Analysis of Genome Survey Sequences and SSR Marker Development for Siamese Mud Carp, Henicorhynchus siamensis, Using 454 Pyrosequencing

Abstract
:1. Introduction
2. Results and Discussion
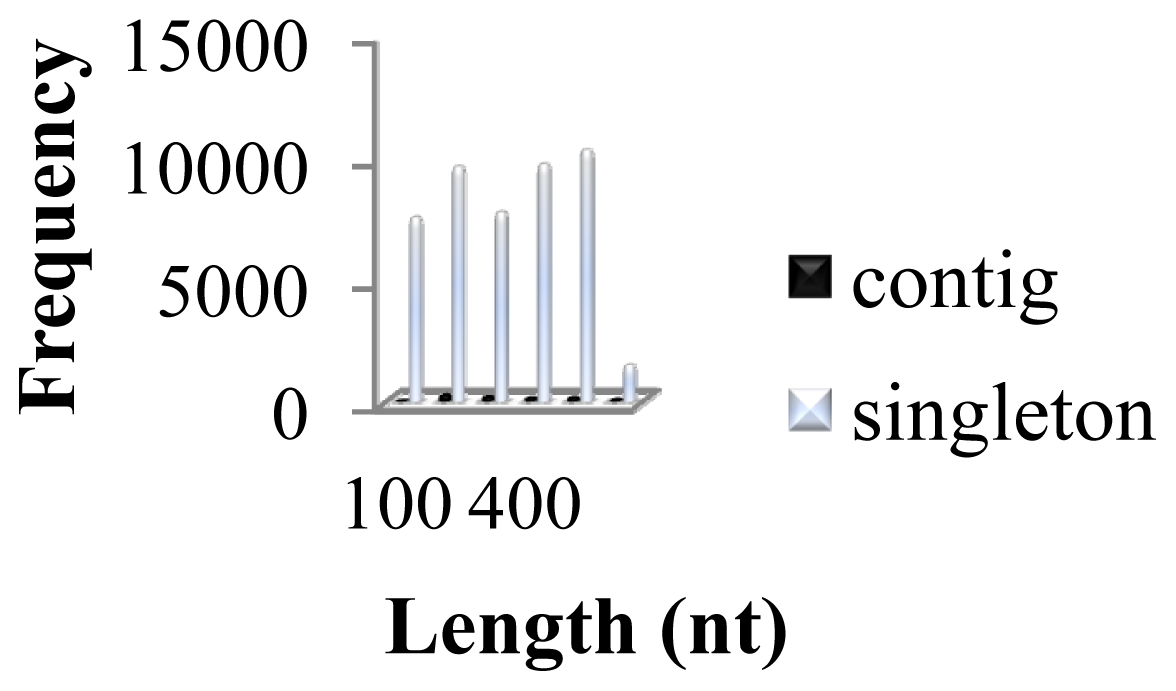
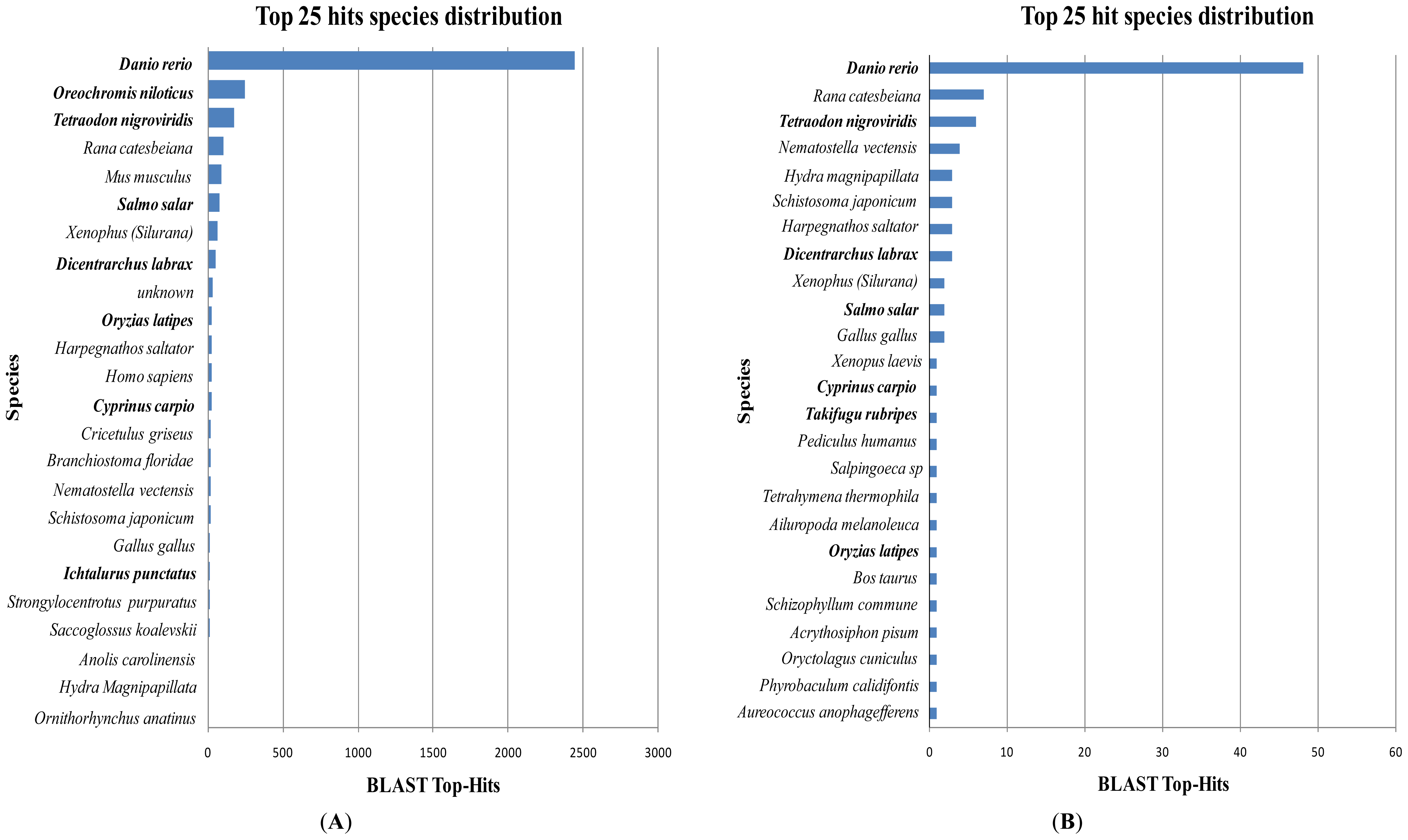
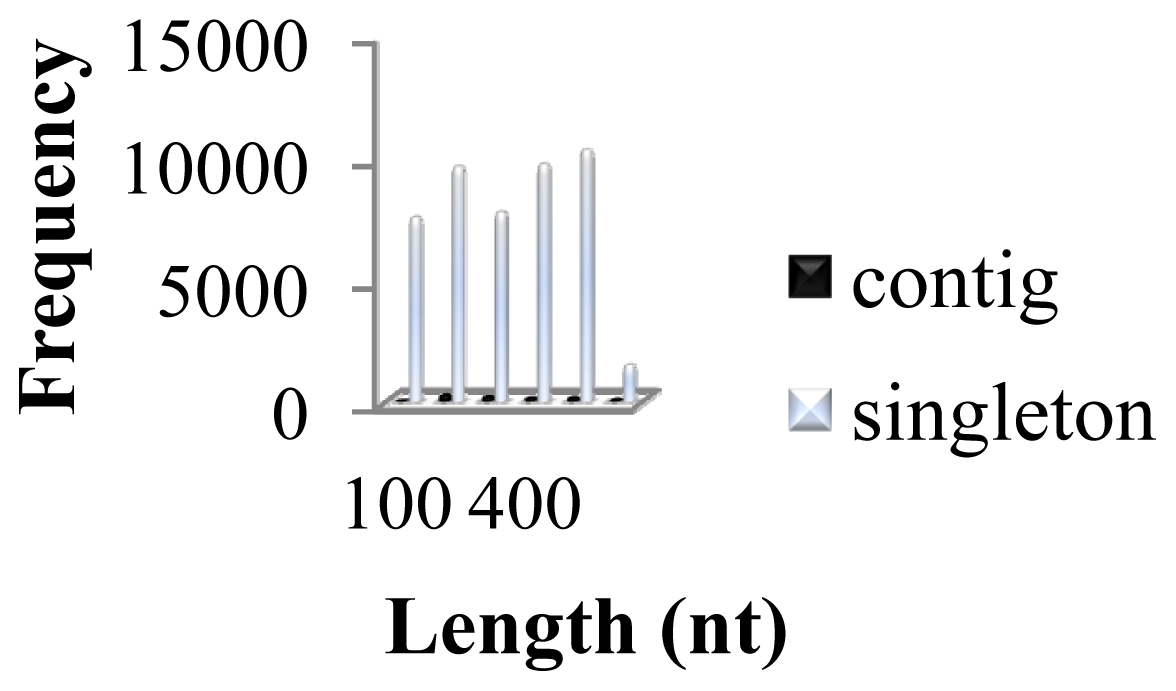
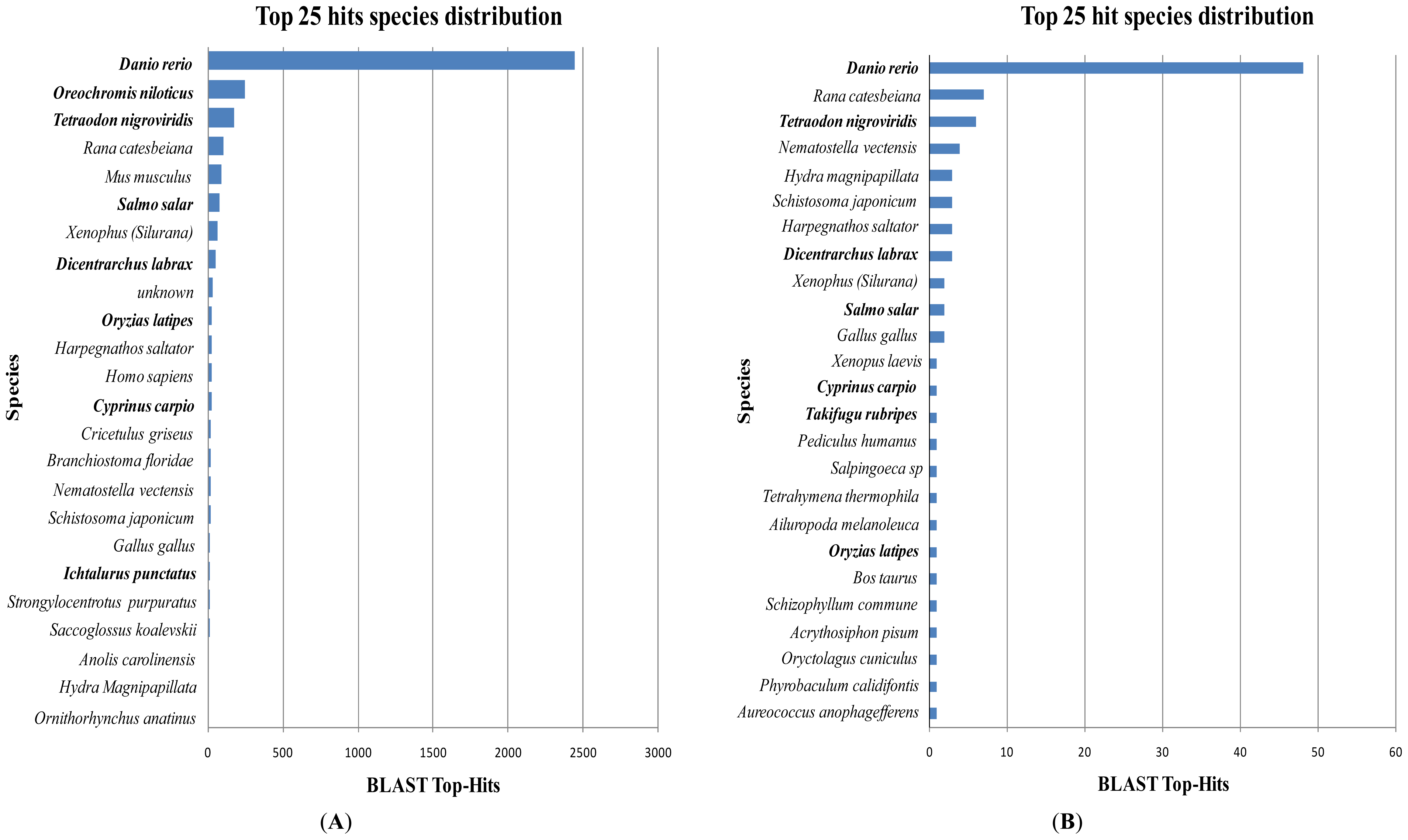
2.1. Comparative Analysis of GSSs
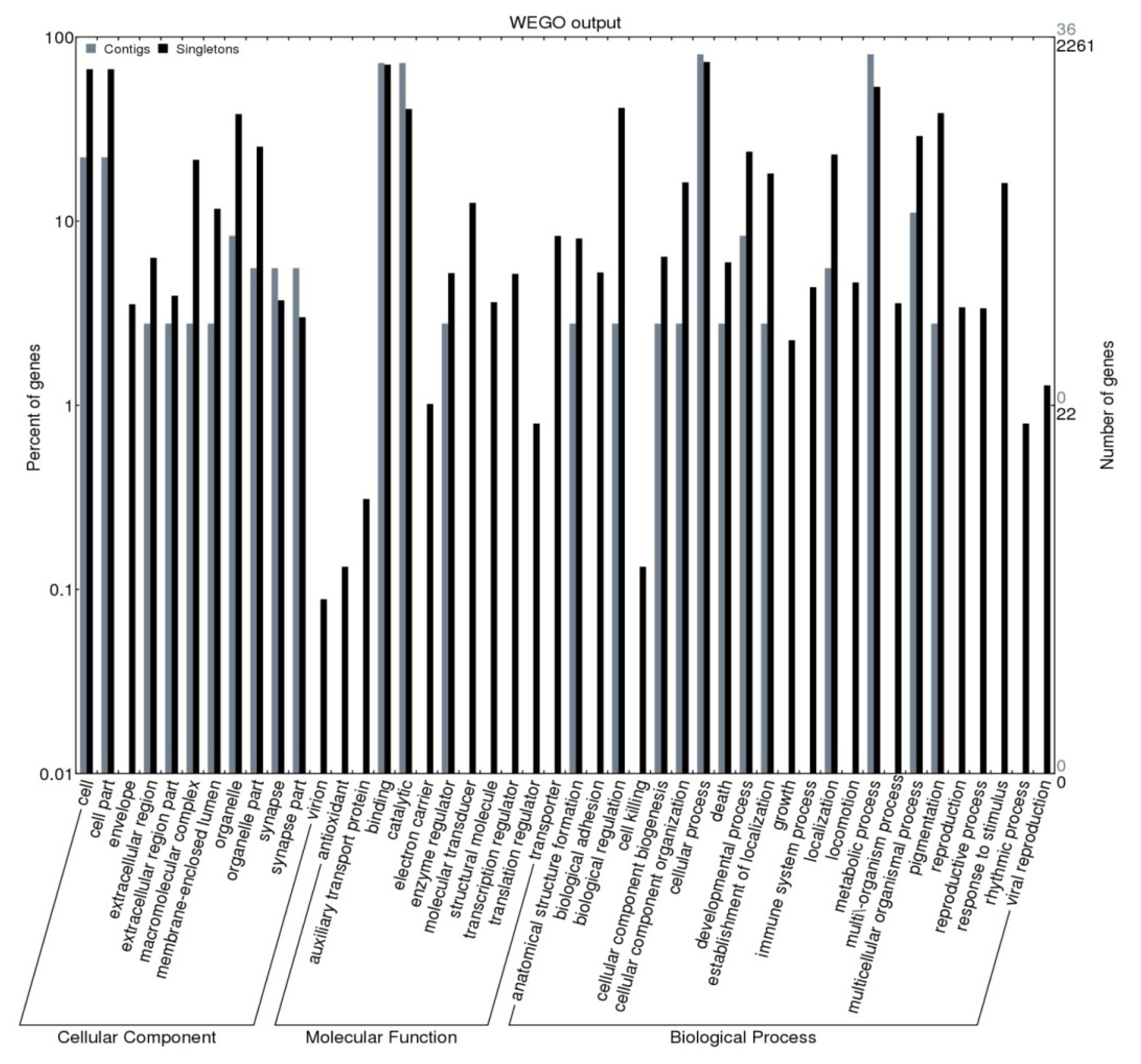
2.2. Gene Ontology Assignment
2.3. KEGG Analysis
2.4. Protein Domains
2.5. Analysis of Genes
2.6. Putative Microsatellite Markers
2.7. GSS-SSR Markers Test
3. Experimental Section
3.1. Samples and DNA Extraction
3.2. Library Construction and 454 Pyrosequencing
3.3. Sequence Cleaning and Assembly
3.4. Annotation
3.5. Identification of GSS-SSR Motifs
3.6. Microsatellite Screening, Amplification and Testing
4. Conclusions
Acknowledgments
References
- Rainboth, W.J. Fishes of the Cambodian Mekong. FAO Species Identification Field Guide for Fishery Purposes; Food and Agriculture Organization of the United Nations: Rome, Italy, 1996; pp. 111–122. [Google Scholar]
- Poulsen, A.F.; Valbo-Jørgensen, J. Fish Migrations and Spawning Habits in the Mekong Mainstream—A Survey Using Local Knowledge; AMFC Technical Report; Mekong River Commission: Phnom Penh, Cambodia, 2000; pp. 38–42. [Google Scholar]
- Lieng, S.; Yim, C.; van Zalinge, N.P. Fisheries of Cambodia I: The bagnet (Dai) fishery in the Tonlesap River. Asian Fish. Sci 1995, 8, 255–262. [Google Scholar]
- Baird, I.G.; Flaherty, M.S.; Phylavanh, B. Rhythms of the river: Lunar phases and migrations of small carp (Cyprinidae) in the Mekong River. Nat. Hist. Bull. Siam Soc 2003, 51, 5–36. [Google Scholar]
- Baran, E.; van Zalinge, N.; Bun, N.P. Floods, Floodplains and Fish Production in the Mekong Basin: Present and Past Trends. Proceeding of the Second Asian Wetland Symposium, Penang, Malaysia, 27–30 August 2001; Penerbit Universiti Sains: Kuala Lumpur, Malaysia, 2001; pp. 920–932. [Google Scholar]
- Adamson, E.A.S.; Hurwood, D.A.; Baker, A.M.; Mather, P.B. Population subdivision in Siamese mud carp Henicorhynchus siamensis in the Mekong River basin: Implications for management. J. Fish Biol 2009, 75, 1371–1392. [Google Scholar]
- Hurwood, D.A.; Adamson, E.A.S.; Mather, P.B. Evidence for strong genetic structure in a regionally important, highly vagile cyprinid (Henicorhynchus lobatus) in the Mekong River Basin. Ecol. Freshw. Fish 2008, 17, 273–283. [Google Scholar]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000; pp. 22–32. [Google Scholar]
- Forister, M.L.; Nice, C.C.; Fordyce, J.A.; Gompert, Z.; Shapiro, A.M. Considering evolutionary processes in the use of single-locus genetic data for conservation, with examples from the Lepidoptera. J. Insect Conserv 2008, 12, 37–51. [Google Scholar]
