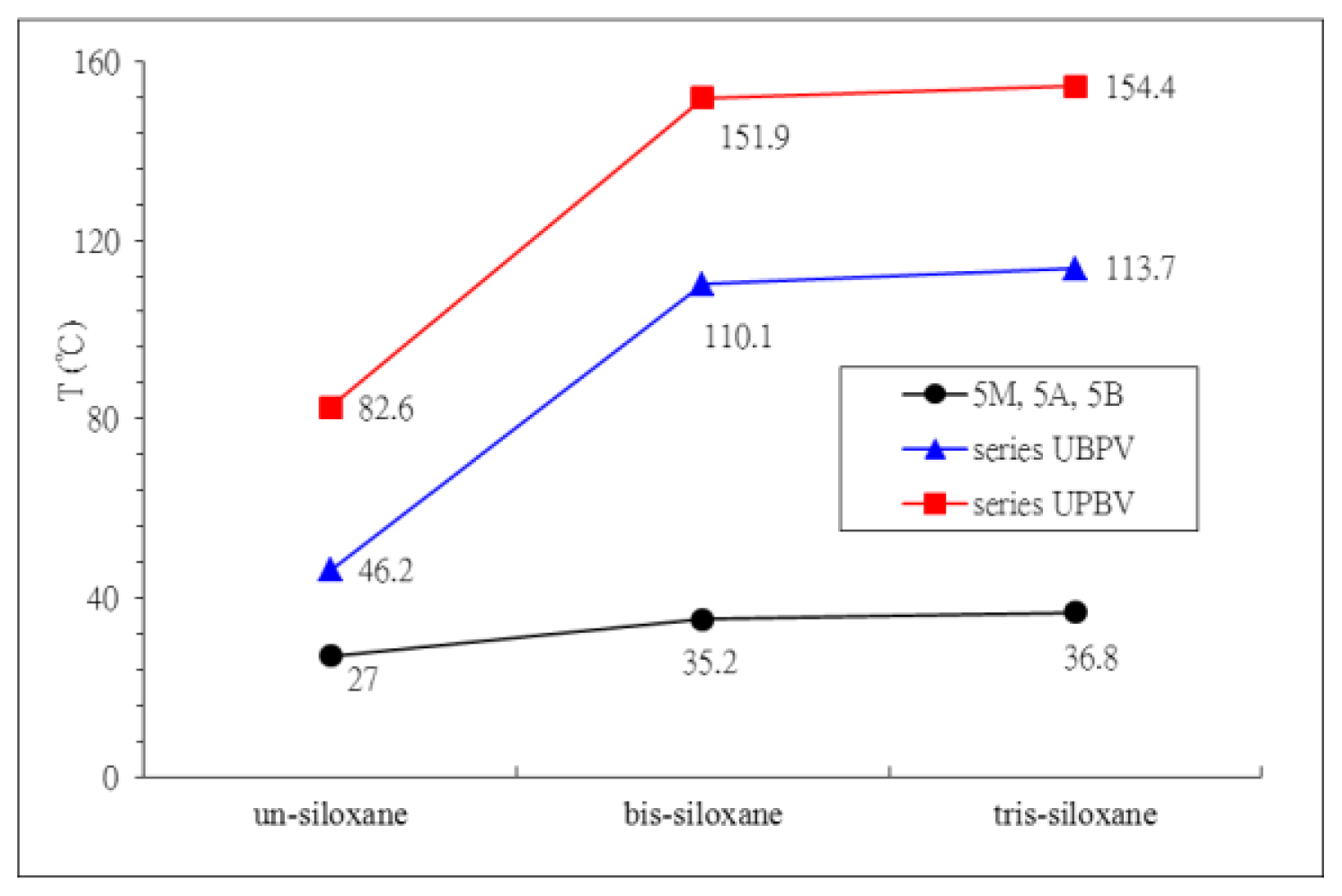
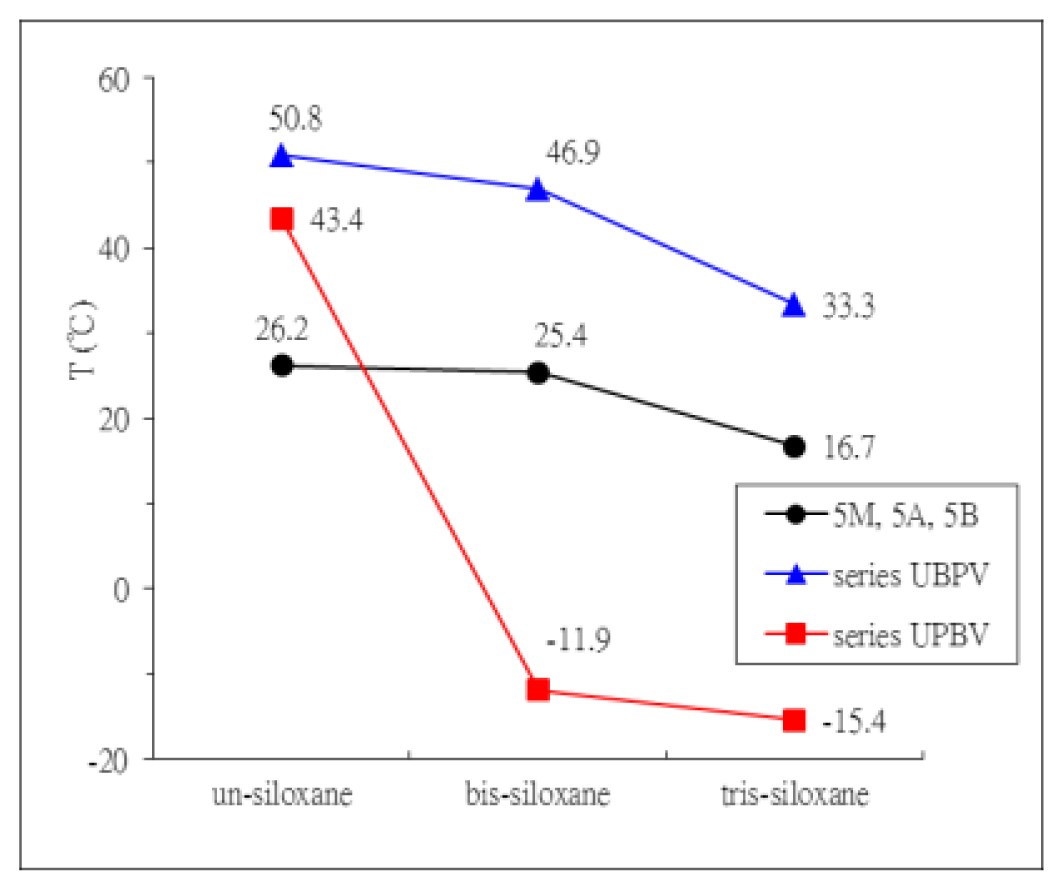
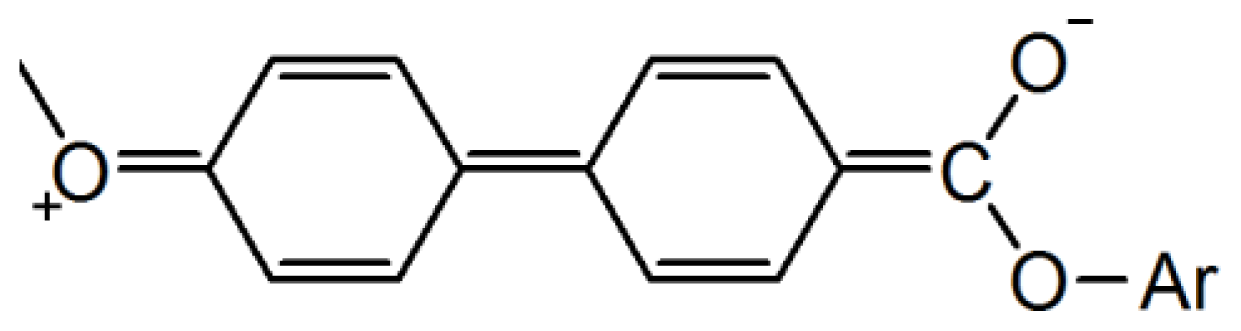
2. Results and Discussion
3. Experimental Section
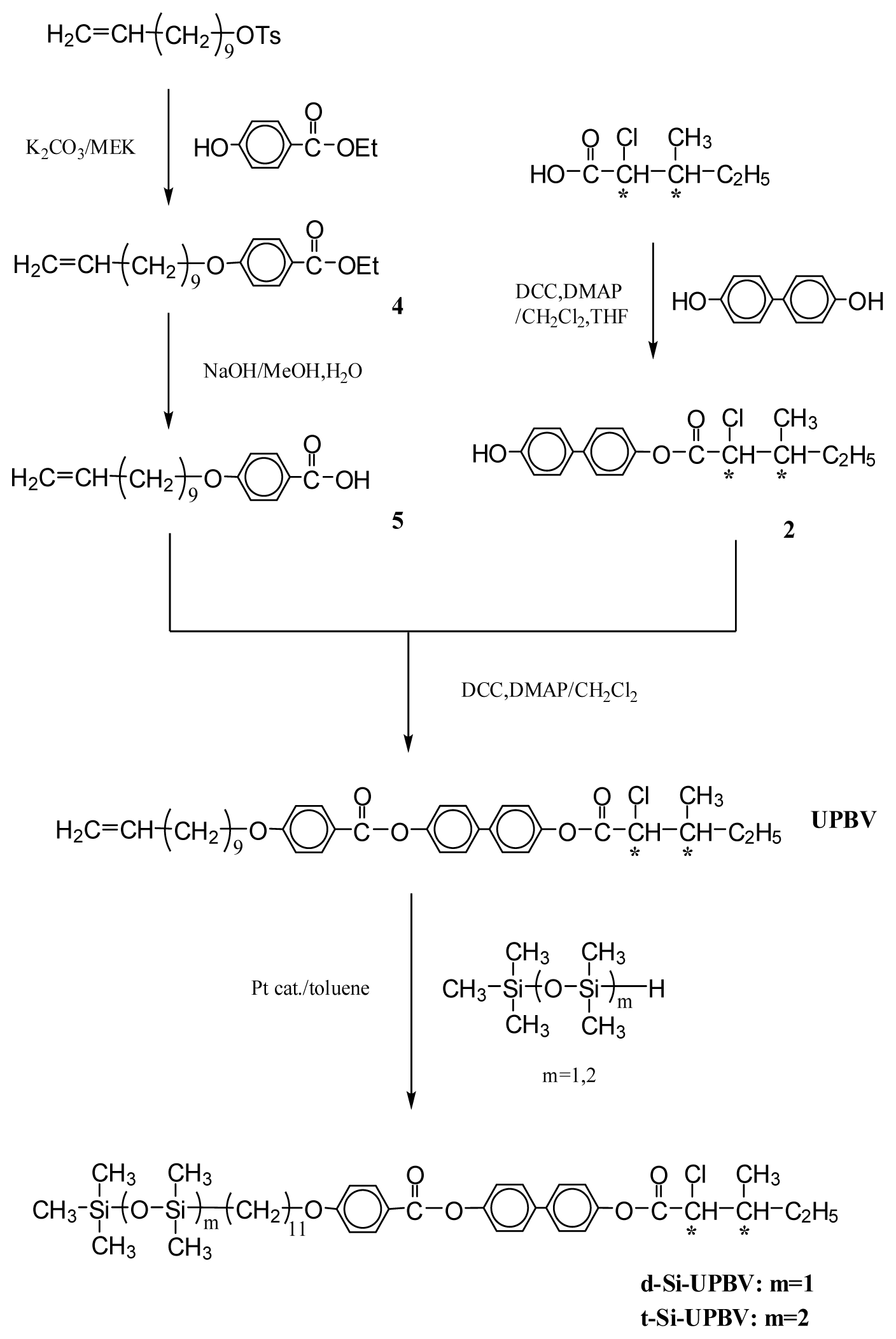
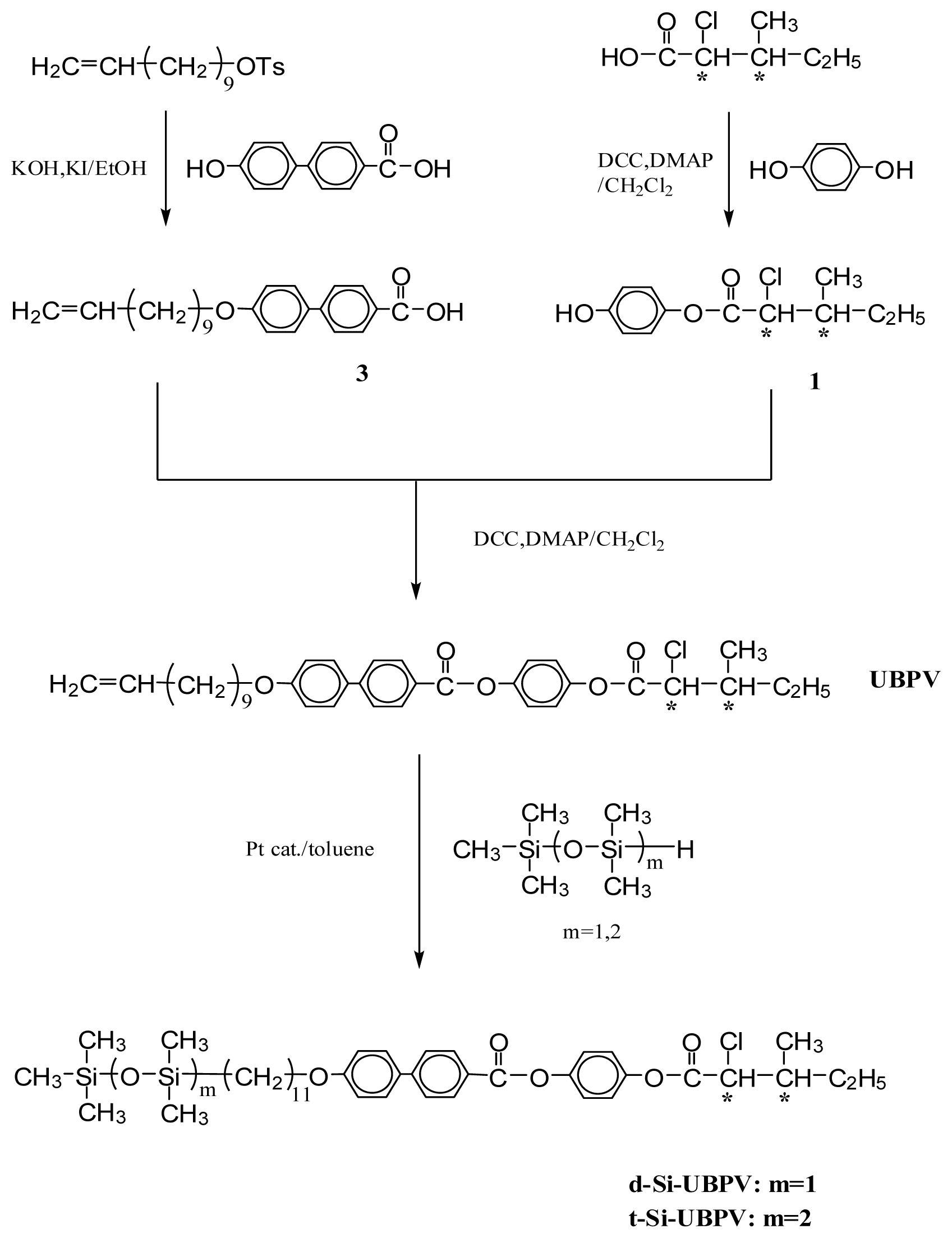
3.1. Synthesis
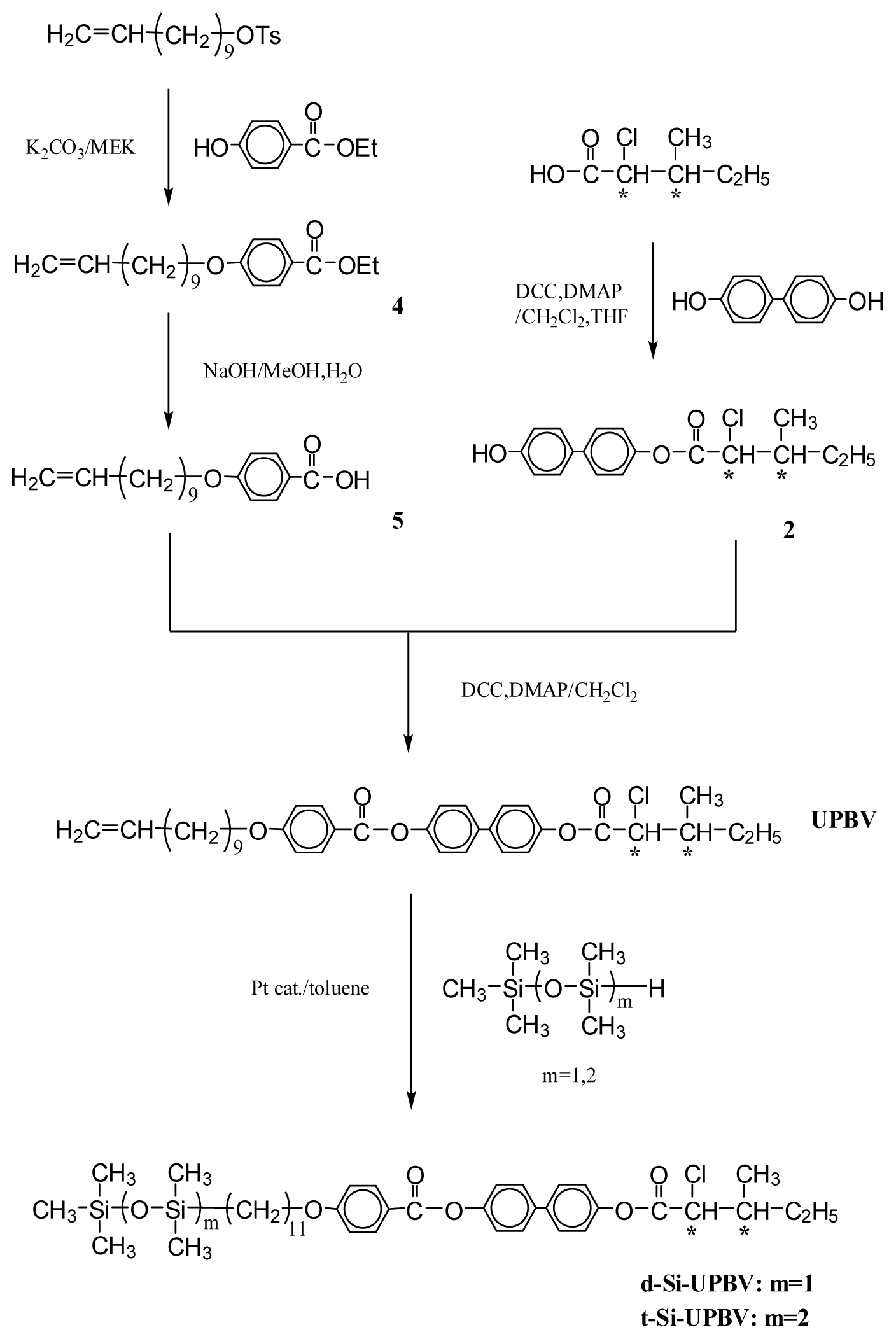
3.1.1. 4-Hydroxyphenyl(2S,3S)-2-chloro-3-methylvalerate (1) and 4-Hydroxybiphenyl-4′-yl(2S,3S)-2-chloro-3-methylvalerate (2)
3.1.2. 4-(10-Undecenyloxy)biphenyl-4′-carboxylic acid (3)
3.1.3. Ethyl-4-(10-undecenyloxy)benzoate (4)
3.1.4. 4-(10-Undecenyloxy)benzoic acid (5)
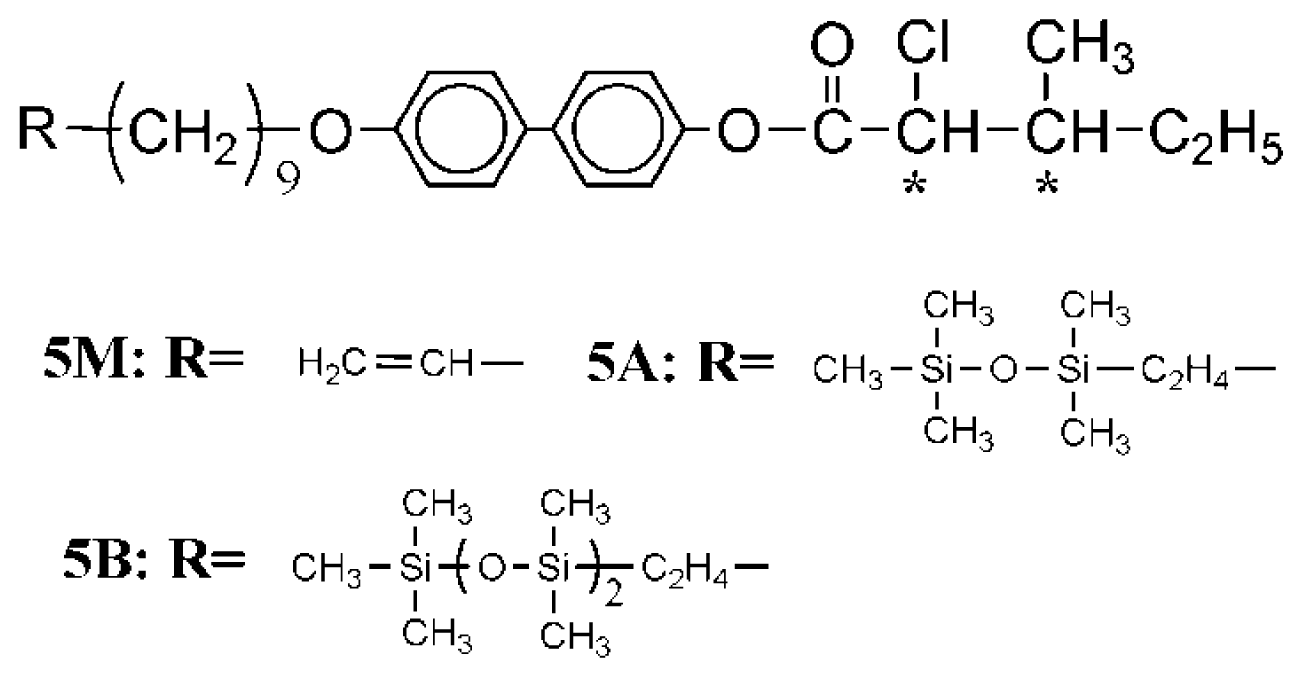
3.1.5. 4-[4-(10-Undecenyloxy)biphenyl-4′-carbonyloxy]phenyl(2S,3S)-2-chloro-3-methylvalerate (UBPV) and 4-[4-(10-Undecenyloxy)phenyl-4′-carbonyloxy]biphenyl(2S,3S)-2-chloro-3-methylvalerate (UPBV)
