Neuroprotection for Ischemic Stroke: Moving Past Shortcomings and Identifying Promising Directions

 ,
,  ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Concept of Neuroprotection in Ischemic Stroke
1.2. Current Status of Neuroprotectant Development: Drug Development Shortcomings
2. Improving Animal Models: Role of Comorbidities & Lifestyle
2.1. Accounting for the Aging Process: Effects on Inflammation
2.1.1. Astrocytes
2.1.2. Macrophages
2.1.3. Microglia
2.1.4. Challenges Associated with Aged Models of Ischemic Stroke
2.2. Effect of Comorbidities on Stroke Risk & Outcome
2.2.1. Diabetic Models
2.2.2. Hypertension Models
2.2.3. Obesity Models
2.3. Accounting for Lifestyle Influences: Effects of Altered Sleep-Wake Patterns
2.4. Identification of Clinically-Relevant Endpoints for Preclinical Studies
3. Promising Directions and Potential Targets in Neuroprotectant Development
3.1. Modulating Astrocyte Activity
3.2. Inhibiting Effects of Microglia
3.3. Modulating the Blood-Brain Barrier (BBB)
4. Targeting Inflammation
4.1. Inflammation: A Deleterious Event or a Beneficial Response?
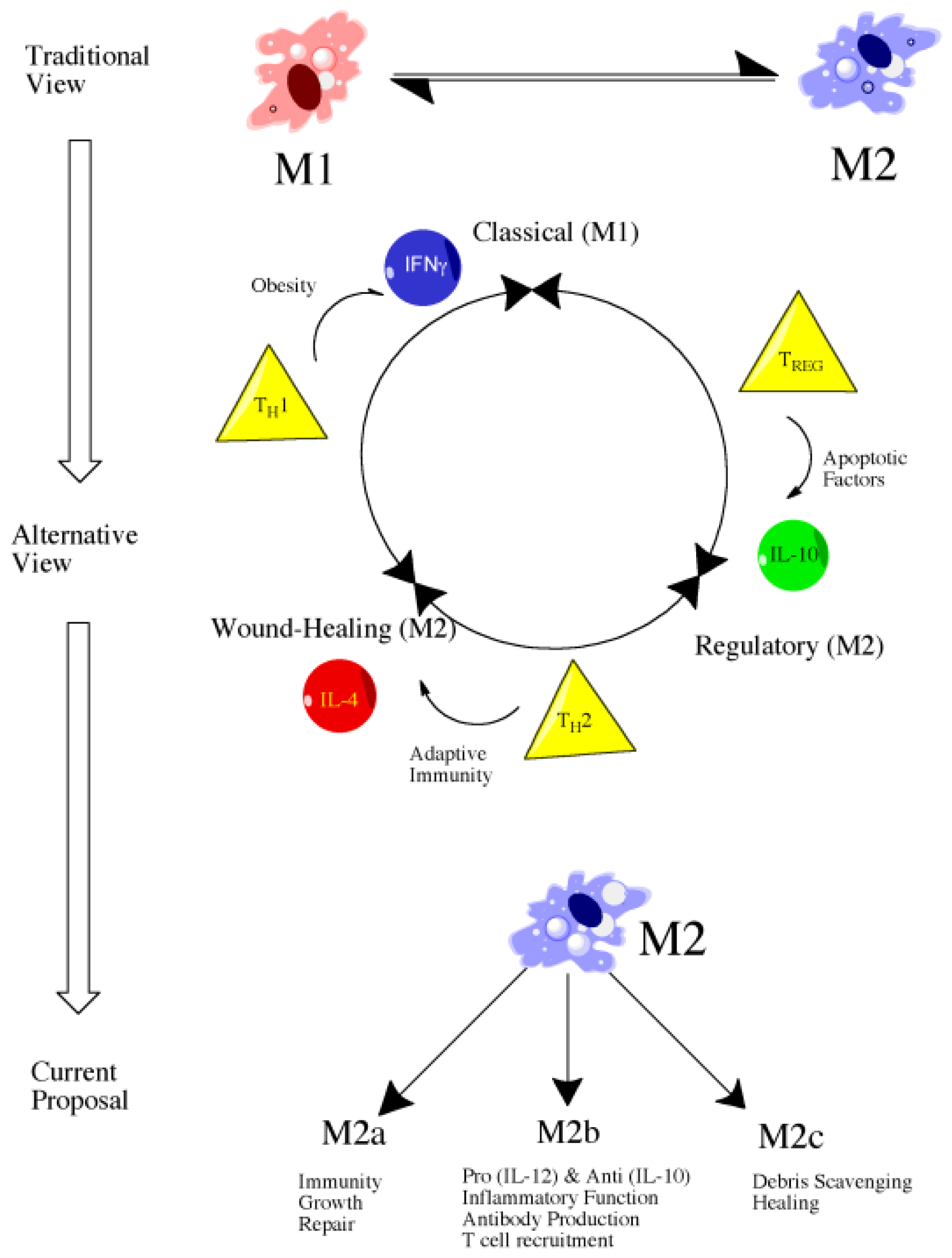
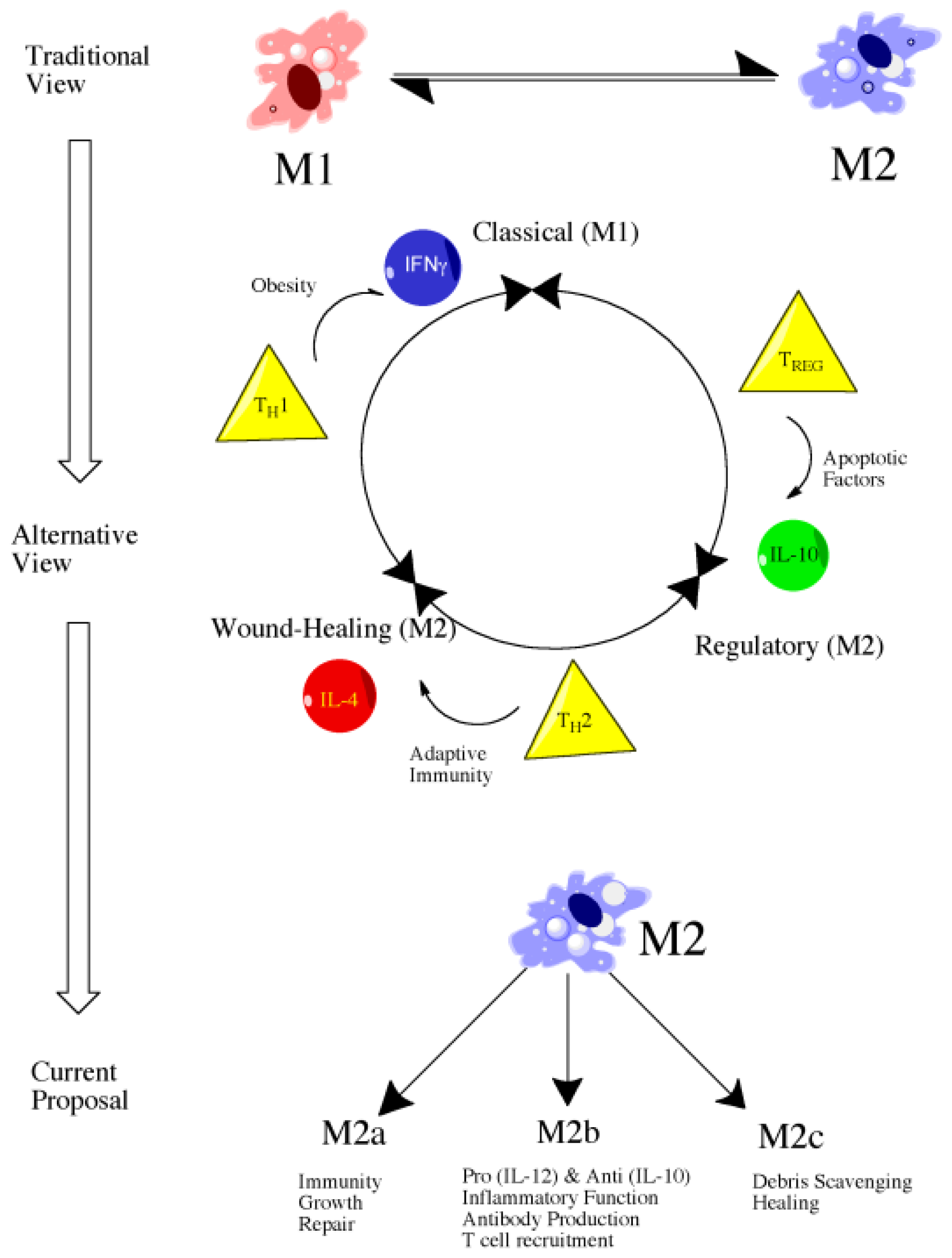
4.2. Introduction to Macrophage Polarization
4.3. M1 versus M2: Classical versus Alternative Activation
4.4. Classically Activated Macrophages (M1)
4.5. Alternatively Activated Macrophages (M2)
4.6. Future Directions: Therapeutic Targeting of Macrophage Subsets
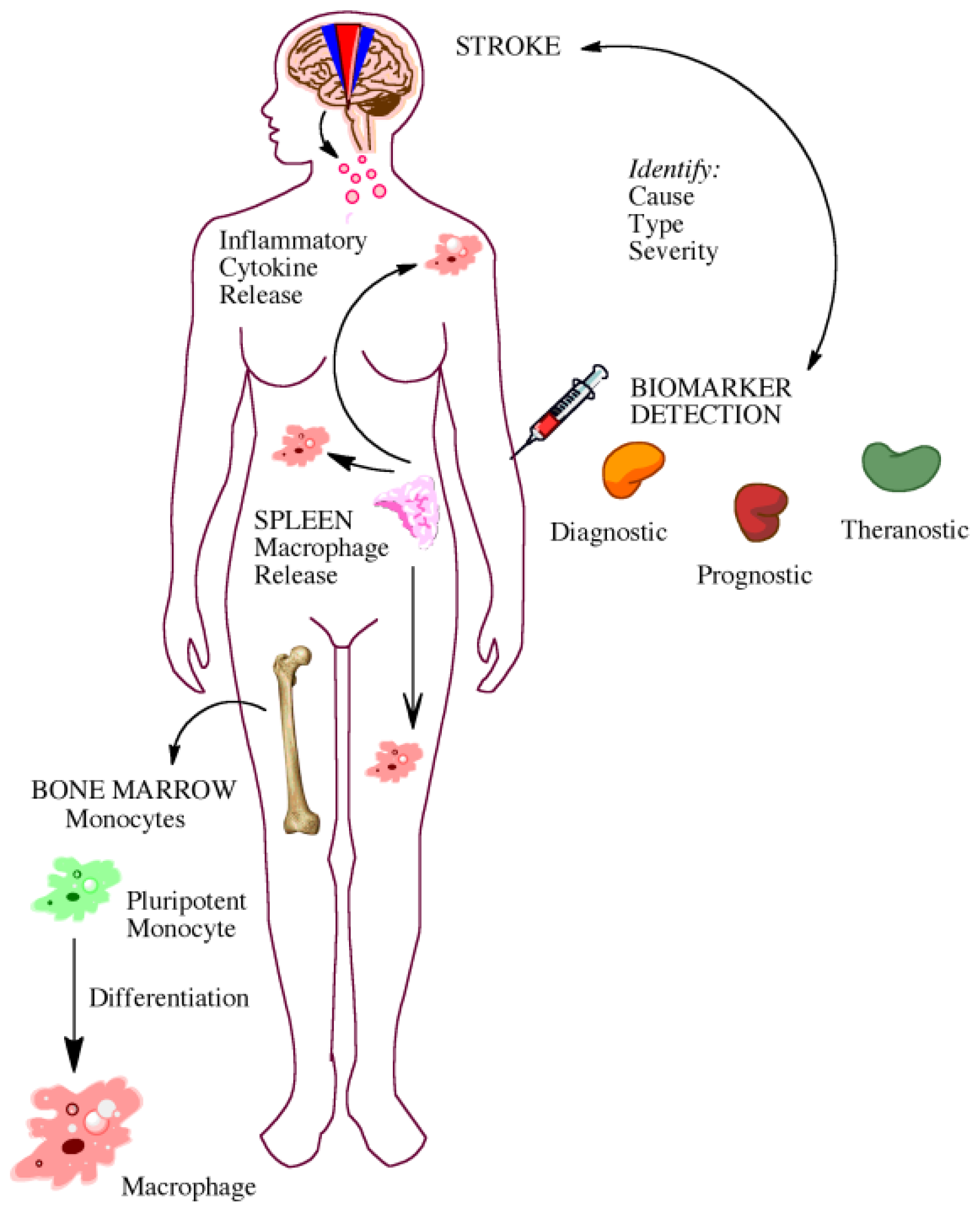
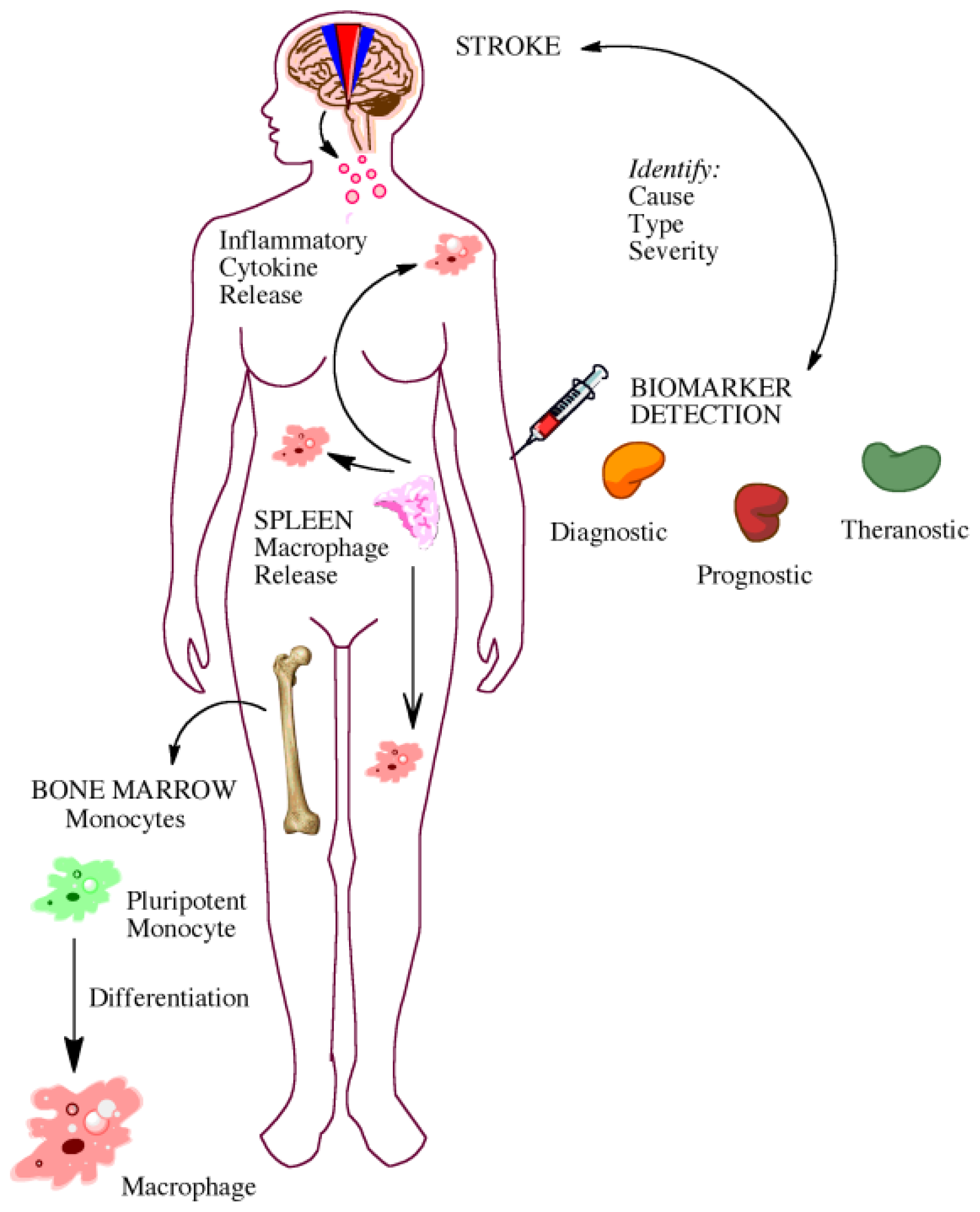
5. Gaining Insight from the Bedside: Utility of Biomarkers
5.1. Biomarkers for Diagnosis and Prognosis
5.2. Biomarkers for Elucidating Disease Pathophysiology
6. Discussion
References
- Astrup, J.; Symon, L.; Branston, N.M.; Lassen, N.A. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 1977, 8, 51–57. [Google Scholar]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med 2008, 14, 497–500. [Google Scholar]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1026 experimental treatments in acute stroke. Ann. Neurol 2006, 59, 467–477. [Google Scholar]
- Fukuda, S.; del Zoppo, G.J. Models of focal cerebral ischemia in the nonhuman primate. ILAR J 2003, 44, 96–104. [Google Scholar]
- Bonafe, M.; Storci, G.; Franceschi, C. Inflamm-aging of the stem cell niche: Breast cancer as a paradigmatic example: Breakdown of the multi-shell cytokine network fuels cancer in aged people. Bioessays 2012, 34, 40–49. [Google Scholar]
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord. Drug Targets 2011, 10, 621–634. [Google Scholar]
- Simpson, R.J.; Lowder, T.W.; Spielmann, G.; Bigley, A.B.; LaVoy, E.C.; Kunz, H. Exercise and the aging immune system. Ageing Res. Rev 2012, 11, 404–420. [Google Scholar]
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflamm-aging and immunosenescence. J. Proteomics 2011, 74, 2313–2323. [Google Scholar]
- DiNapoli, V.A.; Huber, J.D.; Houser, K.; Li, X.; Rosen, C.L. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol. Aging 2008, 29, 753–764. [Google Scholar]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci 2012, 32, 6391–6410. [Google Scholar]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci 2011, 34, 3–11. [Google Scholar]
- Lewis, D.K.; Thomas, K.T.; Selvamani, A.; Sohrabji, F. Age-related severity of focal ischemia in female rats is associated with impaired astrocyte function. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef]
- Gliem, M.; Mausberg, A.K.; Lee, J.I.; Simiantonakis, I.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol 2012, 71, 743–752. [Google Scholar]
- Michalski, D.; Heindl, M.; Kacza, J.H.; Laignel, F.; Küppers-Tiedt, L.; Schneider, D.; Grosche, J.; Boltze, J.; Löhr, M.; Hobohm, C.; et al. Spatio-temporal course of macrophage-like cell accumulation after experimental embolic stroke depending on treatment with tissue plasminogen activator and its combination with hyperbaric oxygenation. Eur. J. Histochem 2012, 56, e14. [Google Scholar]
