Cell Signaling through Protein Kinase C Oxidation and Activation

Abstract
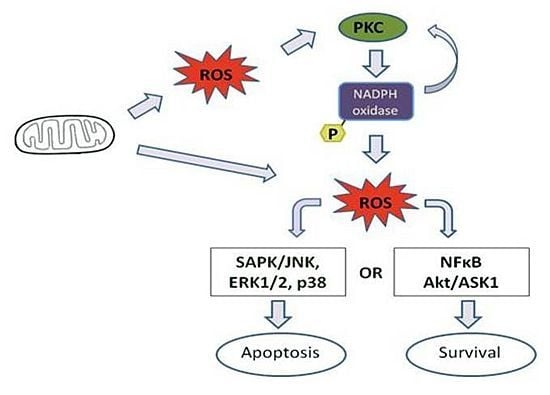
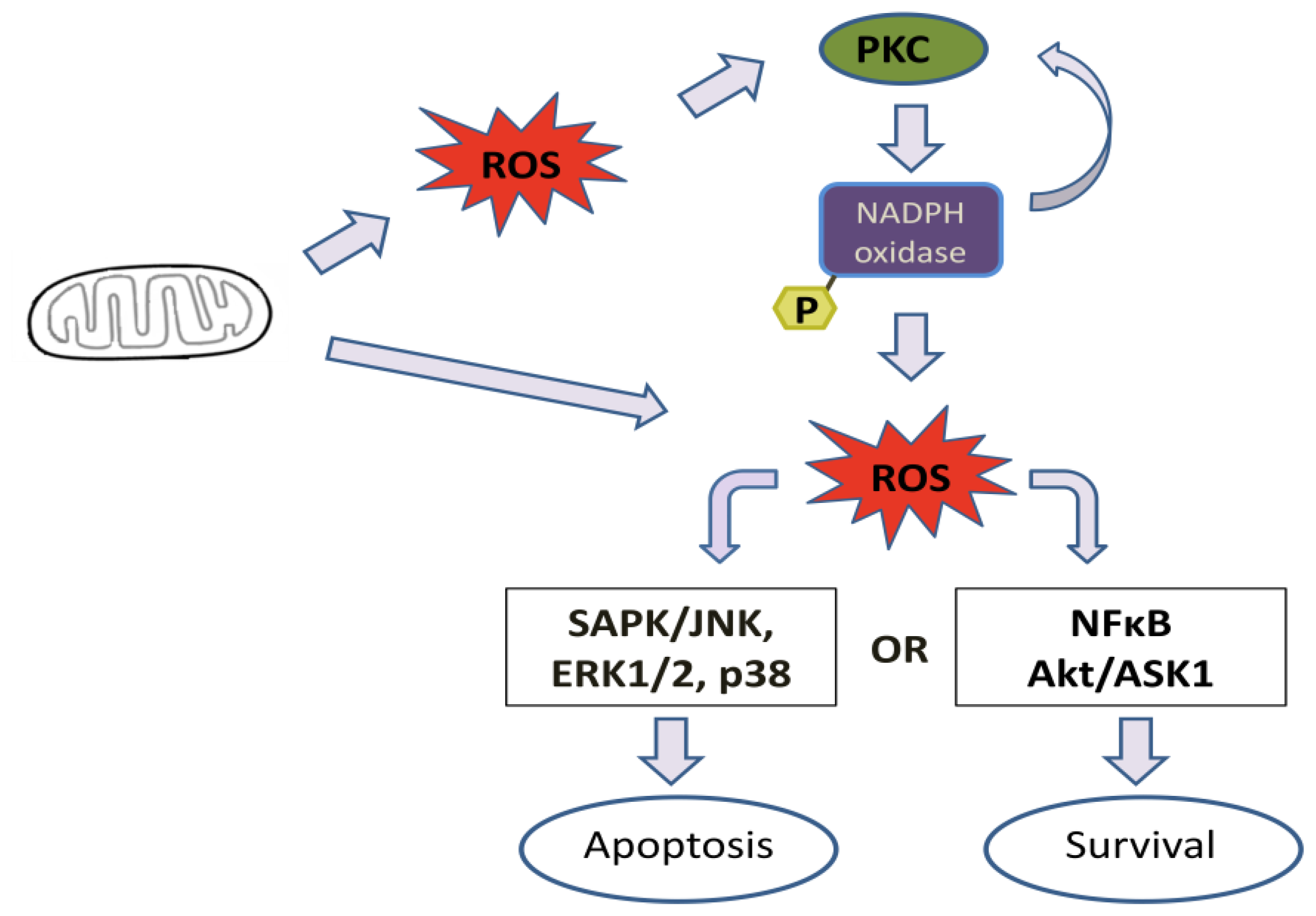
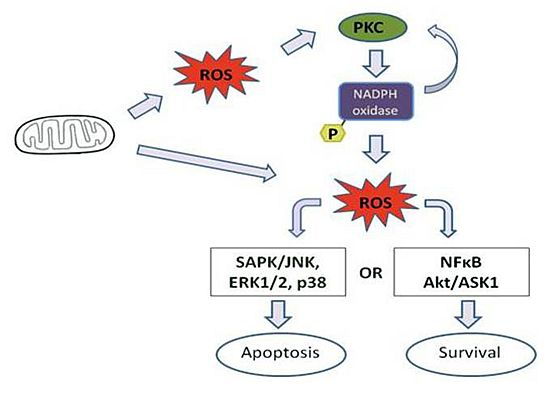
:
1. Introduction
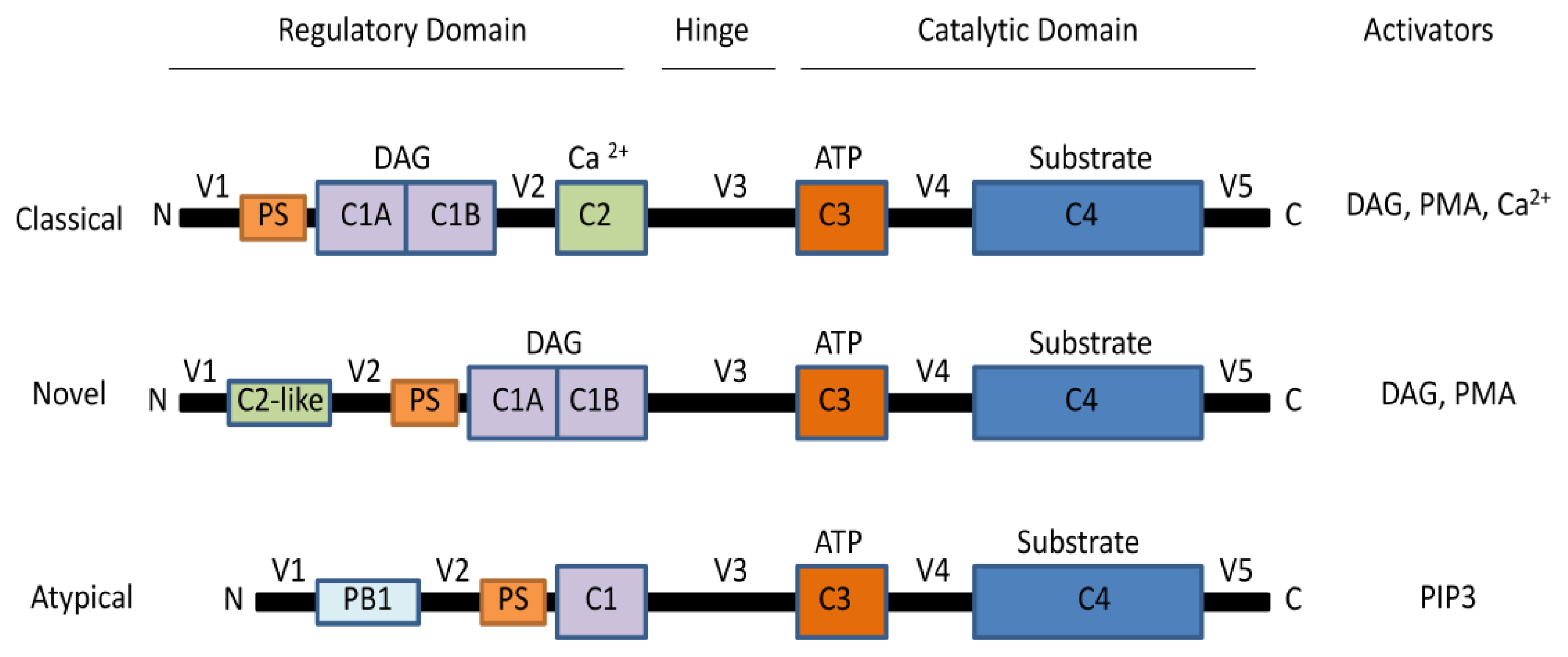
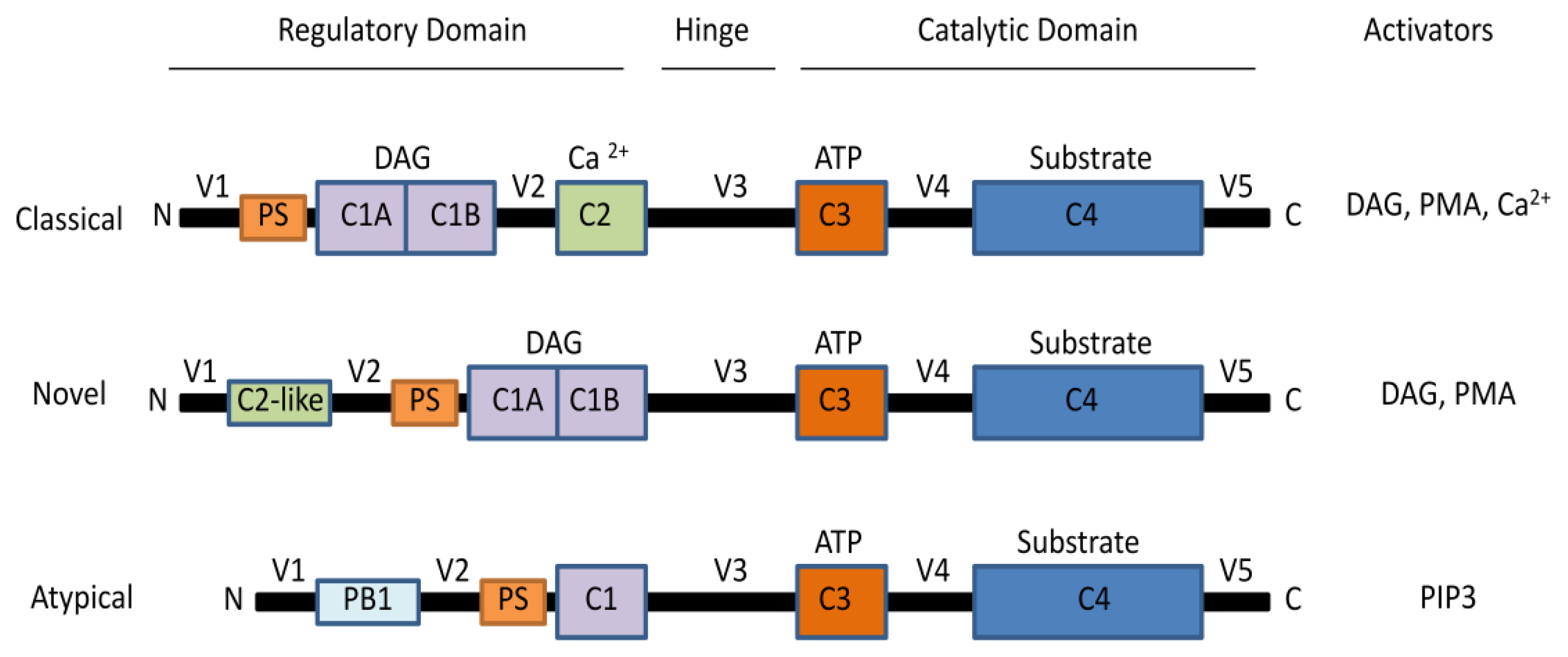
2. The Protein Kinase C Family
2.1. Activation of PKCs
2.2. PKC Receptors and Localization of Isoenzymes
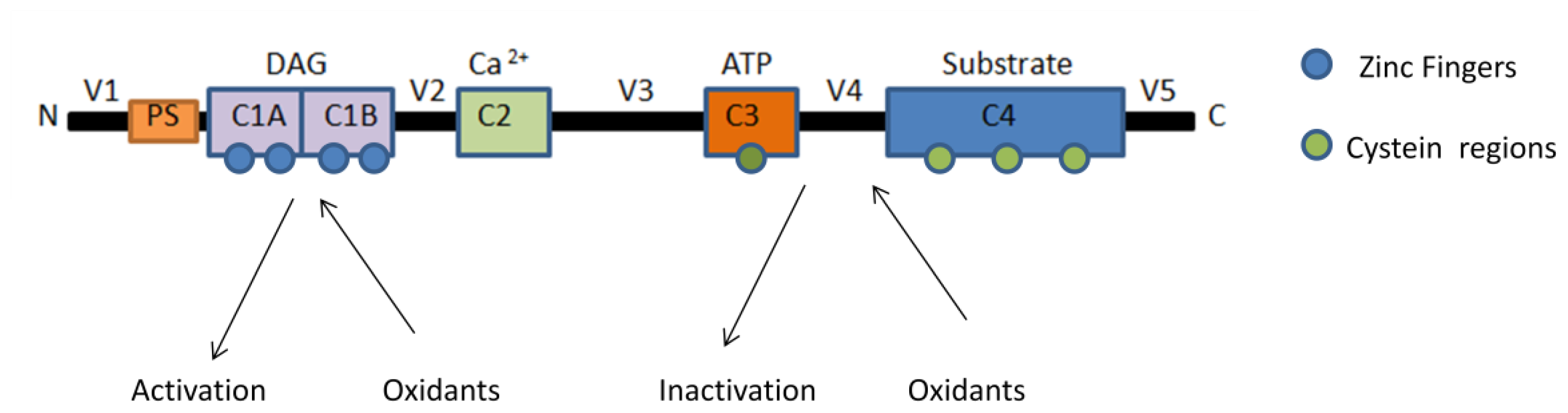
3. Regulation of Protein Kinase C by Reactive Oxygen Species (ROS)
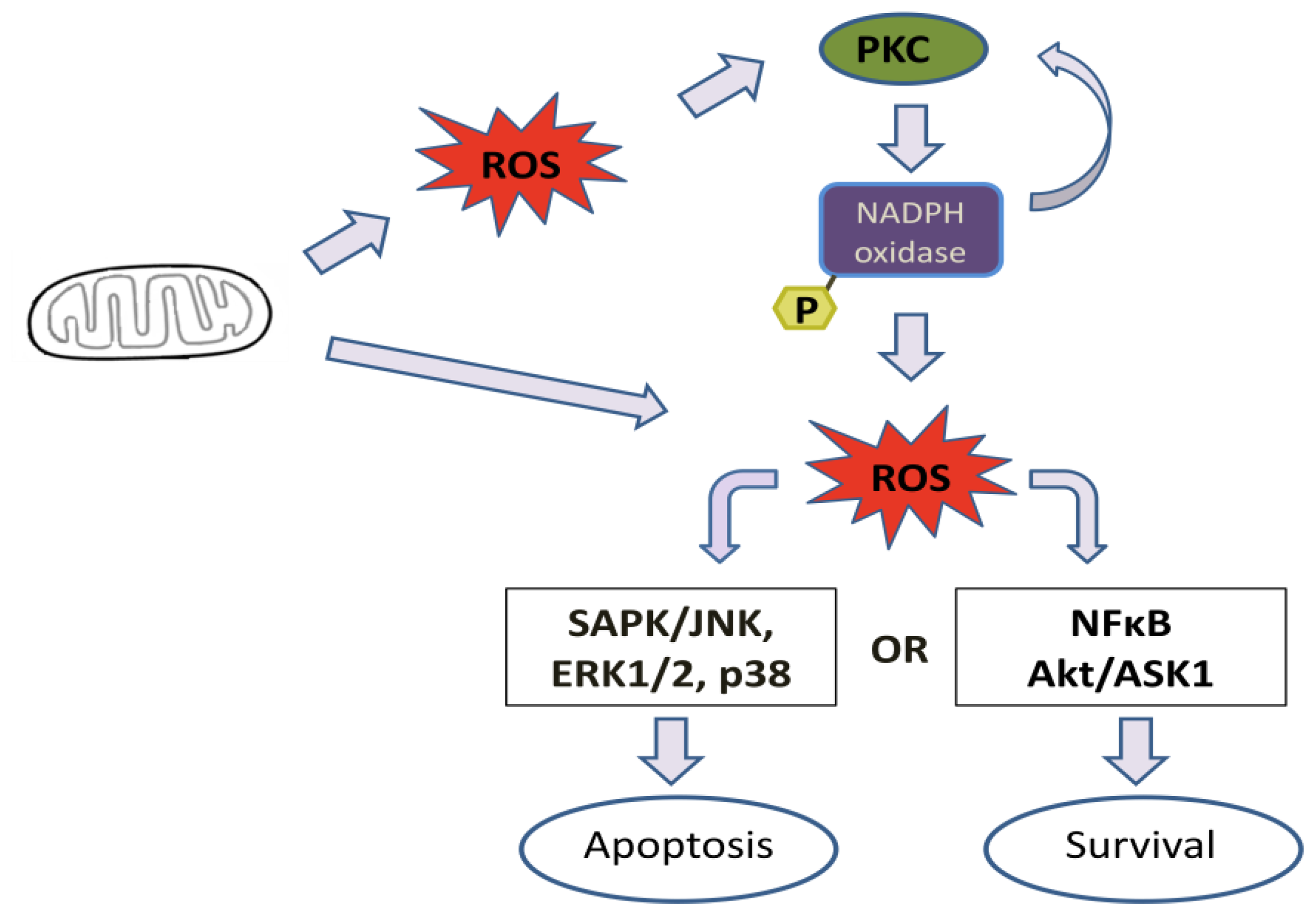
4. Effect of Mitochondrial ROS Generation on PKC Activity
5. PKC Phosphorylates Nox Subunits for ROS Generation
6. PKC-Induced ROS Generation
7. Conclusions
Acknowledgements
References
- Spickett, C.M.; Pitt, A.R.; Morrice, N.; Kolch, W. Proteomic analysis of phosphorylation, oxidation and nitrosylation in signal transduction. Biochim. Biophys. Acta 2006, 1764, 1823–1841. [Google Scholar]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar]
- Leslie, N.R.; Lindsay, Y.; Ross, S.H.; Downes, C.P. Redox regulation of phosphatase function. Biochem. Soc. Trans 2004, 32, 1018–1020. [Google Scholar]
- Suzuki, Y.J.; Forman, H.J.; Sevanian, A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med 1997, 22, 269–285. [Google Scholar]
- Cosentino-Gomes, D.; Russo-Abrahão, T.; Fonseca-de-Souza, A.L.; Ferreira, C.R.; Galina, A.; Meyer-Fernandes, J.R. Modulation of Trypanosoma rangeli ecto-phosphatase activity by hydrogen peroxide. Free Radic. Biol. Med 2009, 47, 152–158. [Google Scholar]
- Cosentino-Gomes, D.; Meyer-Fernandes, J.R. Ecto-phosphatase in protozoan parasites: Possible roles in nutrition, growth and ROS sensing. J. Bioenerg. Biomembr 2011, 43, 89–92. [Google Scholar]
- Knock, G.A.; Ward, J.P.T. Redox regulation of protein kinases as a modulator of vascular function. Antioxid. Redox Signal 2011, 15, 1531–1547. [Google Scholar]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar]
- Nishizuka, Y. Protein Kinase C and lipid signaling for sustained cellular responses. FASEB J 1995, 9, 486–496. [Google Scholar]
- Duquesnes, N.; Lezoualc’h, F.; Crozatier, B. PKC-delta and PKC-epsilon: Foes of the same family or strangers? J. Mol. Cell. Cardiol 2011, 51, 665–673. [Google Scholar]
- Giogi, C.; Agnoletto, C.; Baldini, C.; Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Redox control of Protein Kinase C: Cell and disease-specific aspects. Antioxid. Redox Signal 2010, 13, 1051–1085. [Google Scholar]
- Kikkawa, U.; Kishimoto, A.; Nishizuka, Y. The protein kinase C family: Heterogeneity and its implications. Annu. Rev. Biochem 1989, 58, 31–44. [Google Scholar]
- Svetek, J.; Schara, M.; Pecar, S.; Hergenhahn, M.; Hecker, E. Spectroscopic characterization of specific phorbol ester binding to PKC-receptor sites in membranes in situ. Carcinogenises 1995, 10, 2589–2592. [Google Scholar]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol 2010, 11, 103–112. [Google Scholar]
- Cenni, V.; Döppler, H.; Sonnenburg, E.D.; Maraldi, N.; Newton, A.C.; Toker, A. Regulation of novel protein kinase C epsilon by phosphorylation. Biochem. J 2002, 3, 537–545. [Google Scholar]
- Rozengurt, E.; Rey, O.; Waldron, R.T. Protein kinase D signaling. J. Biol. Chem 2005, 280, 13205–13208. [Google Scholar]
- Wang, Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci 2006, 27, 317–323. [Google Scholar]
- Toker, A.; Newton, A.C. Cellular signaling: Pivoting around PDK-1. Cell 2000, 2, 185–188. [Google Scholar]
- Miyamae, M.; Rodriguez, M.M.; Camacho, S.A.; Diamond, I.; Mochly-Rosen, D.; Figueredo, V.M. Activation of epsilon protein kinase C correlates with a cardioprotective effect of regular ethanol consumption. Proc. Natl. Acad. Sci. USA 1998, 4, 8262–8267. [Google Scholar]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J 2003, 2, 361–371. [Google Scholar]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev 2008, 4, 1341–1378. [Google Scholar]
- Oancea, E.; Meyer, T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 1998, 3, 307–318. [Google Scholar]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free. Radic. Biol. Med 2000, 9, 1349–1361. [Google Scholar]
- Saito, N.; Kikkawa, U.; Nishizuka, Y. The family of protein kinase C and membrane lipid mediators. J. Diabetes Complic 2002, 1, 4–8. [Google Scholar]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol 2000, 6, 1005–1028. [Google Scholar]
- Chiarugi, P.; Buricchi, F. Protein tyrosine phosphorylation and reversible oxidation: Two cross-talking posttranslation modifications. Antioxid. Redox Signal 2007, 1, 1–24. [Google Scholar]