- Zink, R.M.; Barrowclough, G.F. Mitochondrial DNA under siege in avian phylogeography. Mol. Ecol 2008, 17, 2107–2121. [Google Scholar]
- Féral, J.P. How useful are the genetic markers in attempts to understand and manage marine biodiversity? J. Exp. Mar. Biol. Ecol 2002, 268, 121–145. [Google Scholar]
- Zhang, D.; Hewitt, G.M. Nuclear DNA analyses in genetic studies of populations: Practice, problems and prospects. Mol. Ecol 2003, 12, 563–584. [Google Scholar]
- Cheng, L.; Liao, X.; Yu, X.; Tong, J. Development of EST-SSRs by an efficient FIASCO-based strategy: A case study in rare minnow (Gobiocyrpis Rarus). Anim. Biotech 2007, 18, 143–152. [Google Scholar]
- Wang, J.; Yu, X.; Zhao, K.; Zhang, Y.; Tong, J.; Peng, Z. Microsatellite development for an endangered bream Megalobrama pellegrini (Teleostei, Cyprinidae) using 454 sequencing. Int. J. Mol. Sci 2012, 13, 3009–3021. [Google Scholar]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour 2011, 11, 591–611. [Google Scholar]
- Morin, P.A.; Luikart, G.; Wayne, R.K. The SNP workshop group. SNPs in ecology, evolution and conservation. Trends Ecol. Evol 2004, 19, 208–216. [Google Scholar]
- O’Connell, M.; Write, J.M. Microsatellite DNA in fishes. Rev. Fish Biol. Fish 1997, 7, 331–363. [Google Scholar]
- Romana-Eguia, M.R.R.; Ikeda, M.; Basiao, Z.U.; Taniguchi, N. Genetic diversity in farmed Asian Nile and red hybrid tilapia stocks evaluated from microsatellite and mitochondrial DNA analysis. Aquaculture 2004, 236, 131–150. [Google Scholar]
- Vera, J.C.; Wheat, C.W.; Fescemyer, H.W.; Frilander, M.J.; Crawford, D.L.; Hanski, I.; Marden, J.H. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol. Ecol 2008, 17, 1636–1647. [Google Scholar]
- Salem, M.; Rexroad, C.E., III; Wang, J.; Thorgaard, G.H.; Yao, J. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Jung, H.; Lyons, R.E.; Dinh, H.; Hurwood, D.A.; McWilliam, S.; Mather, P.B. Transcriptomics of a giant freshwater prawn (Macrobrachium rosenbergii): De novo assembly, annotation and marker discovery. PLoS One 2011, 6, e27938. [Google Scholar]
- Strong, W.B.; Nelson, R.G. Preliminary profile of the Cryptosporidium parvum genome: An expressed sequence tag and genome survey sequence analysis. Mol. Biochem. Parasitol 2000, 107, 1–32. [Google Scholar]
- Lynch, M.; Sung, W.; Morris, K.; Coffey, N.; Landry, C.R.; Dopman, E.B.; Dickinson, W.J.; Okamoto, K.; Kulkarni, S.; Hartl, D.L.; et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. USA 2008, 105, 9272–9277. [Google Scholar]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; de Pamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar]
- Elmer, K.R.; Fan, S.; Gunter, H.M.; Jones, J.C.; Boekhoff, S.; Kuraku, S.; Meyer, A. Rapid evolution and selection inferred from the transcriptomes of sympatric craterlake cichlid fishes. Mol. Ecol 2010, 19, 197–211. [Google Scholar]