- Guerra-Laso, J.M.; Gonzalez-Garcia, S.; Gonzalez-Cortes, C.; Diez-Tascon, C.; Lopez-Medrano, R.; Rivero-Lezcano, O.M. Macrophages from elders are more permissive to intracellular multiplication of. Mycobacterium tuberculosis. Age (Dordr) 2012. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Buga, A.M.; Kokaia, Z. Perturbed cellular response to brain injury during aging. Ageing Res. Rev 2011, 10, 71–79. [Google Scholar]
- Kassner, S.S.; Kollmar, R.; Bonaterra, G.A.; Hildebrandt, W.; Schwab, S.; Kinscherf, R. The early immunological response to acute ischemic stroke: Differential gene expression in subpopulations of mononuclear cells. Neuroscience 2009, 160, 394–401. [Google Scholar]
- Von Bernhardi, R.; Tichauer, J.E.; Eugenin, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem 2010, 112, 1099–1114. [Google Scholar]
- Bishop, G.M.; Dang, T.N.; Dringen, R.; Robinson, S.R. Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox. Res 2011, 19, 443–451. [Google Scholar]
- Luo, X.G.; Ding, J.Q.; Chen, S.D. Microglia in the aging brain: Relevance to neurodegeneration. Mol. Neurodegener 2010, 5, 12. [Google Scholar]
- Vukovic, J.; Colditz, M.J.; Blackmore, D.G.; Ruitenberg, M.J.; Bartlett, P.F. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci 2012, 32, 6435–6443. [Google Scholar]
- Rosano, C.; Marsland, A.L.; Gianaros, P.J. Maintaining brain health by monitoring inflammatory processes: A mechanism to promote successful aging. Aging Dis 2012, 3, 16–33. [Google Scholar]
- Belayev, L.; Alonso, O.F.; Busto, R.; Zhao, W.; Ginsberg, M.D. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke 1996, 27, 1616–1622, discussion 1623. [Google Scholar]
- Rosen, C.L.; Dinapoli, V.A.; Nagamine, T.; Crocco, T. Influence of age on stroke outcome following transient focal ischemia. J. Neurosurg 2005, 103, 687–694. [Google Scholar]
- Goodman, E.; Li, C.; Tu, Y.K.; Ford, E.; Sun, S.S.; Huang, T.T. Stability of the factor structure of the metabolic syndrome across pubertal development: Confirmatory factor analyses of three alternative models. J. Pediatr 2009, 155, e1–e8. [Google Scholar]
- Chien, K.L.; Hsu, H.C.; Sung, F.C.; Su, T.C.; Chen, M.F.; Lee, Y.T. Metabolic syndrome as a risk factor for coronary heart disease and stroke: An 11-year prospective cohort in Taiwan community. Atherosclerosis 2007, 194, 214–221. [Google Scholar]
- Maruyama, K.; Uchiyama, S.; Iwata, M. Metabolic syndrome and its components as risk factors for first-ever acute ischemic noncardioembolic stroke. J. Stroke Cerebrovasc. Dis 2009, 18, 173–177. [Google Scholar]
- Lambert, G.W.; Straznicky, N.E.; Lambert, E.A.; Dixon, J.B.; Schlaich, M.P. Sympathetic nervous activation in obesity and the metabolic syndrome—Causes, consequences and therapeutic implications. Pharmacol. Ther 2010, 126, 159–172. [Google Scholar]
- Roriz-Filho, J.S.; Roriz, S.T.; Foss, M.P.; Foss-Freitas, M.C.; Ferriolli, E.; Lima, N.K.C.; Santos, A.C.; Moriguti, J.C. Correlation between Alzheimer’s disease, pre-diabetes and diabetes mellitus evaluated by quantitative magnetic resonance imaging. Alzheimer’s Dementia 2009, 5, P261–P262. [Google Scholar]
- Srinivasan, K.; Sharma, S.S. Augmentation of endoplasmic reticulum stress in cerebral ischemia/reperfusion injury associated with comorbid type 2 diabetes. Neurol. Res 2011, 33, 858–865. [Google Scholar]
- Ye, X.; Chopp, M.; Cui, X.; Zacharek, A.; Cui, Y.; Yan, T.; Shehadah, A.; Roberts, C.; Liu, X.; Lu, M.; et al. Niaspan enhances vascular remodeling after stroke in type 1 diabetic rats. Exp. Neurol 2011, 232, 299–308. [Google Scholar]
- Darsalia, V.; Mansouri, S.; Ortsäter, H.; Olverling, A.; Nozadze, N.; Kappe, C.; Iverfeldt, K.; Tracy, L.M.; Grankvist, N.; Sjöholm, A.; et al. Glucagon-like peptide-1 receptor activation reduces ischaemic brain damage following stroke in Type 2 diabetic rats. Clin. Sci. (Lond. ) 2012, 122, 473–483. [Google Scholar]
- Vahtola, E.; Louhelainen, M.; Forstén, H.; Merasto, S.; Raivio, J.; Kaheinen, P.; Kytö, V.; Tikkanen, I.; Levijoki, J.; Mervaala, E. Sirtuin1-p53, forkhead box O3a, p38 and post-infarct cardiac remodeling in the spontaneously diabetic Goto-Kakizaki rat. Cardiovasc. Diabetol 2010, 9, 5. [Google Scholar]