- Almeida-Amaral, E.E.; Caruso-Neves, C.; Lara, L.S.; Pinheiro, C.M.; Meyer-Fernandes, J.R. Leishmania amazonensis: PKC-like protein kinase modulates the (Na+ + K+)ATPase activity. Exp. Parasitol 2007, 116, 419–426. [Google Scholar]
- Breitkreutz, D.; Braiman-Wiksman, L.; Daum, N.; Denning, M.F.; Tennenbaum, T. Protein kinase C family: On the crossroads of cell signaling in skin and tumor epithelium. J. Cancer Res. Clin. Oncol 2007, 11, 793–808. [Google Scholar]
- Mochly-Rosen, D.; Gordon, A.S. Anchoring proteins for protein kinase C: A means for isozyme selectivity. FASEB J 1998, 1, 35–42. [Google Scholar]
- Hug, H.; Sarre, T.F. Protein kinase C isoenzymes: Divergence in signal transduction? Biochem. J. 1993, 2, 329–343. [Google Scholar]
- Benoit, S.C.; Kemp, C.J.; Elias, C.F.; Abplanalp, W.; Herman, J.P.; Migrenne, S.; Lefevre, A.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Yu, F.; et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellularlocalization in rodents. J. Clin. Invest 2009, 9, 2577–2589. [Google Scholar]
- Oyasu, M.; Fujimiya, M.; Kashiwagi, K.; Ohmori, S.; Imaeda, H.; Saito, N. Immunogold electron microscopic demonstration of distinct submembranous localization of theactivated γPKC depending on the stimulation. J. Histochem. Cytochem 2008, 56, 253–265. [Google Scholar]
- Judé, S.; Martel, E.; Vincent, F.; Besson, P.; Couet, C.; Ogilvie, G.K.; Pinault, M.; De Chalendar, C.; Bougnoux, P.; Richard, S.; et al. Dietary long-chain n-3 fatty acids modify blood and cardiac phospholipids and reduce protein kinase-C-delta and protein kinase-C-epsilon translocation. Br. J. Nutr 2007, 6, 1143–1151. [Google Scholar]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem 1997, 272, 217–221. [Google Scholar]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal 2005, 7, 560–577. [Google Scholar]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar]
- Chen, K.; Craige, S.E.; Keaney, J.F., Jr. Downstream targets and intracellular compartmentalization in Nox Signaling. Antioxid. Redox Signal. 2009, 11, 2467–2480. [Google Scholar]
- Lin, Y.L.; Shivji, M.K.; Chen, C.; Kolodner, R.; Wood, R.D.; Dutta, A. The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J. Biol. Chem 1998, 3, 1453–1461. [Google Scholar]
- Martelli, A.M.; Evangelisti, C.; Nyakern, M.; Manzoli, F.A. Nuclear protein kinase C. Biochim. Biophys. Acta 2006, 1761, 542–551. [Google Scholar]
- Storz, P.; Döppler, H.; Toker, A. Activation loop phosphorylation controls protein kinase D-dependent activation of nuclear factor kappaB. Mol. Pharmacol 2004, 66, 870–879. [Google Scholar]
- Storz, P.; Döppler, H.; Toker, A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol. Cell. Biol 2004, 24, 2614–2626. [Google Scholar]
- Storz, P.; Toker, A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J 2003, 22, 109–120. [Google Scholar]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys 1996, 29, 169–202. [Google Scholar]
- Lambert, A.J.; Brand, M.D. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: Ubiquinone oxidoreductase (complex I). J. Biol. Chem 2004, 17, 39414–39420. [Google Scholar]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal 2011, 1, 459–468. [Google Scholar]
- Korchak, H.M.; Rossi, M.W.; Kilpatrick, L.E. Selective role for beta-protein kinase C in signaling for O-2 generation but not degranulation or adherence in differentiated HL60 cells. J. Biol. Chem 1998, 16, 27292–27299. [Google Scholar]
- Wang, Y.; Biswas, G.; Prabu, S.K.; Avadhani, N.G. Modulation of mitochondrial metabolic function by phorbol 12-myristate 13-acetate through increased mitochondrial translocation of protein kinase Calpha in C2C12 myocytes. Biochem. Pharmacol 2006, 28, 881–892. [Google Scholar]
- DelCarlo, M.; Loeser, R.F. Chondrocyte cell death mediated by reactive oxygen species-dependent activation of PKC-betaI. Am. J. Physiol. Cell Physiol 2006, 290, 802–811. [Google Scholar]
- Noland, T.A., Jr; Dimino, M.J. Characterization and distribution of protein kinase C in ovarian tissue. Biol. Reprod. 1986, 35, 863–872. [Google Scholar]
- Li, L.; Lorenzo, P.S.; Bogi, K.; Blumberg, P.M.; Yuspa, S.H. Protein kinase C delta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol. Cell. Biol 1999, 12, 8547–8558. [Google Scholar]
- Majumder, P.K.; Pandey, P.; Sun, X.; Cheng, K.; Datta, R.; Saxena, S.; Kharbanda, S.; Kufe, D. Mitochondrial translocation of protein kinase C deltain phorbol ester-induced cytochrome c release and apoptosis. J. Biol. Chem 2000, 21, 21793–21796. [Google Scholar]