- Burge, C.B.; Karlin, S. Finding the genes in genomic DNA. Curr. Opin. Struct. Biol 1998, 8, 346–354. [Google Scholar]
- Shikano, T.; Ramadevi, J.; Shimada, Y.; Merila, J. Utility of sequenced genomes for microsatellite marker development in non-model organisms: A case study of functionally important genes in nine-spined sticklebacks (Pungitius pungitius). BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Milano, I.; Babbucci, M.; Panitz, F.; Ogden, R.; Nielsen, R.O.; Taylor, M.I.; Helyar, S.J.; Carvalho, G.R.; Espineira, M.; Atanassova, M.; et al. Novel tools for conservation genomics: Comparing two high-throughput approaches for SNP discovery in the transcriptome of the European hake. PLoS One 2011, 6, e28008. [Google Scholar]
- Jiang, Y.; Lu, J.; Peatman, E.; Kucuktas, H.; Liu, S.; Wang, S.; Sun, F.; Liu, Z. A pilot study for channel catfish whole genome sequencing and de novo assembly. BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Coppe, A.; Pujolar, J.M.; Maes, G.E.; Larsen, P.F.; Hansen, M.M.; Bernatchez, L.; Zane, L.; Bortoluzzi, S. Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EelBase opens new perspectives for the study of the critically endangered European eel. BMC Genomics 2010, 11, 635. [Google Scholar]
- Panhuis, T.M.; Broitman-Maduro, G.; Uhrig, J.; Maduro, M.; Reznick, D.N. Analysis of expressed sequence tags from the placenta of the live-bearing fish Poeciliopsis (Poeciliidae). J. Hered 2011, 102, 353–361. [Google Scholar]
- Wang, J.-P.Z.; Lindsay, B.G.; Leebens-Mack, J.; Cui, L.; Wall, K.; Miller, W.C.; depamphilis, C.W. EST clustering error evaluation and correction. Bioinformatics 2004, 20, 2973–2984. [Google Scholar]
- Mittapalli, O.; Bai, X.; Mamidala, P.; Rajarapu, S.P.; Bonello, P.; Herms, D.A. Tissue-specific transcriptomics of the exotic invasive insect pest emerald ash borer. PLoS One 2010, 5, e13708. [Google Scholar]
- Ng, A.; Uribe, R.A.; Yieh, L.; Nuckels, R.; Gross, J.M. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 2009, 136, 2601–2611. [Google Scholar]
- Mommsen, T.P.; Hochachka, P.W. The purine nucleotide cycle as two temporally separated metabolic units. A study on trout muscle. Metabolism 1988, 37, 552–556. [Google Scholar]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase. The energy charge hypothesis revisited. BioEssays 2001, 23, 1112–1119. [Google Scholar]
- Tiong, S.Y.; Keizer, C.; Nash, D.; Bleskan, J.; Patterson, D. Drosophila purine auxotrophy: New alleles of adenosine 2 exhibiting a complex visible phenotype. Biochem. Genet 1989, 27, 333–348. [Google Scholar]
- Weber, G.; Sullivan, C. In vitro hormone induction of final oocyte maturation in striped bass (Morone saxatilis) follicles is inhibited by blockers of phosphatidylinositol 3-kinase activity. Comp. Biochem. Physiol. B 2001, 129, 467–473. [Google Scholar]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar]