- Kumari, R.; Willing, L.B.; Patel, S.D.; Baskerville, K.A.; Simpson, I.A. Increased cerebral matrix metalloprotease-9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J. Neurochem. 2011, 119, 1029–1040. [Google Scholar]
- Rees, D.A.; Alcolado, J.C. Animal models of diabetes mellitus. Diabet. Med 2005, 22, 359–370. [Google Scholar]
- Bergerat, A.; Decano, J.; Wu, C.J.; Choi, H.; Nesvizhskii, A.I.; Moran, A.M.; Ruiz-Opazo, N.; Steffen, M.; Herrera, V.L. Prestroke proteomic changes in cerebral microvessels in stroke-prone, transgenic[hCETP]-Hyperlipidemic, Dahl salt-sensitive hypertensive rats. Mol. Med 2011, 17, 588–598. [Google Scholar]
- Henning, E.C.; Warach, S.; Spatz, M. Hypertension-induced vascular remodeling contributes to reduced cerebral perfusion and the development of spontaneous stroke in aged SHRSP rats. J. Cereb. Blood Flow Metab 2010, 30, 827–836. [Google Scholar]
- Schmidt, H.; Zeginigg, M.; Wiltgen, M.; Freudenberger, P.; Petrovic, K.; Cavalieri, M.; Gider, P.; Enzinger, C.; Fornage, M.; Debette, S.; et al. Genetic variants of the NOTCH3 gene in the elderly and magnetic resonance imaging correlates of age-related cerebral small vessel disease. Brain 2011, 134, 3384–3397. [Google Scholar]
- Krafft, P.R.; Bailey, E.L.; Lekic, T.; Rolland, W.B.; Altay, O.; Tang, J.; Wardlaw, J.M.; Zhang, J.H.; Sudlow, C.L. Etiology of stroke and choice of models. Int. J. Stroke 2012, 7, 398–406. [Google Scholar]
- Phillips, S.J. Pathophysiology and management of hypertension in acute ischemic stroke. Hypertension 1994, 23, 131–136. [Google Scholar]
- Slemmer, J.E.; Shaughnessy, K.S.; Scanlan, A.P.; Sweeney, M.I.; Gottschall-Pass, K.T. Choice of diet impacts the incidence of stroke-related symptoms in the spontaneously hypertensive stroke-prone rat model. Can. J. Physiol. Pharmacol 2012, 90, 243–248. [Google Scholar]
- Gatti, R.R.; Santos, P.S.; Sena, A.A.; Marangoni, K.; Araujo, M,A.; Goulart, L.R. The interaction of AGT and NOS3 gene polymorphisms with conventional risk factors increases predisposition to hypertension. J. Renin Angiotensin Aldosterone Syst. 2012. [Google Scholar] [CrossRef]
- Cohen, L.; Curhan, G.C.; Forman, J.P. Influence of age on the association between lifestyle factors and risk of hypertension. J. Am. Soc. Hypertens 2012, 6, 284–290. [Google Scholar]
- Yang, C.; Zhang, X.; Song, S.W.; Yu, J.G.; Cai, G.J. Cerebral artery remodeling in stroke-prone spontaneously hypertensive rats. CNS Neurosci. Ther 2011, 17, 785–786. [Google Scholar]
- Abrahamsen, C.T.; Barone, F.C.; Campbell, W.G., Jr; Nelson, A.H.; Contino, L.C.; Pullen, M.A.; Grygielko, E.T.; Edwards, R.M.; Laping, N.J.; Brooks, D.P. The angiotensin type 1 receptor antagonist, eprosartan, attenuates the progression of renal disease in spontaneously hypertensive stroke-prone rats with accelerated hypertension. J. Pharmacol. Exp. Ther. 2002, 301, 21–28. [Google Scholar]
- Jalal, F.Y.; Yang, Y.; Thompson, J.; Lopez, A.C.; Rosenberg, G.A. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke 2012, 43, 1115–1122. [Google Scholar]
- Bailey, E.L.; Smith, C.; Sudlow, C.L.; Wardlaw, J.M. Is the spontaneously hypertensive stroke prone rat a pertinent model of sub cortical ischemic stroke? A systematic review. Int. J. Stroke 2011, 6, 434–444. [Google Scholar]
- Zhang, X.H.; Lei, H.; Liu, A.J.; Zou, Y.X.; Shen, F.M.; Su, D.F. Increased oxidative stress is responsible for severer cerebral infarction in stroke-prone spontaneously hypertensive rats. CNS Neurosci. Ther 2011, 17, 590–598. [Google Scholar]
- Mansoorali, K.P.; Prakash, T.; Kotresha, D.; Prabhu, K.; Rama Rao, N. Cerebroprotective effect of Eclipta alba against global model of cerebral ischemia induced oxidative stress in rats. Phytomedicine 2012, 19, 1108–1116. [Google Scholar]
- Baretic, M. Targets for medical therapy in obesity. Dig. Dis 2012, 30, 168–172. [Google Scholar]