- Majumder, P.K.; Mishra, N.C.; Sun, X.; Bharti, A.; Kharbanda, S.; Saxena, S.; Kufe, D. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ 2001, 12, 465–470. [Google Scholar]
- Baines, C.P.; Zhang, J.; Wang, G.W.; Zheng, Y.T.; Xiu, J.X.; Cardwell, E.M.; Bolli, R.; Ping, P. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: Enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ. Res 2002, 8, 390–397. [Google Scholar]
- Jiang, Y.Y.; Huang, H.; Wang, H.J.; Wu, D.; Yang, R.; Tashiro, S.; Onodera, S.; Ikejima, T. Interruption of mitochondrial complex IV activity and cytochrome c expression activated O2 −-mediated cell survival in silibinin-treated human melanoma A375-S2 cells via IGF-1R-PI3K-Akt and IGF-1R-PLC γ-PKC pathways. Eur. J. Pharmacol 2011, 668, 78–87. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 13, 813–820. [Google Scholar]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol 2006, 15, 167–178. [Google Scholar]
- Nishikawa, T.; Kukidome, D.; Sonoda, K.; Fujisawa, K.; Matsuhisa, T.; Motoshima, H.; Matsumura, T.; Araki, E. Impact of mitochondrial ROS production on diabetic vascular complications. Diabetes Res. Clin. Pract 2007, 77, 41–45. [Google Scholar]
- Rathore, R.; Zheng, Y.M.; Li, X.Q.; Wang, Q.S.; Liu, Q.H.; Ginnan, R.; Singer, H.A.; Ho, Y.S.; Wang, Y.X. Mitochondrial ROS-PKC epsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+] in pulmonary artery smooth muscle cells. Biochem. Biophys. Res. Commun 2006, 351, 784–790. [Google Scholar]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Protective role of taurine against arsenic-induced mitochondria-dependent hepatic apoptosis via the inhibition of PKCdelta-JNK pathway. PloS One 2010, 5, e12602. [Google Scholar]
- Kunwar, A.; Narang, H.; Priyadarsini, K.I.; Krishna, M.; Pandey, R.; Sainis, K.B. Delayed activation of PKCdelta and NFkappaB and higher radioprotection in splenic lymphocytes by copper (II)-Curcumin (1:1) complex as compared to curcumin. J. Cell. Biochem 2007, 102, 1214–1224. [Google Scholar]
- Weber, N.C.; Schlack, W. The concept of anaesthetic-induced cardioprotection: Mechanisms of action. Best. Pract. Res. Clin. Anaesthesiol 2005, 19, 429–443. [Google Scholar]
- Bouwman, R.A.; Musters, R.J.; van Beek-Harmsen, B.J.; de Lange, J.J.; Lamberts, R.R.; Loer, S.A.; Boer, C. Sevoflurane-induced cardioprotection depends on PKC-alpha activation via production of reactive oxygen species. Br. J. Anaesth 2007, 99, 639–645. [Google Scholar]
- Kim, H.J.; Chakravarti, N.; Oridate, N.; Choe, C.; Claret, F.X.; Lotan, R. N-(4-hydroxyphenyl) retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells. Oncogene 2006, 25, 2785–2794. [Google Scholar]
- Cheng, J.J.; Wung, B.S.; Chao, Y.J.; Wang, D.L. Cyclic strain-induced reactive oxygen species involved in ICAM-1 gene induction in endothelial cells. Hypertension 1998, 31, 125–130. [Google Scholar]
- Wung, B.S.; Cheng, J.J.; Hsieh, H.J.; Shyy, Y.J.; Wang, D.L. Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ. Res 1997, 81, 1–7. [Google Scholar]
- Ali, M.H.; Pearlstein, D.P.; Mathieu, C.E.; Schumacker, P.T. Mitochondrial requirement for endothelial responses to cyclic strain: Implications for mechanotransduction. Am. J. Physiol. Lung Cell. Mol. Physiol 2004, 287, 486–496. [Google Scholar]
- Ali, M.H.; Mungai, P.T.; Schumacker, P.T. Stretch-induced phosphorylation of focal adhesion kinase in endothelial cells: Role of mitochondrial oxidants. Am. J. Physiol. Lung Cell. Mol. Physiol 2006, 291, 38–45. [Google Scholar]
- Fontayne, A.; Dang, P.M.; Gougerot-Pocidalo, M.A.; El-Benna, J. Phosphorylation of p47phox sites by PKC alpha, betaII, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 2002, 18, 7743–7750. [Google Scholar]
- Lambeth, J.D. Nox enzymes and the biology of reactive oxygen. Nat. Rev. Immunol 2004, 4, 181–189. [Google Scholar]
- El Benna, J.; Faust, L.P.; Babior, B.M. The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J. Biol. Chem 1994, 23, 23431–234316. [Google Scholar]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/Nox2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med 2009, 30, 217–225. [Google Scholar]
- Faust, L.R.; El Benna, J.; Babior, B.M.; Chanock, S.J. The phosphorylation targets of p47phox, a subunit of the respiratory burst oxidase. Functions of the individual target serines as evaluated by site-directed mutagenesis. J. Clin. Invest 1995, 96, 1499–1505. [Google Scholar]
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leukoc. Biol 2004, 76, 76760–76781. [Google Scholar]
- Kawahara, T.; Lambeth, J.D. Molecular evolution of Phox-related regulatory subunits for NADPH oxidase enzymes. BMC Evol. Biol. 2007, 7, 178. [Google Scholar]