3.1.6. Liquid Crystal Siloxane Compounds d-Si-UBPV, t-Si-UBPV, d-Si-UPBV, and t-Si-UPBV
4. Conclusions





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Phase transitions, °C (corresponding enthalpy changes, Kcal/mole) |
|---|---|
| UBPV | |
| d-Si-UBPV | |
| t-Si-UBPV |
| Compound | Phase transitions, °C (corresponding enthalpy changes, Kcal/mole) |
|---|---|
| UPBV | |
| d-Si-UPBV | |
| t-Si-UPBV |
Acknowledgments
Conflicts of Interest
References
- Corsellis, E.A.; Coles, H.J. Thermal behaviour and electro-optic properties of a nematic exhibiting cyclic siloxane. Mol. Cryst. Liq. Cryst 1995, 261, 71–78. [Google Scholar]
- Newton, J.; Coles, H.J.; Hodge, P.; Hannington, J. Synthesis and properties of low-molar liquid-crystalline siloxane derivatives. J. Mater. Chem 1994, 4, 869–874. [Google Scholar]
- Coles, H.J.; Owen, H.; Newton, J.; Hodge, P. New low molar mass organosiloxanes with unusual ferroelectric properties. Liq. Cryst 1993, 15, 739–744. [Google Scholar]
- Coles, H.J.; Owen, H.; Newton, J.; Hodge, P. Electro-optic effects in novel siloxane containing oligomeric liquid crystals II: Smectic C materials. Proc. SPIE 1995, 2408, 22–29. [Google Scholar]
- Kloess, P.; McComb, J.; Coles, H.J.; Zentel, R. Synthesis and properties of a new series of low-molar-mass organo-siloxane derivatives. Ferroelectrics 1996, 180, 233–243. [Google Scholar]
- Scherowsky, G.; Schliwa, A.; Springer, J.; Kuhnpast, K.; Trapp, W. Fast switching ferroelectric liquid-crystalline polymers. Liq. Cryst 1989, 5, 1281–1295. [Google Scholar]
- Shibaev, V.P.; Kozlovsky, M.V.; Plate, N.A. Ferroelectric liquid-crystalline polymethacrylates. Liq. Cryst 1990, 8, 545–551. [Google Scholar]