- Kantorova, E.; Chomova, M.; Kurca, E.; Sivak, S.; Zelenak, K.; Kučera, P.; Galajda, P. Leptin, adiponectin and ghrelin, new potential mediators of ischemic stroke. Neuro Endocrinol. Lett 2011, 32, 716–721. [Google Scholar]
- Gerdes, S.; Osadtschy, S.; Rostami-Yazdi, M.; Buhles, N.; Weichenthal, M.; Mrowietz, U. Leptin, adiponectin, visfatin and retinol-binding protein-4—Mediators of comorbidities in patients with psoriasis? Exp. Dermatol 2012, 21, 43–47. [Google Scholar]
- Katsiki, N.; Ntaios, G.; Vemmos, K. Stroke, obesity and gender: A review of the literature. Maturitas 2011, 69, 239–243. [Google Scholar]
- Prugger, C.; Luc, G.; Haas, B.; Arveiler, D.; Machez, E.; Ferrieres, J.; Ruidavets, J.B.; Bingham, A.; Montaye, M.; Amouyel, P.; et al. Adipocytokines and the risk of ischemic stroke: The PRIME study. Ann. Neurol 2012, 71, 478–486. [Google Scholar]
- Pan, W.; Kastin, A.J. Tumor necrosis factor and stroke: Role of the blood-brain barrier. Prog. Neurobiol 2007, 83, 363–374. [Google Scholar]
- De Artinano, A.A.; Miguel, C.M. Experimental rat models to study the metabolic syndrome. Br. J. Nutr 2009, 102, 1246–1253. [Google Scholar]
- Osmond, J.M.; Mintz, J.D.; Stepp, D.W. Preventing increased blood pressure in the obese Zucker rat improves severity of stroke. Am. J. Physiol. Heart Circ. Physiol 2010, 299, H55–H61. [Google Scholar]
- Osmond, J.M.; Mintz, J.D.; Dalton, B.; Stepp, D.W. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension 2009, 53, 381–386. [Google Scholar]
- Katakam, P.V.; Snipes, J.A.; Steed, M.M.; Busija, D.W. Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J. Cereb. Blood Flow Metab 2012, 32, 792–804. [Google Scholar]
- Zheng, P.; Ji, G.; Ma, Z.; Liu, T.; Xin, L.; Wu, H.; Liang, X.; Liu, J. Therapeutic effect of puerarin on non-alcoholic rat fatty liver by improving leptin signal transduction through JAK2/STAT3 pathways. Am. J. Chin. Med 2009, 37, 69–83. [Google Scholar]
- Dronne, M.A.; Grenier, E.; Chapuisat, G.; Hommel, M.; Boissel, J.P. A modelling approach to explore some hypotheses of the failure of neuroprotective trials in ischemic stroke patients. Prog. Biophys. Mol. Biol 2008, 97, 60–78. [Google Scholar]
- Ayas, N.T.; White, D.P.; Manson, J.E.; Stampfer, M.J.; Speizer, F.E.; Malhotra, A.; Hu, F.B. A prospective study of sleep duration and coronary heart disease in women. Arch. Intern. Med 2003, 163, 205–209. [Google Scholar]
- Ferrie, J.E.; Shipley, M.J.; Cappuccio, F.P.; Brunner, E.; Miller, M.A.; Kumari, M.; Marmot, M.G. A prospective study of change in sleep duration: Associations with mortality in the Whitehall II cohort. Sleep 2007, 30, 1659–1666. [Google Scholar]
- Qureshi, A.I.; Giles, W.H.; Croft, J.B.; Bliwise, D.L. Habitual sleep patterns and risk for stroke and coronary heart disease: A 10-year follow-up from NHANES I. Neurology 1997, 48, 904–911. [Google Scholar]
- Ikehara, S.; Iso, H.; Date, C.; Kikuchi, S.; Watanabe, Y.; Wada, Y.; Inaba, Y.; Tamakoshi, A.; JACC Study Group. Association of sleep duration with mortality from cardiovascular disease and other causes for Japanese men and women: The JACC study. Sleep 2009, 32, 295–301. [Google Scholar]
- Sabanayagam, C.; Shankar, A. Sleep duration and cardiovascular disease: Results from the national health interview survey. Sleep 2010, 33, 1037–1042. [Google Scholar]
- Cappuccio, F.; Taggart, F.M.; Kandala, N.B.; Currie, A.; Peile, E.; Stranges, S.; Miller, M.A. Meta-analysis of short sleep duration and obesity in children and adults. Sleep 2008, 31, 619–626. [Google Scholar]
- Chaput, J.P.; Despres, J.P.; Bouchard, C.; Tremblay, A. Association of sleep duration with type 2 diabetes and impaired glucose tolerance. Diabetologia 2007, 50, 2298–2304. [Google Scholar]
- Schultes, B.; Schmid, S.; Peters, A.; Born, J.; Fehm, H.L. Sleep loss and the development of diabetes: A review of current evidence. Exp. Clin. Endocrinol. Diabetes 2005, 113, 563–567. [Google Scholar]
- Spiegel, K.; Knutson, K.; Leproult, R.; Tasali, E.; van Cauter, E. Sleep loss: A novel risk factor for insulin resistance and Type 2 diabetes. J. Appl. Physiol 2005, 99, 2008–2019. [Google Scholar]