- Bey, E.A.; Xu, B.; Bhattacharjee, A.; Oldfield, C.M.; Zhao, X.; Li, Q.; Subbulakshmi, V.; Feldman, G.M.; Wientjes, F.B.; Cathcart, M.K. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J. Immunol 2004, 1, 5730–5738. [Google Scholar]
- Harrigan, T.J.; Abdullaev, I.F.; Jourd’heuil, D.; Mongin, A.A. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: The role of NADPH oxidases. J. Neurochem 2008, 106, 2449–2462. [Google Scholar]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J 2005, 15, 401–416. [Google Scholar]
- Okamura, N.; Curnutte, J.T.; Roberts, R.L.; Babior, B.M. Relationship of protein phosphorylation to the activation of the respiratory burst in human neutrophils. Defects in the phosphorylation of a group of closely related 48-kDa proteins in two forms of chronic granulomatous disease. J. Biol. Chem 1988, 15, 6777–6782. [Google Scholar]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal 2006, 18, 69–82. [Google Scholar]
- Fulton, DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal 2009, 11, 2443–2452. [Google Scholar]
- Herrera, M.; Silva, G.B.; Garvin, J.L. Angiotensin II stimulates thick ascending limb superoxide production via protein kinase C(α)-dependent NADPH oxidase activation. J. Biol. Chem 2010, 285, 21323–21328. [Google Scholar]
- Dekker, L.V.; Leitges, M.; Altschuler, G.; Mistry, N.; McDermott, A; Roes, J.; Segal, A.W. Protein kinase C-beta contributes to NADPH oxidase activation in neutrophils. Biochem. J. 2000, 1, 285–289. [Google Scholar]
- Korchak, H.M.; Corkey, B.E.; Yaney, G.C.; Kilpatrick, L.E. Negative regulation of ligand-initiated Ca2+ uptake by PKC-beta II in differentiated HL60 cells. Am. J. Physiol. Cell Physiol 2001, 281, 514–523. [Google Scholar]
- Majumdar, S.; Kane, L.H.; Rossi, M.W.; Volpp, B.D.; Nauseef, W.M.; Korchak, H.M. Protein kinase C isotypes and signal-transduction in human neutrophils: Selective substrate specificity of calcium-dependent beta-PKC and novel calcium-independent nPKC. Biochim. Biophys. Acta 1993, 16, 276–286. [Google Scholar]
- Siow, Y.L.; Au-Yeung, K.K.; Woo, C.W.; O, K. Homocysteine stimulates phosphorylation of NADPH oxidase p47phox and p67phox subunits in monocytes via protein kinase C beta activation. Biochem. J 2006, 15, 73–82. [Google Scholar]
- Brown, G.E.; Stewart, M.Q.; Liu, H.; Ha, V.L.; Yaffe, M.B. A novel assay system implicates PtdIns(3,4)P(2), PtdIns(3)P, and PKC delta in intracellular production of reactive oxygen species by the NADPH oxidase. Mol. Cell 2003, 11, 35–47. [Google Scholar]
- Cheng, N.; He, R.; Tian, J.; Dinauer, M.C.; Ye, R.D. A critical role of protein kinase C delta activation loop phosphorylation in formyl-methionyl-leucyl-phenylalanine-induced phosphorylation of p47(phox) and rapid activation of nicotinamide adenine dinucleotide phosphate oxidase. J. Immunol 2007, 1179, 7720–7728. [Google Scholar]
- Nitti, M.; Furfaro, A.L.; Traverso, N.; Odetti, P.; Storace, D.; Cottalasso, D.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. PKC delta and NADPH oxidase in AGE-induced neuronal death. Neurosci. Lett 2007, 18, 261–265. [Google Scholar]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med 2008, 1, 1223–1231. [Google Scholar]
- White, C.N.; Figtree, G.A.; Liu, C.C.; Garcia, A.; Hamilton, E.J.; Chia, K.K.; Rasmussen, H.H. Angiotensin II inhibits the Na+–K+ pump via PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Cell Physiol 2009, 296, 693–700. [Google Scholar]
- Dang, P.M.; Fontayne, A.; Hakim, J.; El Benna, J.; Périanin, A. Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J. Immunol 2001, 15, 1206–1213. [Google Scholar]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci 2009, 12, 857–863. [Google Scholar]
- Wu, D.M.; Lu, J.; Zheng, Y.L.; Zhang, Y.Q.; Hu, B.; Cheng, W.; Zhang, Z.F.; Li, M.Q. Small interfering RNA-mediated knockdown of protein kinase C zeta attenuates domoic acid-induced cognitive deficits in mice. Toxicol. Sci. 2012. [Google Scholar] [CrossRef]
- Leverence, J.T.; Medhora, M.; Konduri, G.G.; Sampath, V. Lipopolysaccharide-induced cytokine expression in alveolar epithelial cells: Role of PKCζ-mediated p47phox phosphorylation. Chem. Biol. Interact 2011, 15, 72–81. [Google Scholar]
- El Benna, J.; Faust, R.P.; Johnson, J.L.; Babior, B.M. Phosphorylation of the respiratory burst oxidase subunit p47phox as determined by two-dimensional phosphopeptide mapping. Phosphorylation by protein kinase C, protein kinase A, and a mitogen-activated protein kinase. J. Biol. Chem 1996, 15, 6374–6378. [Google Scholar]