- Vallerien, S.U.; Kremer, F.; Fischer, E.W. Experimental proof of piezoelectricity in cholesteric and chiral smectic C*-phases of LC-elastomers. Makromol. Chem. Rapid Commun 1990, 11, 593–598. [Google Scholar]
- Kapitza, H.; Zentel, R. Chiral liquid-crystalline polymers by polymer-analogous reactions. Makromol. Chem 1991, 192, 1859–1872. [Google Scholar]
- Endo, H.; Hachiya, S.; Uchida, S.; Hashimoto, K.; Kawasaki, K. Helical pitch of ferroelectric liquid-crystalline polymers and copolymers. Liq. Cryst 1991, 9, 635–641. [Google Scholar]
- Vallerien, S.U.; Kremer, F.; Kapitza, H.; Zentel, R.; Fischer, E.W. Softmode at the ferroelectric/non ferroelectric phase-transition in a liquid crystalline polymer. Ferroelectrics 1990, 109, 273–278. [Google Scholar]
- Lagerwall, S.T.; Dahl, I. Ferroelectric liquid crystals. Mol. Cryst. Liq. Cryst 1984, 114, 151–187. [Google Scholar]
- Lagerwall, S.T.; Otterholm, B.; Skarp, K. Material properties of ferroelectric liquid crystals and their relevance for applications and devices. Mol. Cryst. Liq. Cryst 1987, 152, 503–587. [Google Scholar]
- Roberts, J.C.; Kapernaum, N.; Giesselmann, F.; Wand, M.D.; Lemieux, R.P. Fast switching organosiloxane ferroelectric liquid crystals. J. Mater. Chem 2008, 18, 5301–5306. [Google Scholar]
- Grüneberg, K.; Naciri, J.; Shashidhar, R. Ferroelectric properties of a fast switching cyclic siloxane oligomer. Chem. Mater 1996, 8, 2486–2498. [Google Scholar]
- Keith, C.; Reddy, R.A.; Baumeister, U.; Tschierske, C. Banana-shaped liquid crystals with two oligosiloxane end-groups: Field-induced switching of supramolecular chirality. J. Am. Chem. Soc 2004, 126, 14312–14313. [Google Scholar]