- Pace, M.C.; Thomas, P. Steroid-induced oocyte maturation in Atlantic croaker (Micropogonias undulatus) is dependent on activation of the phosphatidylinositol 3-kinase/akt signal transduction pathway. Biol. Reprod 2005, 73, 988–996. [Google Scholar]
- Kavanaugh, S.I.; Nozaki, M.; Sower, S.A. Origins of gonadotropin-releasing hormone (GnRH) in vertebrates: Identification of a novel GnRH in a basal vertebrate, the sea lamprey. Endocrinology 2008, 149, 3860–3869. [Google Scholar]
- Pemberton, J.G.; Stafford, J.L.; Yu, Y.; Chang, J.P. Differential involvement of phosphoinositide 3-kinase in gonadotrophin-releasing hormone actions in gonadotrophs and somatotrophs of goldfish, Carassius auratus. J. Neuroendocrinol 2011, 23, 660–674. [Google Scholar]
- Wallace, K.B.; Starkov, A.A. Mitochondrial targets of drug toxicity. Annu. Rev. Pharmacol. Toxicol 2000, 40, 353–388. [Google Scholar]
- Birceanu, O.; McClelland, G.B.; Wang, Y.S.; Brown, J.C.L.; Wilkie, M.P. The lampricide 3-trifluoromethyl-4-nitrophenol (TFM) uncouples mitochondrial oxidative phosphorylation in both sea lamprey (Petromyzon marinus) and TFM-tolerant rainbow trout (Oncorhynchus mykiss). Comp. Biochem. Physiol. C 2011, 153, 342–349. [Google Scholar]
- Tocher, D.R. Metabolism and functions of lipids and fatty acids in teleost fish. Rev. Fish Sci 2003, 11, 107–184. [Google Scholar]
- Leaver, M.J.; Bautista, J.M.; Björnsson, T.; Jönsson, E.; Krey, G.; Tocher, D.R.; Torstensen, B.E. Towards fish lipid nutrigenomics: Current state and prospects for fin-fish aquaculture. Rev. Fish Sci 2008, 16, 73–94. [Google Scholar]
- Minghetti, M.; Leaver, M.J.; Tocher, D.R. Transcriptional control mechanisms of genes of lipid and fatty acid metabolism in the Atlantic salmon (Salmo salar L.) established cell line, SHK-1. Biochim. et Biophys. Acta 2011, 1811, 194–202. [Google Scholar]
- Kock, H.; Fischer, U. A novel immunoglobulin-like transcript from rainbow trout with two Ig-like domains and two isoforms. Mol. Immunol 2008, 45, 1612–1622. [Google Scholar]
- Østergaard, A.E.; Lubieniecki, K.P.; Martin, S.A.M.; Stet, R.J.M.; Davidson, W.S.; Secombes, C.J. Genomic organisation analysis of novel immunoglobulin-like transcripts in Atlantic salmon (Salmo salar) reveals a tightly clustered and multigene family. BMC Genomics 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Secombes, C.J. Fish immunity: The potential impact on vaccine development and performance. Aqua Res 2011, 42, 90–92. [Google Scholar]
- Teichmann, S.A.; Chothia, C. Immunoglobulin superfamily proteins in Caenorhabditis elegans. J. Mol. Biol 2000, 296, 1367–1383. [Google Scholar]