- Kim, J.; Jo, I. Age-dependent association between sleep duration and hypertension in the adult Korean population. Am. J. Hypertens 2010, 23, 1286–1291. [Google Scholar]
- Gottlieb, D.J.; Redline, S.; Nieto, F.J.; Baldwin, C.M.; Newman, A.B.; Resnick, H.E.; Punjabi, N.M. Association of usual sleep duration with hypertension: The sleep heart health study. Sleep 2006, 29, 1009–1014. [Google Scholar]
- Chen, J.C.; Brunner, R.L.; Ren, H.; Wassertheil-Smoller, S.; Larson, J.C.; Levine, D.W.; Allison, M.; Naughton, M.J.; Stefanick, M.L. Sleep duration and risk of ischemic stroke in postmenopausal women. Stroke 2008, 39, 3185–3192. [Google Scholar]
- Nurminen, M.; Karjalainen, A. Epidemiologic estimate of the proportion of fatalities related to occupational factors in Finland. Scand. J. Work Environ. Health 2001, 27, 161–213. [Google Scholar]
- Luckhaupt, S.E.; Tak, S.; Calvert, G.M. The prevalence of short sleep duration by industry and occupation in the national health interview survey. Sleep 2010, 33, 149–159. [Google Scholar]
- Fenzl, T.; Romanowski, C.P.; Flachskamm, C.; Honsberg, K.; Boll, E.; Hoehne, A.; Kimura, M. Fully automated sleep deprivation in mice as a tool in sleep research. J. Neurosci. Methods 2007, 166, 229–235. [Google Scholar]
- Rechtschaffen, A.; Bergmann, B.M.; Gilliland, M.A.; Bauer, K. Effects of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep 1999, 22, 11–31. [Google Scholar]
- Coenen, A.M.; van Luijtelaar, E.L. Stress induced by three procedures of deprivation of paradoxical sleep. Physiol. Behav 1985, 35, 501–504. [Google Scholar]
- Hsu, J.C.; Lee, Y.S.; Chang, C.N.; Ling, E.A.; Lan, C.T. Sleep deprivation prior to transient global cerebral ischemia attenuates glial reaction in the rat hippocampal formation. Brain Res 2003, 984, 170–181. [Google Scholar]
- Moldovan, M.; Constantinescu, A.O.; Balseanu, A.; Oprescu, N.; Zagrean, L.; Popa-Wagner, A. Sleep deprivation attenuates experimental stroke severity in rats. Exp. Neurol 2010, 222, 135–143. [Google Scholar]
- Bonnet, M.H.; Arand, D.L. Clinical effects of sleep fragmentation versus sleep deprivation. Sleep Med. Rev 2003, 7, 297–310. [Google Scholar]
- Saver, J.L.; Johnston, K.C.; Homer, D.; Wityk, R.; Koroshetz, W.; Truskowski, L.L.; Haley, E.C. Infarct volume as a surrogate or auxiliary outcome measure in ischemic stroke clinical trials. The RANTTAS Investigators. Stroke 1999, 30, 293–298. [Google Scholar]
- Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999, 30, 2752–2758.
- Takano, T.; Oberheim, N.; Cotrina, M.L.; Nedergaard, M. Astrocytes and ischemic injury. Stroke 2009, 40, S8–S12. [Google Scholar]
- Bambrick, L.; Kristian, T.; Fiskum, G. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem. Res 2004, 29, 601–608. [Google Scholar]
- Carmignoto, G.; Gomez-Gonzalo, M. The contribution of astrocyte signalling to neurovascular coupling. Brain Res. Rev 2010, 63, 138–148. [Google Scholar]
- Rossi, D.J.; Brady, J.D.; Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci 2007, 10, 1377–1386. [Google Scholar]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.A.; Pozzan, T.; Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci 2003, 6, 43–50. [Google Scholar]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Stahlberg, A.; Aprico, K.; Larsson, K.; Yabe, T.; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab 2008, 28, 468–481. [Google Scholar]
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med 2006, 12, 829–834. [Google Scholar]
- Voskuhl, R.R.; Peterson, R.S.; Song, B.; Ao, Y.; Morales, L.B.; Tiwari-Woddruff, S.; Sofroniew, M.V. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci 2009, 29, 11511–11522. [Google Scholar]
- Cotrina, M.L.; Nedergaard, M. Astrocytes in the aging brain. J. Neurosci. Res 2002, 67, 1–10. [Google Scholar]
- Dinapoli, V.A.; Benkovic, S.A.; Li, X.; Kelly, K.A.; Miller, D.B.; Rosen, C.L.; Huber, J.D.; O’Callaghan, J.P. Age exaggerates proinflammatory cytokine signaling and truncates signal transducers and activators of transcription 3 signaling following ischemic stroke in the rat. Neuroscience 2010, 170, 633–644. [Google Scholar]