- Kramer, I.M.; van der Bend, R.L.; Verhoeven, A.J.; Roos, D. The 47-kDa protein involved in the NADPH:O2 oxidoreductase activity of human neutrophils is phosphorylated by cyclic AMP-dependent protein kinase without induction of a respiratory burst. Biochim. Biophys. Acta 1988, 16, 189–196. [Google Scholar]
- El Benna, J.; Han, J.; Park, J.W.; Schmid, E.; Ulevitch, R.J.; Babior, B.M. Activation of p38 in stimulated human neutrophils: Phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch. Biochem. Biophys 1996, 15, 395–400. [Google Scholar]
- Dewas, C.; Fay, M.; Gougerot-Pocidalo, M.A.; El-Benna, J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucylphenylalanine- induced p47phox phosphorylation in human neutrophils. J. Immunol 2000, 165, 5238–5244. [Google Scholar]
- Park, H.S.; Lee, S.M.; Lee, J.H.; Kim, Y.S.; Bae, Y.S.; Park, J.W. Phosphorylation of the leucocyte NADPH oxidase subunit p47(phox) by casein kinase 2: Conformation-dependent phosphorylation and modulation of oxidase activity. Biochem. J 2001, 15, 783–790. [Google Scholar]
- Chen, Q.; Powell, D.W.; Rane, M.J.; Singh, S.; Butt, W.; Klein, J.B.; McLeish, K.R. Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J. Immunol 2003, 15, 5302–5308. [Google Scholar]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 29, 5130–5135. [Google Scholar]
- Martyn, K.D.; Kim, M.J.; Quinn, M.T.; Dinauer, M.C.; Knaus, U.G. p21-activated kinase (Pak) regulates NADPH oxidase activation in human neutrophils. Blood 2005, 1, 3962–3969. [Google Scholar]
- Waite, K.A.; Wallin, R.; Qualliotine-Mann, D.; McPhail, L.C. Phosphatidic acid-mediated phosphorylation of the NADPH oxidase component p47-phox. Evidence that phosphatidic acid may activate a novel protein kinase. J. Biol. Chem 1997, 13, 15569–15578. [Google Scholar]
- Chowdhury, A.K.; Watkins, T.; Parinandi, N.L.; Saatian, B.; Kleinberg, M.E.; Usatyuk, P.V.; Natarajan, V. Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J. Biol. Chem 2005, 27, 20700–20711. [Google Scholar]
- Joglar, B.; Rodriguez-Pallares, J.; Rodriguez-Perez, A.I.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: Relevance to progression of the disease. J. Neurochem 2009, 109, 656–669. [Google Scholar]
- Tsai, C.T.; Wang, D.L.; Chen, W.P.; Hwang, J.J.; Hsieh, C.S.; Hsu, K.L.; Tseng, C.D.; Lai, L.P.; Tseng, Y.Z.; Chiang, F.T.; et al. Angiotensin II increases expression of alpha1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element-binding protein-dependent pathway in HL-1 myocytes. Circ. Res 2007, 25, 1476–1485. [Google Scholar]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar]
- Devaraj, S.; Dasu, M.R.; Singh, U.; Rao, L.V.; Jialal, I. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis 2009, 203, 67–74. [Google Scholar]
- Paletas, K.; Sailer, X.; Rizeq, L.; Dimitriadi, A.; Koliakos, G.; Kaloyianni, M. Angiotensin-II-dependent NHE1 activation in human monocytes. J. Am. Soc. Hypertens 2008, 2, 173–181. [Google Scholar]
- Romero, M.; Jiménez, R.; Sánchez, M.; López-Sepúlveda, R.; Zarzuelo, M.J.; O’Valle, F.; Zarzuelo, A.; Pérez-Vizcaíno, F.; Duarte, J. Quercetin inhibits vascular superoxide production induced by endothelin-1: Role of NADPH oxidase, uncoupled eNOS and PKC. Atherosclerosis 2009, 202, 58–67. [Google Scholar]
- Koliakos, G.; Befani, C.; Paletas, K.; Kaloyianni, M. Effect of endothelin on sodium/hydrogen exchanger activity of human monocytes and atherosclerosis-related functions. Ann. N. Y. Acad. Sci 2007, 1095, 274–291. [Google Scholar]
- Dikalov, S.I.; Li, W.; Doughan, A.K.; Blanco, R.R.; Zafari, A.M. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am. J. Physiol. Regul. Integr. Comp. Physiol 2012, 302, 1134–1142. [Google Scholar]
- Yang, J.; Lane, P.H.; Pollock, J.S.; Carmines, P.K. Protein kinase C-dependent NAD(P)H oxidase activation induced by type 1 diabetes in renal medullary thick ascending limb. Hypertension 2010, 55, 468–473. [Google Scholar]
- Plumb, R.D.; El-Sherbeeny, N.A.; Dixon, L.J.; Hughes, S.M.; Devine, A.B.; Leahey, W.J.; McVeigh, G.E. NAD(P)H-dependent superoxide production in platelets: The role of angiotensin II and protein kinase C. Clin. Biochem 2005, 38, 607–613. [Google Scholar]
- Soetikno, V.; Watanabe, K.; Sari, F.R.; Harima, M.; Thandavarayan, R.A.; Veeraveedu, P.T.; Arozal, W.; Sukumaran, V.; Lakshmanan, A.P.; Arumugam, S.; et al. Curcumin attenuates diabetic nephropathy by inhibiting PKC-α and PKC-β1 activity in streptozotocin-induced type I diabetic rats. Mol. Nutr. Food. Res 2011, 55, 1655–1665. [Google Scholar]