- Potapov, V.; Sobolev, V.; Edelman, M.; Kister, A.; Gelfand, I. Protein-protein recognition: Juxtaposition of domain and interface cores in immunoglobulins and other sandwich-like proteins. J. Mol. Biol. 2004, 342, 665–679. [Google Scholar]
- Brown, R.S. Zinc finger proteins: Getting a grip on RNA. Curr. Opin. Struct. Biol 2005, 15, 94–98. [Google Scholar]
- Wolfe, S.A.; Nekludova, L.; Pabo, C.O. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct 2000, 29, 183–212. [Google Scholar]
- Brayer, K.J.; Segal, D.J. Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys 2008, 50, 111–131. [Google Scholar]
- Craigie, R. Retroviral DNA Integration. In Mobile DNA II; Craig, N.L., Craigie, R., Gellert, M., Lambowitz, A.M., Eds.; ASM Press: Washington. DC, USA, 2002; pp. 613–630. [Google Scholar]
- Gao, X.; Voytas, D.F. A eukaryotic gene family related to retroelement integrases. Trends Genetics 2005, 21, 133–137. [Google Scholar]
- Gumbiner, B.M. Regulation of cadherin adhesive activity. J. Cell Biol 2000, 148, 399–404. [Google Scholar]
- Wheelock, M.J.; Johnson, K.R. Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol 2003, 19, 207–35. [Google Scholar]
- Langin, T.; Capy, P.; Daboussi, M.J. The transposable element impala, a fungal member of the Tc1-mariner superfamily. Mol. Gen. Genet 1995, 246, 19–28. [Google Scholar]
- Robertson, H.M. The Tc1-mariner superfamily of transposons in animals. J. Insect Physiol 1995, 41, 99–105. [Google Scholar]
- Avancini, R.; Walden, K.; Robertson, H. The genomes of most animals have multiple members of the Tc1 family of transposable elements. Genetica 1996, 98, 131–140. [Google Scholar]
- Capriglione, T.; Odierna, G.; Caputo, V.; Canapa, A.; Olmo, E. Characterization of a Tc1-like transposon in the Antarctic ice-fish, Chionodraco hamatus. Gene 2002, 295, 193–198. [Google Scholar]
- Clark, K.J.; Carlson, D.F.; Leaver, M.J.; Foster, L.K.; Fahrenkrug, S.C. Passport, a native Tc1 transposon from flatfish, is functionally active in vertebrate cells. Nucleic Acids Res 2009, 37, 1239–1247. [Google Scholar]
- Tafalla, C.; Estepa, A.; Coll, J.M. Fish transposons and their potential use in aquaculture. J. Biotechnol 2006, 123, 397–412. [Google Scholar]
- Collier, L.S.; Carlson, C.M.; Ravimohan, S.; Dupuy, A.J.; Largaespada, D.A. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 2005, 436, 272–276. [Google Scholar]
- Dupuy, A.J.; Akagi, K.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A. Cancer biology: Sleeping beauty awakens. Nature 2005, 436, 184–186. [Google Scholar]
- Krücken, J.; Schroetel, R.M.; Müller, I.U.; Saïdani, N.; Marinovski, P.; Benten, W.P.; Stamm, O.; Wunderlich, F. Comparative analysis of the human gimap gene cluster encoding a novel GTPase family. Gene 2004, 341, 291–304. [Google Scholar]