- Pettigrew, L.C.; Kasner, S.E.; Albers, G.W.; Gorman, M.; Grotta, J.C.; Sherman, D.G.; Funakoshi, Y.; Ishibashi, H. Arundic Acid (ONO-2506); Stroke Study Group. Safety and tolerability of arundic acid in acute ischemic stroke. J. Neurol. Sci. 2006, 251, 50–56. [Google Scholar]
- Tateishi, N.; Mori, T.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Matsui, T.; Asano, T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: Suppression of astrocytic activation by a novel agent (R)-(–)-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J. Cereb. Blood Flow Metab 2002, 22, 723–734. [Google Scholar]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar]
- Lalancette-Hebert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci 2007, 27, 2596–2605. [Google Scholar]
- Hayakawa, K.; Mishima, K.; Nozako, M.; Hazekawa, M.; Mishima, S.; Fujioka, M.; Orito, K.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke 2008, 39, 951–958. [Google Scholar]
- Sakata, H.; Niizuma, K.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Narasimhan, P.; Maier, C.M.; Nishiyama, Y.; Chan, P.H. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J. Neurosci 2012, 32, 3462–3473. [Google Scholar]
- Xu, L.; Fagan, S.C.; Waller, J.L.; Edwards, D.; Borlongan, C.V.; Zheng, J.; Hill, W.D.; Feuerstein, G.; Hess, D.C. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol 2004, 4, 7. [Google Scholar]
- Campbell, J.H.; Burdo, T.H.; Autissier, P.; Bombardier, J.P.; Westmoreland, S.V.; Soulas, C.; Gonzalez, R.G.; Ratai, E.-M.; Williams, K.C. Minocycline inhibition of monocyte activation correlates with neuronal protection in SIV neuroAIDS. PLoS One 2011, 6, e18688. [Google Scholar]
- Yenari, M.A.; Xu, L.; Tang, X.N.; Qiao, Y.; Giffard, R.G. Microglia potentiate damage to blood-brain barrier constituents: Improvement by minocycline in vivo and in vitro. Stroke 2006, 37, 1087–1093. [Google Scholar]
- Lampl, Y.; Boaz, M.; Gilad, R.; Lorberboym, M.; Dabby, R.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410. [Google Scholar]
- Fagan, S.C.; Cronic, L.E.; Hess, D.C. Minocycline development for acute ischemic stroke. Transl. Stroke Res 2011, 2, 202–208. [Google Scholar]
- Switzer, J.A.; Hess, D.C.; Ergul, A.; Waller, J.L.; Machado, L.S.; Dobos-Portik, V.; Pettigrew, L.C.; Clark, W.M.; Fagan, S.C. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke 2011, 42, 2633–2635. [Google Scholar]
- Lee, S.R.; Lo, E.H. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. J. Cereb. Blood Flow Metab 2004, 24, 720–727. [Google Scholar]
- Lee, S.R.; Tsuji, K.; Lo, E.H. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J. Neurosci 2004, 24, 671–678. [Google Scholar]
- Lo, E.H.; Wang, X.; Cuzner, M.L. Extracellular proteolysis in brain injury and inflammation: Role for plasminogen activators and matrix metalloproteinases. J. Neurosci. Res 2002, 69, 1–9. [Google Scholar]
- Rosell, A.; Lo, E.H. Multiphasic roles for matrix metalloproteinases after stroke. Curr. Opin. Pharmacol 2008, 8, 82–89. [Google Scholar]
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med 2006, 12, 441–445. [Google Scholar]
- Pfefferkorn, T.; Rosenberg, G.A. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke 2003, 34, 2025–2030. [Google Scholar]
- Mbye, L.H.; Keles, E.; Tao, L.; Zhang, J.; Chung, J.; Larvie, M.; Koppula, R.; Lo, E.H.; Whalen, M.J. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab 2012, 32, 515–524. [Google Scholar]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci 2011, 12, 388–399. [Google Scholar]
- Polazzi, E.; Monti, B. Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol 2010, 92, 293–315. [Google Scholar]
- Popovich, P.G.; Longbrake, E.E. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci 2008, 9, 481–493. [Google Scholar]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol 2008, 8, 958–969. [Google Scholar]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol 2011, 11, 723–737. [Google Scholar]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol 2011, 11, 750–761. [Google Scholar]