- Ha, H.; Lee, H.B. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology (Carlton) 2005, 10, S7–S10. [Google Scholar]
- Lin, R.Z.; Wang, T.P.; Hung, R.J.; Chuang, Y.J.; Chien, C.C.; Chang, H.Y. Tumor-induced endothelial cell apoptosis: Roles of NAD(P)H oxidase-derived reactive oxygen species. J. Cell. Physiol 2011, 226, 1750–1762. [Google Scholar]
- Wei, X.F.; Zhou, Q.G.; Hou, F.F.; Liu, B.Y.; Liang, M. Advanced oxidation protein products induce mesangial cell perturbation through PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Ren. Physiol 2009, 296, 427–437. [Google Scholar]
- Griendling, K.K.; Ushio-Fukai, M. Reactive oxygen species as mediators of angiotensin II signaling. Regul. Pept 2000, 28, 21–27. [Google Scholar]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res 2002, 90, 1205–1213. [Google Scholar]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 2004, 24, 677–683. [Google Scholar]
- Shi, G.; Fu, Y.; Jiang, W.; Yin, A.; Feng, M.; Wu, Y.; Kawai, Y.; Miyamori, I.; Fan, C. Activation of Src-ATF1 pathway is involved in upregulation of Nox1, a catalytic subunit of NADPH oxidase, by aldosterone. Endocr. J. 2011, 58, 491–499. [Google Scholar]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res 2001, 88, 888–894. [Google Scholar]
- Wingler, K.; Wünsch, S.; Kreutz, R.; Rothermund, L.; Paul, M.; Schmidt, H.H. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic. Biol. Med 2001, 31, 1456–1464. [Google Scholar]
- Wei, H.; Mi, X.; Ji, L.; Yang, L.; Xia, Q.; Wei, Y.; Miyamori, I.; Fan, C. Protein kinase C-delta is involved in induction of Nox1 gene expression by aldosterone in rat vascular smooth muscle cells. Biochemistry (Mosc) 2010, 75, 304–309. [Google Scholar]
- Fan, C.Y.; Katsuyama, M.; Yabe-Nishimura, C. PKCdelta mediates up-regulation of Nox1, a catalytic subunit of NADPH oxidase, via transactivation of the EGF receptor: Possible involvement of PKCdelta in vascular hypertrophy. Biochem. J 2005, 390, 761–767. [Google Scholar]
- San José, G.; Bidegain, J.; Robador, P.A.; Díez, J.; Fortuño, A.; Zalba, G. Insulin-induced NADPH oxidase activation promotes proliferation and matrix metalloproteinase activation in monocytes/macrophages. Free Radic. Biol. Med 2009, 46, 1058–1067. [Google Scholar]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and Nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol 2006, 26, 333–339. [Google Scholar]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol 2004, 24, 1844–1854. [Google Scholar]
- Schröder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol 2009, 29, 239–245. [Google Scholar]
- Colston, J.T.; de la Rosa, S.D.; Strader, J.R.; Anderson, M.A.; Freeman, G.L. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett 2005, 579, 2533–2540. [Google Scholar]
- Gorin, Y.; Ricono, J.M.; Wagner, B.; Kim, N.H.; Bhandari, B.; Choudhury, G.G.; Abboud, H.E. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochem. J 2004, 381, 231–239. [Google Scholar]
- Ren, Y.; D’Ambrosio, M.A.; Wang, H.; Peterson, E.L.; Garvin, J.L.; Carretero, O.A. Mechanisms of angiotensin II-enhanced connecting tubule glomerular feedback. Am. J. Physiol. Ren. Physiol 2012. [Google Scholar] [CrossRef]
- Tan, Y.; Li, X.; Prabhu, S.D.; Brittian, K.R.; Chen, Q.; Yin, X.; McClain, C.J.; Zhou, Z.; Cai, L. Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. J. Am. Coll. Cardiol 2012, 59, 1477–1486. [Google Scholar]
- Sun, P.; Yue, P.; Wang, W.H. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J. Physiol. Ren. Physiol 2012, 302, 679–687. [Google Scholar]
- Silva, G.B.; Ortiz, P.A.; Hong, N.J.; Garvin, J.L. Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension 2006, 48, 467–472. [Google Scholar]
- Cabello-Verrugio, C.; Acuña, M.J.; Morales, M.G.; Becerra, A.; Simon, F.; Brandan, E. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem. Biophys. Res. Commun 2011, 8, 665–670. [Google Scholar]
- Silva, J.; Pastorello, M.; Arzola, J.; Zavala, L.E.; De Jesús, S.; Varela, M.; Matos, M.G.; del Rosario Garrido, M.; Israel, A. AT1 receptor and NAD(P)H oxidase mediate angiotensin II-stimulated antioxidant enzymes and mitogen-activated protein kinase activity in the rat hypothalamus. J. Renin-Angiotensin-Aldosterone Syst 2010, 11, 234–242. [Google Scholar]
- Hogarty, D.C.; Speakman, E.A.; Puig, V.; Phillips, M.I. The role of angiotensin, AT1 and AT2 receptors in the pressor, drinking and vasopressin responses to central angiotensin. Brain Res 1992, 586, 289–294. [Google Scholar]
- Phillips, M.I.; Sumners, C. Angiotensin II in central nervous system physiology. Regul. Pept 1998, 78, 1–11. [Google Scholar]