- Saunders, A.; Lamb, T.; Pascall, J.; Hutchings, A.; Dion, C.; Carter, C.; Hepburn, L.; Langhorne, J.; Butcher, G.W. Expression of GIMAP1, a GTPase of the immunity-associated protein family, is not up-regulated in malaria. Malar. J 2009, 8, 53. [Google Scholar]
- Leveelahti, L.; Leskinen, P.; Leder, E.H.; Waser, W.; Nikinmaa, M. Responses of threespine stickleback (Gasterosteus aculeatus, L.) transcriptome to hypoxia. Comp. Biochem. Physiol. D 2011, 6, 370–381. [Google Scholar]
- Timmerhaus, G.; Krasnov, A.; Nilsen, P.; Alarcon, M.; Afanasyev, S.; Rode, M.; Takle, H.; Jøgensen, S.M. Transcriptome profiling of immune responses to cardiomyopathy syndrome (CMS) in Atlantic salmon. BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar]
- Zheng, X.H.; Lu, C.Y.; Zhao, Y.Y.; Lee, C.; Cao, D.C.; Chang, Y.M.; Liang, L.Q.; Sun, X.W. A set of polymorphic trinucleotide and tetranucleotide microsatellite markers for silver Crucian carp (Carassius auratus gibelio) and cross-amplification in Crucian carp. Biochem. Genet 2010, 48, 624–635. [Google Scholar]
- Ma, H.; Chen, S. Development of polymorphic microsatellite markers in barfin flounder (Verasper moseri) and spotted halibut (Verasper variegates) by the cross-species amplification. Mol. Biol. Rep 2011, 38, 4545–4551. [Google Scholar]
- Saarinen, E.V.; Austin, J.D. When technology meets conservation: Increased microsatellite marker production using 454 genome sequencing on the endangered Okaloosa Darter (Ethostoma okaloosae). J. Hered 2010, 101, 784–788. [Google Scholar]
- Castaño-Sanchez, C.; Fuji, K.; Hayashida, K.; Tagami, M.; Ozaki, A.; Hasegawa, O.; Sakamoto, T.; Kawai, J.; Hayashizaki, Y.; Okamoto, N. A set of polymorphic trinucleotide and tetranucleotide microsatellite markers for the Japanese flounder (Paralichthys olivaceus). Anim. Genet 2007, 38, 75–76. [Google Scholar]
- Chambers, G.K.; MacAvoy, E.S. Microsatellites: Consensus and controversy. Comp. Biochem. Physiol. B 2000, 126, 455–476. [Google Scholar]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Genetics 2004, 5, 435–445. [Google Scholar]
- Bertozzi, T.; Sanders, K.L.; Sistrom, M.J.; Gardner, M.G. Anonymous nuclear loci in non-model organisms: Making the most of high-throughput genome surveys. Bioinformatics 2012, 28, 1807–1810. [Google Scholar]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1998, 16, 1215. [Google Scholar]
- Gardner, M.G.; Fitch, A.J.; Bertozzi, T.; Lowe, A.J. Rise of the machines, recommendations for ecologists using next generation sequencing for microsatellite development. Mol. Ecol. Resour 2011, 11, 1093–1101. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Miller, W. Gapped BLAST and PSIBLAST: A new generation of protein database search programs. Nucleic. Acids Res 1997, 25, 3389–3402. [Google Scholar]
- National Center for Biotechnology Information (NCBI). Available online: http://www.ncbi.nlm.nih.gov/ accessed on 27 September 2011.