- Bettermann, K. Biomarkers for stroke: In search of fingerprints. J. Stroke Cerebrovasc. Dis 2011, 20, 173–176. [Google Scholar]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar]
- Than, M.; Cullen, L.; Aldous, S.; Parsonage, W.A.; Reid, C.M.; Greenslade, J.; Flaws, D.; Hammett, C.J.; Beam, D.M.; Ardagh, M.W.; et al. 2-Hour accelerated diagnostic protocol to assess patients with chest pain symptoms using contemporary troponins as the only biomarker: The ADAPT trial. J. Am. Coll. Cardiol 2012, 59, 2091–2098. [Google Scholar]
- Weant, K.A.; Baker, S.N. New windows, same old house: An update on acute stroke management. Adv. Emerg. Nurs. J 2012, 34, 112–121. [Google Scholar]
- Lansberg, M.G.; Bluhmki, E.; Thijs, V.N. Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke: A metaanalysis. Stroke 2009, 40, 2438–2441. [Google Scholar]
- Montaner, J.; Mendioroz, M.; Ribó, M.; Delgado, P.; Quintana, M.; Penalba, A.; Chacón, P.; Molina, C.; Fernández-Cadenas, I.; Rosell, A.; et al. A panel of biomarkers including caspase-3 and D-dimer may differentiate acute stroke from stroke-mimicking conditions in the emergency department. J Intern Med 2011, 270, 166–174. [Google Scholar]
- Navarro-Sobrino, M.; Rosell, A.; Hernandez-Guillamon, M.; Penalba, A.; Boada, C.; Domingues-Montanari, S.; Ribo, M.; Alvarez-Sabin, J.; Montaner, J. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis 2011, 216, 205–211. [Google Scholar]
- Qian, L.; Ding, L.; Cheng, L.; Zhu, X.; Zhao, H.; Jin, J.; Guan, D.; Zhang, B.; Chen, X.; Xu, Y. Early biomarkers for post-stroke cognitive impairment. J. Neurol 2012, 259, 2111–2118. [Google Scholar]
- Tsai, T.H.; Chen, Y.L.; Lin, H.S.; Liu, C.F.; Chang, H.W.; Lu, C.H.; Chang, W.N.; Chen, S.F.; Wu, C.J.; Leu, S.; et al. Link between lipoprotein-associated phospholipase A2 gene expression of peripheral-blood mononuclear cells and prognostic outcome after acute ischemic stroke. J. Atheroscler. Thromb 2012, 19, 523–531. [Google Scholar]
- Ramos-Fernandez, M.; Bellolio, M.F.; Stead, L.G. Matrix metalloproteinase-9 as a marker for acute ischemic stroke: A systematic review. J. Stroke Cerebrovasc. Dis 2011, 20, 47–54. [Google Scholar]
- Vangilder, R.L.; Huber, J.D.; Rosen, C.L.; Barr, T.L. The transcriptome of cerebral ischemia. Brain Res. Bull 2012, 88, 313–319. [Google Scholar]
- Denes, A.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Interleukin-1 and stroke: Biomarker, harbinger of damage, and therapeutic target. Cerebrovasc. Dis 2011, 32, 517–527. [Google Scholar]
- Montaner, J.; Alvarez-Sabín, J.; Molina, C.; Anglés, A.; Abilleira, S.; Arenillas, J.; González, M.A.; Monasterio, J. Matrix metalloproteinase expression after human cardioembolic stroke: Temporal profile and relation to neurological impairment. Stroke 2001, 32, 1759–1766. [Google Scholar]
- Castellanos, M.; Leira, R.; Serena, J.; Pumar, J.M.; Lizasoain, I.; Castillo, J.; Dávalos, A. Plasma metalloproteinase-9 concentration predicts hemorrhagic transformation in acute ischemic stroke. Stroke 2003, 34, 40–46. [Google Scholar]
- Montaner, J.; Molina, C.A.; Monasterio, J.; Abilleira, S.; Arenillas, J.F.; Ribó, M.; Quintana, M.; Alvarez-Sabín, J. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation 2003, 107, 598–603. [Google Scholar]
- Tayebjee, M.H.; Lim, H.S.; MacFadyen, R.J.; Lip, G.Y. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 and -2 in type 2 diabetes: Effect of 1 year’s cardiovascular risk reduction therapy. Diabetes Care 2004, 27, 2049–2051. [Google Scholar]
- Sundstrom, J.; Vasan, R.S. Circulating biomarkers of extracellular matrix remodeling and risk of atherosclerotic events. Curr. Opin. Lipidol 2006, 17, 45–53. [Google Scholar]
- Hasan, N.; McColgan, P.; Bentley, P.; Edwards, R.J.; Sharma, P. Towards the identification of blood biomarkers for acute stroke in humans: A comprehensive systematic review. Br. J. Clin. Pharmacol 2012, 74, 230–240. [Google Scholar]