- Wang, G.; Anrather, J.; Glass, M.J.; Tarsitano, M.J.; Zhou, P.; Frys, K.A.; Pickel, V.M.; Iadecola, C. Nox2, Ca2+, and protein kinase C play a role in angiotensin II-induced free radical production in nucleus tractus solitarius. Hypertension 2006, 48, 482–489. [Google Scholar]
- Chung, Y.W.; Kim, H.K.; Kim, I.Y.; Yim, M.B.; Chock, P.B. Dual function of protein kinase C (PKC) in 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced manganese superoxide dismutase (MnSOD) expression: Activation of CREB and FOXO3a by PKC-alpha phosphorylation and by PKC-mediated inactivation of Akt, respectively. J. Biol. Chem 2011, 286, 29681–29690. [Google Scholar]
- Kamiya, T.; Makino, J.; Hara, H.; Inagaki, N.; Adachi, T. Extracellular-superoxide dismutase expression during monocytic differentiation of U937 cells. J. Cell. Biochem 2011, 112, 244–255. [Google Scholar]
- Deng, B.; Xie, S.; Wang, J.; Xia, Z.; Nie, R. Inhibition of protein kinase C β(2) prevents tumor necrosis factor-α-induced apoptosis and oxidative stress in endothelial cells: The role of NADPH oxidase subunits. J. Vasc. Res 2012, 49, 144–159. [Google Scholar]
- Pandey, D.; Fulton, D.J. Molecular regulation of NADPH oxidase 5 via the MAPK pathway. Am. J. Physiol. Heart Circ. Physiol 2011, 300, 1336–1344. [Google Scholar]
- Serrander, L.; Jaquet, V.; Bedard, K.; Plastre, O.; Hartley, O.; Arnaudeau, S.; Demaurex, N.; Schlegel, W.; Krause, K.H. Nox5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie 2007, 89, 1159–1167. [Google Scholar]
- Jagnandan, D.; Church, J.E.; Banfi, B.; Stuehr, D.J.; Marrero, M.B.; Fulton, D.J. Novel mechanism of activation of NADPH oxidase 5. Calcium sensitization via phosphorylation. J. Biol. Chem 2007, 282, 6494–6507. [Google Scholar]
- Gupte, S.A.; Kaminski, P.M.; George, S.; Kouznestova, L.; Olson, S.C.; Mathew, R.; Hintze, T.H.; Wolin, M.S. Peroxide generation by p47phox-Src activation of Nox2 has a key role in protein kinase C-induced arterial smooth muscle contraction. Am. J. Physiol. Heart Circ. Physiol 2009, 296, 1048–1057. [Google Scholar]
- Park, J.W.; Babior, B.M. Activation of the leukocyte NADPH oxidase subunit p47phox by protein kinase C. A phosphorylation-dependent change in the conformation of the C-terminal end of p47phox. Biochemistry 1997, 36, 7474–7480. [Google Scholar]
- Baggiolini, M.; Boulay, F.; Badwey, J.A.; Curnutte, J.T. Activation of neutrophil leukocytes: Chemoattractant receptors and respiratory burst. FASEB J 1993, 7, 1004–1010. [Google Scholar]
- Martelli, A.M.; Sang, N.; Borgatti, P.; Capitani, S.; Neri, L.M. Multiple biological responses activated by nuclear protein kinase C. J. Cell. Biochem 1999, 74, 499–521. [Google Scholar]
- Divecha, N.; Banfić, H.; Irvine, R.F. The polyphosphoinositide cycle exists in the nuclei of Swiss 3T3 cells under the control of a receptor (for IGF-I) in the plasma membrane, and stimulation of the cycle increases nuclear diacylglycerol and apparently induces translocation of protein kinase C to the nucleus. EMBO J 1991, 10, 3207–3214. [Google Scholar]
- Neri, L.M.; Borgatti, P.; Capitani, S.; Martelli, A.M. Nuclear diacylglycerol produced by phosphoinositide-specific phospholipase C is responsible for nuclear translocation of protein kinase C-alpha. J. Biol. Chem 1998, 273, 29738–29744. [Google Scholar]
- Sun, B.; Murray, N.R.; Fields, A.P. A role for nuclear phosphatidylinositol-specific phospholipase C in the G2/M phase transition. J. Biol. Chem 1997, 272, 26313–26317. [Google Scholar]
- Deacon, E.M.; Pettitt, T.R.; Webb, P.; Cross, T.; Chahal, H.; Wakelam, M.J.; Lord, J.M. Generation of diacylglycerol molecular species through the cell cycle: A role for 1-stearoyl, 2-arachidonyl glycerol in the activation of nuclear protein kinase C-betaII at G2/M. J. Cell. Sci 2002, 115, 983–989. [Google Scholar]
- Neri, L.M.; Borgatti, P.; Capitani, S.; Martelli, A.M. The nuclear phosphoinositide 3-kinase/AKT pathway: A new second messenger system. Biochim. Biophys. Acta 2002, 1584, 73–80. [Google Scholar]
- Neri, L.M.; Martelli, A.M.; Borgatti, P.; Colamussi, M.L.; Marchisio, M.; Capitani, S. Increase in nuclear phosphatidylinositol 3-kinase activity and phosphatidylinositol (3,4,5) trisphosphate synthesis precede PKC-zeta translocation to the nucleus of NGF-treated PC12 cells. FASEB J 1999, 13, 2299–2310. [Google Scholar]
- Wooten, M.W.; Zhou, G.; Wooten, M.C.; Seibenhener, M.L. Transport of protein kinase C isoforms to the nucleus of PC12 cells by nerve growth factor: Association of atypical zeta-PKC with the nuclear matrix. J. Neurosci. Res 1997, 49, 393–403. [Google Scholar]