- Götz, S.; García-Gómez, J.M.; Terol, J.; William, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with Blast2GO suite. Nucleic Acids Res 2008, 36, 3420–3435. [Google Scholar]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res 2006, 34, D354–D357. [Google Scholar]
- InterPro Databases; European Molecular Biology Laboratory: Cambridgeshire, UK, 1999. Available online: http://www.ncbi.nlm.nih.gov/ accessed on 25 January 2012.
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duguenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res 2009, 37, D211–D215. [Google Scholar]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res 2006, 34, W293–W297. [Google Scholar]
- Faircloth, B.C. Msatcommander: Detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol. Ecol. Resour 2008, 8, 92–94. [Google Scholar]
- Rozen, S.; Skaletsky, H. Primer3 in the WWW for general users and for biologist programmers. Methods Mol. Biol 2000, 132, 365–386. [Google Scholar]
- Meglécz, E. MICROFAMILY (version 1): A computer program for detecting flanking-region similarities among different microsatellite loci. Mol. Ecol. Notes 2007, 7, 18–20. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar]
- Park, S.D.E. Trypanotolerance in West African cattle and the population genetic effects of selection. Ph.D. thesis, University of Dublin, Dublin, Ireland, 2001. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Dataset |
|---|---|
| Total number of bases (Mb) | 17.44 Mb |
| Average read length (nt) | 264 nt |
| Number of reads | |
| Total reads | 65,954 |
| Assembled | 5,297 |
| Singleton | 46,393 |
| Repeat | 280 |
| Number of contig | |
| Total contigs | 857 |
| Average contig read length (nt) | 352 nt |
| Largest contig (nt) | 2,373 nt |
| Number of large contigs > 500 nt | 143 |
| IPR accession | Domain name | Domain description | Total of occurrence (Con/Sing) |
|---|---|---|---|
| IPR006130 | Asp/Orn_carbamoylTrfase | Aspartate/ornithine carbamoyltransferase | 1 (1/0) |
| IPR006132 | Asp/Orn_carbamoyltranf_P-bd | Aspartate/ornithine carbamoyltransferase carbamoyl-P binding, | 1 (1/0) |
| IPR002126 | Cadherin | Cadherin | 15 (0/15) |
| IPR005135 | Exo_endo_phos | Endonuclease/exonuclease/phosphatase | 1 (1/0) |
| IPR001845 | HTH_ArsR_DNA-bd_dom | HTH arsR-type DNA-binding domain | 1 (1/0) |
| IPR013098 | Ig_I-set | Immunoglobulin I-set | 14 (1/13) |
| IPR007110 | Ig_like | Immunoglobulin-like | 26 (0/26) |
| IPR013783 | Ig-like_fold | Immunoglobulin-like fold | 84 (2/82) |
| IPR013106 | Ig_V-set | Immunoglobulin V-set | 24 (0/24) |
| IPR001584 | Integrase_cat-core | Integrase, catalytic core | 23 (1/22) |
| IPR011009 | Kinase-like_dom | Protein kinase-like domain | 14 (0/14) |
| IPR000719 | Prot_kinase_cat_dom | Protein kinase, catalytic domain | 15 (0/15) |
| IPR012337 | RNaseH-like | Ribonuclease H-like | 29 (1/28) |
| IPR000477 | RVT | Reverse transcriptase | 37 (2/35) |
| IPR000276 | 7TM_GPCR_Rhodpsn | GPCR, rhodopsin-like, 7TM | 18 (0/18) |
| IPR002492 | Transposase_Tc1-like | Transposase, Tc1-like | 2 (2/0) |
| IPR002035 | VWF_A | Von Willebrand factor, type A | 2 (2/0) |
| IPR006612 | Znf_C2CH | Zinc finger, C2CH-type | 1 (1/0) |
| IPR013087 | Znf_C2H2/integrase_DNA-bd 5 | Zinc finger, C2H2-type/ integrase, DNA-binding | 28 (0/28) |
| IPR007087 | zf-C2H2 | Zinc finger, C2H2 | 52 (0/52) |
| Candidate genes | E value range | Matched species | Length range (nt) | Total of occurrence (Con/Sing) |
|---|---|---|---|---|
| Enzymatic poly | 3.12 × 10−91–4.80 × 10−13 | Tetraodon nigroviridis | 269–1037 | 15 (3/12) |