- Marginean, I.C.; Stanca, D.M.; Vacaras, V.; Soritau, O.; Margiean, M.; Muresanu, D.F. Plasmatic markers in hemorrhagic stroke. J. Med. Life 2011, 4, 148–150. [Google Scholar]
- Metting, Z.; Wilczak, N.; Rodiger, L.A.; Schaaf, J.M.; van der Naalt, J. GFAP and S100B in the acute phase of mild traumatic brain injury. Neurology 2012, 78, 1428–1433. [Google Scholar]
- Wiesmann, M.; Steinmeier, E.; Magerkurth, O.; Linn, J.; Gottmann, D.; Missler, U. Outcome prediction in traumatic brain injury: Comparison of neurological status, CT findings, and blood levels of S100B and GFAP. Acta Neurol. Scand 2010, 121, 178–185. [Google Scholar]
- Montaner, J.; Mendioroz, M.; Delgado, P.; García-Berrocoso, T.; Giralt, D.; Merino, C.; Ribó, M.; Rosell, A.; Penalba, A.; Fernández-Cadenas, I. Differentiating ischemic from hemorrhagic stroke using plasma biomarkers: The S100B/RAGE pathway. J. Proteomics 2012, 75, 4758–4765. [Google Scholar]
- Jonsson, H.; Johnsson, P.; Birch-Iensen, M.; Alling, C.; Westaby, S.; Blomquist, S. S100B as a predictor of size and outcome of stroke after cardiac surgery. Ann. Thorac. Surg 2001, 71, 1433–1437. [Google Scholar]
- Glickman, S.W.; Phillips, S.; Anstrom, K.J.; Laskowitz, D.T.; Cairns, C.B. Discriminative capacity of biomarkers for acute stroke in the emergency department. J. Emerg. Med 2011, 41, 333–339. [Google Scholar]
- Peric, B.; Zagar, I.; Novakovic, S.; Zgajnar, J.; Hocevar, M. Role of serum S100B and PET-CT in follow-up of patients with cutaneous melanoma. BMC Cancer 2011, 11, 328. [Google Scholar]
- Strobel, K.; Skalsky, J.; Kalff, V.; Baumann, K.; Seifert, B.; Joller-Jemelka, H.; Dummer, R.; Steinert, H.C. Tumour assessment in advanced melanoma: Value of FDG-PET/CT in patients with elevated serum S-100B. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1366–1375. [Google Scholar]
- Hirano, K.; Takashima, S.; Dougu, N.; Taguchi, Y.; Nukui, T.; Konishi, H.; Toyoda, S.; Kitajima, I.; Tanaka, K. Study of hemostatic biomarkers in acute ischemic stroke by clinical subtype. J. Stroke Cerebrovasc. Dis 2012, 21, 404–410. [Google Scholar]
- Alvarez-Perez, F.J.; Castelo-Branco, M.; Alvarez-Sabin, J. Usefulness of measurement of fibrinogen, D-dimer, D-dimer/fibrinogen ratio, C reactive protein and erythrocyte sedimentation rate to assess the pathophysiology and mechanism of ischaemic stroke. J. Neurol. Neurosurg. Psychiatry 2011, 82, 986–992. [Google Scholar]
- Ning, M.; Sarracino, D.A.; Buonanno, F.S.; Krastins, B.; Chou, S.; McMullin, D.; Wang, X.; Lopez, M.; Lo, E.H. Proteomic protease substrate profiling of tPA treatment in acute ischemic stroke patients: A step toward individualizing thrombolytic therapy at the bedside. Transl. Stroke Res 2010, 1, 268–275. [Google Scholar]
- Hayakawa, K.; Pham, L.D.; Katusic, Z.S.; Arai, K.; Lo, E.H. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc. Natl. Acad. Sci. USA 2012, 109, 7505–7510. [Google Scholar]
- Chu, L.F.; Wang, W.T.; Ghanta, V.K.; Lin, C.H.; Chiang, Y.Y.; Hsueh, C.M. Ischemic brain cell-derived conditioned medium protects astrocytes against ischemia through GDNF/ERK/NF-kB signaling pathway. Brain Res 2008, 1239, 24–35. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Turner, R.C.; Lucke-Wold, B.; Lucke-Wold, N.; Elliott, A.S.; Logsdon, A.F.; Rosen, C.L.; Huber, J.D. Neuroprotection for Ischemic Stroke: Moving Past Shortcomings and Identifying Promising Directions. Int. J. Mol. Sci. 2013, 14, 1890-1917. https://doi.org/10.3390/ijms14011890
Turner RC, Lucke-Wold B, Lucke-Wold N, Elliott AS, Logsdon AF, Rosen CL, Huber JD. Neuroprotection for Ischemic Stroke: Moving Past Shortcomings and Identifying Promising Directions. International Journal of Molecular Sciences. 2013; 14(1):1890-1917. https://doi.org/10.3390/ijms14011890
Chicago/Turabian StyleTurner, Ryan C., Brandon Lucke-Wold, Noelle Lucke-Wold, Alisa S. Elliott, Aric F. Logsdon, Charles L. Rosen, and Jason D. Huber. 2013. "Neuroprotection for Ischemic Stroke: Moving Past Shortcomings and Identifying Promising Directions" International Journal of Molecular Sciences 14, no. 1: 1890-1917. https://doi.org/10.3390/ijms14011890