- Calcerrada, M.C.; Miguel, B.G.; Martín, L.; Catalán, R.E.; Martínez, A.M. Involvement of phosphatidylinositol 3-kinase in nuclear translocation of protein kinase C zeta induced by C2-ceramide in rat hepatocytes. FEBS Lett 2002, 514, 361–365. [Google Scholar]
- Borgatti, P.; Mazzoni, M.; Carini, C.; Neri, L.M.; Marchisio, M.; Bertolaso, L.; Previati, M.; Zauli, G.; Capitani, S. Changes of nuclear protein kinase C activity and isotype composition in PC12 cell proliferation and differentiation. Exp. Cell Res 1996, 224, 72–78. [Google Scholar]
- Buchner, K. The role of protein kinase C in the regulation of cell growth and in signalling to the cell nucleus. J. Cancer Res. Clin. Oncol 2000, 126, 1–11. [Google Scholar]
- Ushio-Fukai, M. Localizing NADPH oxidase-derived ROS. Sci. STKE 2006, 2006, re8. [Google Scholar]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem 2004, 279, 45935–45941. [Google Scholar]
- Van Buul, J.D.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and localization of Nox2 and Nox4 in primary human endothelial cells. Antioxid. Redox Signal 2005, 7, 308–317. [Google Scholar]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar]
- Spencer, N.Y.; Yan, Z.; Boudreau, R.L.; Zhang, Y.; Luo, M.; Li, Q.; Tian, X.; Shah, A.M.; Davisson, R.L.; Davidson, B.; et al. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. J. Biol. Chem 2011, 286, 8977–8987. [Google Scholar]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar]
- Shiose, A.; Kuroda, J.; Tsuruya, K.; Hirai, M.; Hirakata, H.; Naito, S.; Hattori, M.; Sakaki, Y.; Sumimoto, H. A novel superoxide-producing NAD(P)H oxidase in kidney. J. Biol. Chem 2001, 276, 1417–1423. [Google Scholar]
- Pendergrass, K.D.; Gwathmey, T.M.; Michalek, R.D.; Grayson, J.M.; Chappell, M.C. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem. Biophys. Res. Commun 2009, 384, 149–154. [Google Scholar]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar]
- Becker, K.P.; Kitatani, K.; Idkowiak-Baldys, J.; Bielawski, J.; Hannun, Y.A. Selective inhibition of juxtanuclear translocation of protein kinase C betaII by a negative feedback mechanism involving ceramide formed from the salvage pathway. J. Biol. Chem 2005, 280, 2606–2612. [Google Scholar]
- Voris, J.P.; Sitailo, L.A.; Rahn, H.R.; Defnet, A.; Gerds, A.T.; Sprague, R.; Yadav, V.; Caroline Le Poole, I.; Denning, M.F. Functional alterations in protein kinase C beta II expression in melanoma. Pigment. Cell Melanoma. Res 2010, 23, 216–224. [Google Scholar]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar]
- Datta, R.; Yoshinaga, K.; Kaneki, M.; Pandey, P.; Kufe, D. Phorbol ester-induced generation of reactive oxygen species is protein kinase cbeta -dependent and required for SAPK activation. J. Biol. Chem 2000, 275, 41000–41003. [Google Scholar]
- Agudo-López, A.; Miguel, B.G.; Fernández, I.; Martínez, A.M. Role of protein kinase C and mitochondrial permeability transition pore in the neuroprotective effect of ceramide in ischemia-induced cell death. FEBS Lett 2011, 585, 99–103. [Google Scholar]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal 2009, 11, 791–810. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKC group | PKC isoform | Cell/tissue type | References |
|---|---|---|---|
| Classical | PKCα | Kidney | [81] |
| PKCβ | Leukemic cells; Neutrophils; and Monocytes. | [46,82–85] | |
| Novel | PKCδ | Monocytes; Neuroblastoma; Neutrophils; and Fibroblast. | [75,86–88] |
| PKCɛ | Pulmonary artery smooth muscle; and Myocytes. | [89,90] | |
| Atypical | PKCζ | Leukocytes; Neurons; Hippocampus of mice; and Alveolus. | [91–94] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell Signaling through Protein Kinase C Oxidation and Activation. Int. J. Mol. Sci. 2012, 13, 10697-10721. https://doi.org/10.3390/ijms130910697
Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. Cell Signaling through Protein Kinase C Oxidation and Activation. International Journal of Molecular Sciences. 2012; 13(9):10697-10721. https://doi.org/10.3390/ijms130910697
Chicago/Turabian StyleCosentino-Gomes, Daniela, Nathália Rocco-Machado, and José Roberto Meyer-Fernandes. 2012. "Cell Signaling through Protein Kinase C Oxidation and Activation" International Journal of Molecular Sciences 13, no. 9: 10697-10721. https://doi.org/10.3390/ijms130910697
APA StyleCosentino-Gomes, D., Rocco-Machado, N., & Meyer-Fernandes, J. R. (2012). Cell Signaling through Protein Kinase C Oxidation and Activation. International Journal of Molecular Sciences, 13(9), 10697-10721. https://doi.org/10.3390/ijms130910697