| Lrr and pyd domainscontaining protein 12 | 3.42 × 10−35–1.72 × 10−8 | Danio rerio | 123–439 | 19 (0/19) |
| Novel protein | 1.18 × 10−65–5.42 × 10−6 | Danio rerio | 120–595 | 43 (7/36) |
| Orf2-encoded protein | 4.63 × 10−97–3.33 × 10−8 | Danio rerio | 143–1679 | 27 (7/20) |
| Protein nlrc3-like | 7.76 × 10−71–1.32 × 10−9 | Danio rerio | 220–528 | 13 (2/11) |
| Retrotransposable element tf2 | 3.95 × 10−97–4.63 × 10−6 | Takifugu rubripes | 145–527 | 120 (2/118) |
| Reverse transcriptase-like protein | 7.74 × 10−52–1.34 × 10−11 | Danio rerio | 151–544 | 22 (4/18) |
| Sjchgc01974 protein | 5.82 × 10−27–5.28 × 10−7 | Mus musculus | 139–430 | 18 (0/18) |
| Transposable element tc1 transposase 155 kda protein type 1-like | 6.41 × 10−23–1.80 × 10−8 | Danio rerio | 128–233 | 5 (5/0) |
| Transposase | 1.65 × 10−47–5.92 × 10−12 | Rana catesbeiana | 196–507 | 21 (4/17) |
| Locus | Primer sequence | Repeat Motif | Pop * | Na * | Ho * | He * | PIC * | PHWE * | Percent missing |
|---|---|---|---|---|---|---|---|---|---|
| HS2 | GTGGCGGAAATGGGCTTC | (ATCT)^14 | BB | 15 | 0.868 | 0.913 | 0.907 | 0.602 | 7% |
| CCTGAGGCATTTCATAAACTCCG | UB | 18 | 0.619 | 0.902 | 0.889 | 0.000 | 10% | ||
| HS4 | CTCATCACCCGCTGTGTTTC | (ATCT)^11 | BB | 35 | 0.775 | 0.962 | 0.961 | 0.006 | 0% |
| CACACACTGACAGGCAGAC | UB | 37 | 0.894 | 0.940 | 0.938 | 0.125 | 0% | ||
| HS5 | TGTCGTTCTCTGGCTGTCC | (ATCT)^13 | BB | 23 | 0.976 | 0.932 | 0.928 | 0.081 | 0% |
| CCCAGATACAGGAGTGGGATG | UB | 19 | 0.787 | 0.919 | 0.913 | 0.078 | 0% | ||
| HS12 | TTGCCTGGAGGACAAGACC | (ATCT)^9 | BB | 22 | 0.725 | 0.936 | 0.932 | 0.003 | 0% |
| TGCCACTGCACAGTAAACG | UB | 27 | 0.711 | 0.954 | 0.952 | 0.001 | 0% | ||
| HS14 | ACACGAGTGAGGAGTGCTG | (CTGT)^9 | BB | 14 | 0.806 | 0.846 | 0.832 | 0.608 | 12% |
| AGGCCACAAACTTCTGCTTG | UB | 15 | 0.810 | 0.822 | 0.812 | 0.550 | 10% | ||
| HS21 | CAACAAGCAGAGCGACAGG | (ACTC)^8 | BB | 7 | 0.730 | 0.705 | 0.657 | 0.981 | 0% |
| TGTTGATAACGCGCCACAG | UB | 11 | 0.596 | 0.750 | 0.722 | 0.569 | 0% | ||
| HS23 | TGAATGGAATGAGAGGTTCAGC | (GAGT)^8 | BB | 12 | 0.878 | 0.830 | 0.810 | 0.303 | 0% |
| TGCTGCTTGTGTGTTCAAAG | UB | 13 | 0.957 | 0.873 | 0.860 | 0.000 | 0% | ||
| HS24 | AACACCATACACCTGCACC | (AAAC)^8 | BB | 6 | 0.341 | 0.504 | 0.477 | 0.007 | 9% |
| ACTCCTGTGGTGGAAGAAAGG | UB | 5 | 0.467 | 0.528 | 0.483 | 0.000 | 0% |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iranawati, F.; Jung, H.; Chand, V.; Hurwood, D.A.; Mather, P.B. Analysis of Genome Survey Sequences and SSR Marker Development for Siamese Mud Carp, Henicorhynchus siamensis, Using 454 Pyrosequencing. Int. J. Mol. Sci. 2012, 13, 10807-10827. https://doi.org/10.3390/ijms130910807
Iranawati F, Jung H, Chand V, Hurwood DA, Mather PB. Analysis of Genome Survey Sequences and SSR Marker Development for Siamese Mud Carp, Henicorhynchus siamensis, Using 454 Pyrosequencing. International Journal of Molecular Sciences. 2012; 13(9):10807-10827. https://doi.org/10.3390/ijms130910807
Chicago/Turabian StyleIranawati, Feni, Hyungtaek Jung, Vincent Chand, David A. Hurwood, and Peter B. Mather. 2012. "Analysis of Genome Survey Sequences and SSR Marker Development for Siamese Mud Carp, Henicorhynchus siamensis, Using 454 Pyrosequencing" International Journal of Molecular Sciences 13, no. 9: 10807-10827. https://doi.org/10.3390/ijms130910807
APA StyleIranawati, F., Jung, H., Chand, V., Hurwood, D. A., & Mather, P. B. (2012). Analysis of Genome Survey Sequences and SSR Marker Development for Siamese Mud Carp, Henicorhynchus siamensis, Using 454 Pyrosequencing. International Journal of Molecular Sciences, 13(9), 10807-10827. https://doi.org/10.3390/ijms130910807