- Lin, C.H. Synthesis and characterization of ferroelectric liquid crystalline siloxanes containing 4-hydroxyphenyl(2S,3S)-2-chloro-3-methylvalerate. Mol. Cryst. Liq. Cryst 2012, 552, 33–42. [Google Scholar]
- Naciri, J.; Ruth, J.; Grawford, G.; Shashidhar, R.; Ratna, B.R. Novel ferroelectric and electroclinic organosiloxane liquid crystals. Chem. Mater 1995, 7, 1397–1402. [Google Scholar]
- Hsu, C.S.; Her, B.S. Synthesis of ferroelectric liquid-crystalline polymethacrylates containing 1,2-diphenylethane based mesogens. Macromol. Chem. Phys 1996, 197, 4105–4118. [Google Scholar]
- Liao, C.T.; Wu, Z.L.; Wu, N.C.; Liu, J.Y.; Jiang, M.H.; Zou, S.F.; Lee, J.Y. Low-temperature and wide ferroelectric phase in mixtures of chiral and non-chiral tilted smectic C-type liquid crystals. Mol. Cryst. Liq. Cryst 2010, 533, 3–15. [Google Scholar]
- Leadbetter, A.J.; Forst, J.C.; Mazid, M.A. Interlayer correlations in smectic B phases. J. Physique Lett 1979, 40, 325–329. [Google Scholar]
- Goodby, J.W.; Gray, G.W.; McDonnell, D.G. Dipole moments and the smectic C phase. Mol. Cryst. Liq. Cryst 1976, 34, 183–188. [Google Scholar]
- Goodby, J.W.; Gray, G.W. A natural progression from smectic C to tilted smectic B properties in the n-Alkyl 4′-n-Alkoxybiphenyl-4-carboxylates. Mol. Cryst. Liq. Cryst 1978, 48, 127–149. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, C.-H. Synthesis and Characterization of Ferroelectric Liquid Crystalline Organosiloxanes Containing 4-(4-undecanyloxy bi-phenyl-1-carboxyloxy)phenyl (2S,3S)-2-chloro-3-methylvalerate and 4-(4-undecanyloxybenzoyloxy)biphenyl (2S,3S)-2-chloro-3-methylvalerate. Int. J. Mol. Sci. 2013, 14, 21306-21318. https://doi.org/10.3390/ijms141121306
Lin C-H. Synthesis and Characterization of Ferroelectric Liquid Crystalline Organosiloxanes Containing 4-(4-undecanyloxy bi-phenyl-1-carboxyloxy)phenyl (2S,3S)-2-chloro-3-methylvalerate and 4-(4-undecanyloxybenzoyloxy)biphenyl (2S,3S)-2-chloro-3-methylvalerate. International Journal of Molecular Sciences. 2013; 14(11):21306-21318. https://doi.org/10.3390/ijms141121306
Chicago/Turabian StyleLin, Chih-Hung. 2013. "Synthesis and Characterization of Ferroelectric Liquid Crystalline Organosiloxanes Containing 4-(4-undecanyloxy bi-phenyl-1-carboxyloxy)phenyl (2S,3S)-2-chloro-3-methylvalerate and 4-(4-undecanyloxybenzoyloxy)biphenyl (2S,3S)-2-chloro-3-methylvalerate" International Journal of Molecular Sciences 14, no. 11: 21306-21318. https://doi.org/10.3390/ijms141121306