The Apoptotic Volume Decrease Is an Upstream Event of MAP Kinase Activation during Staurosporine-Induced Apoptosis in HeLa Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
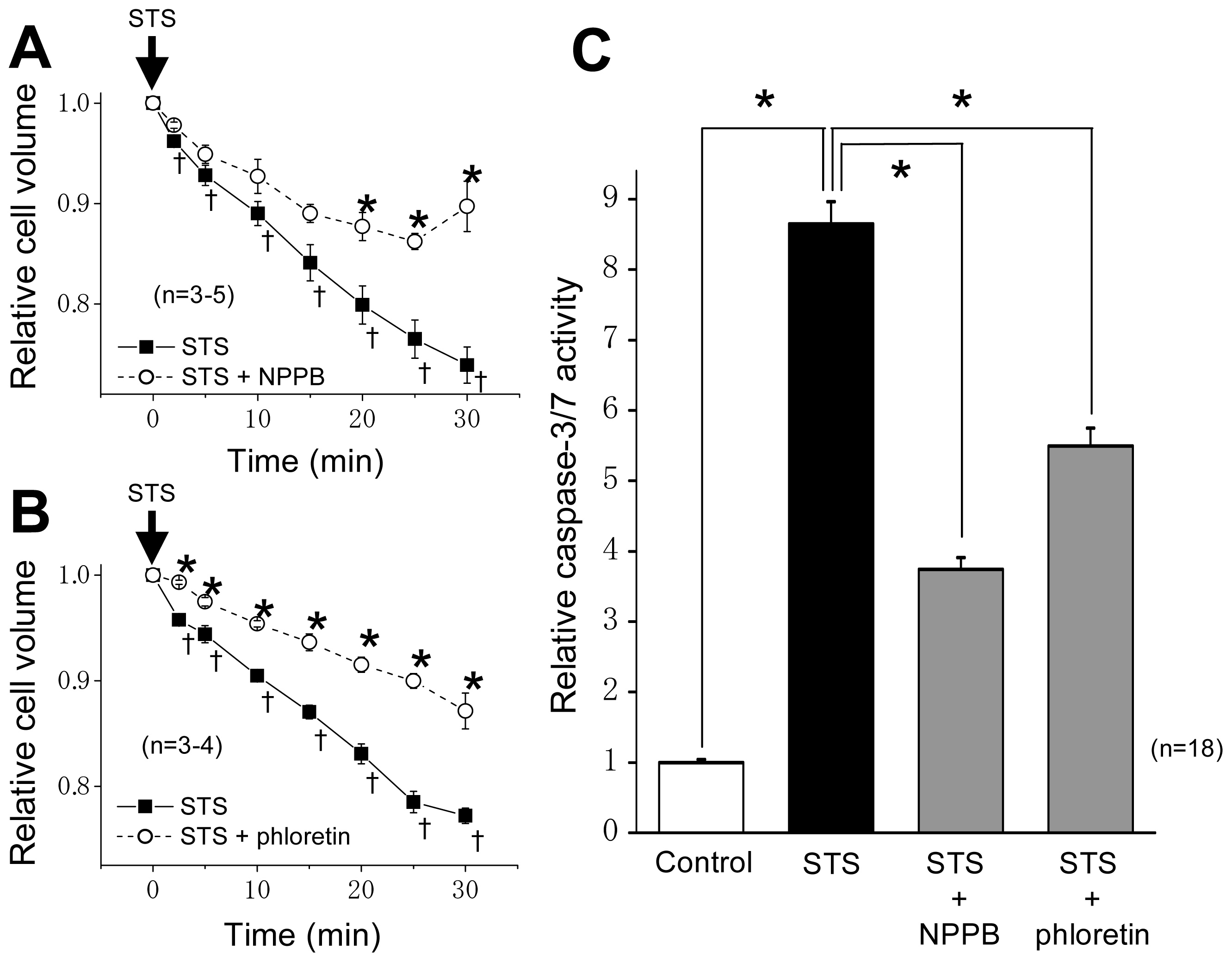
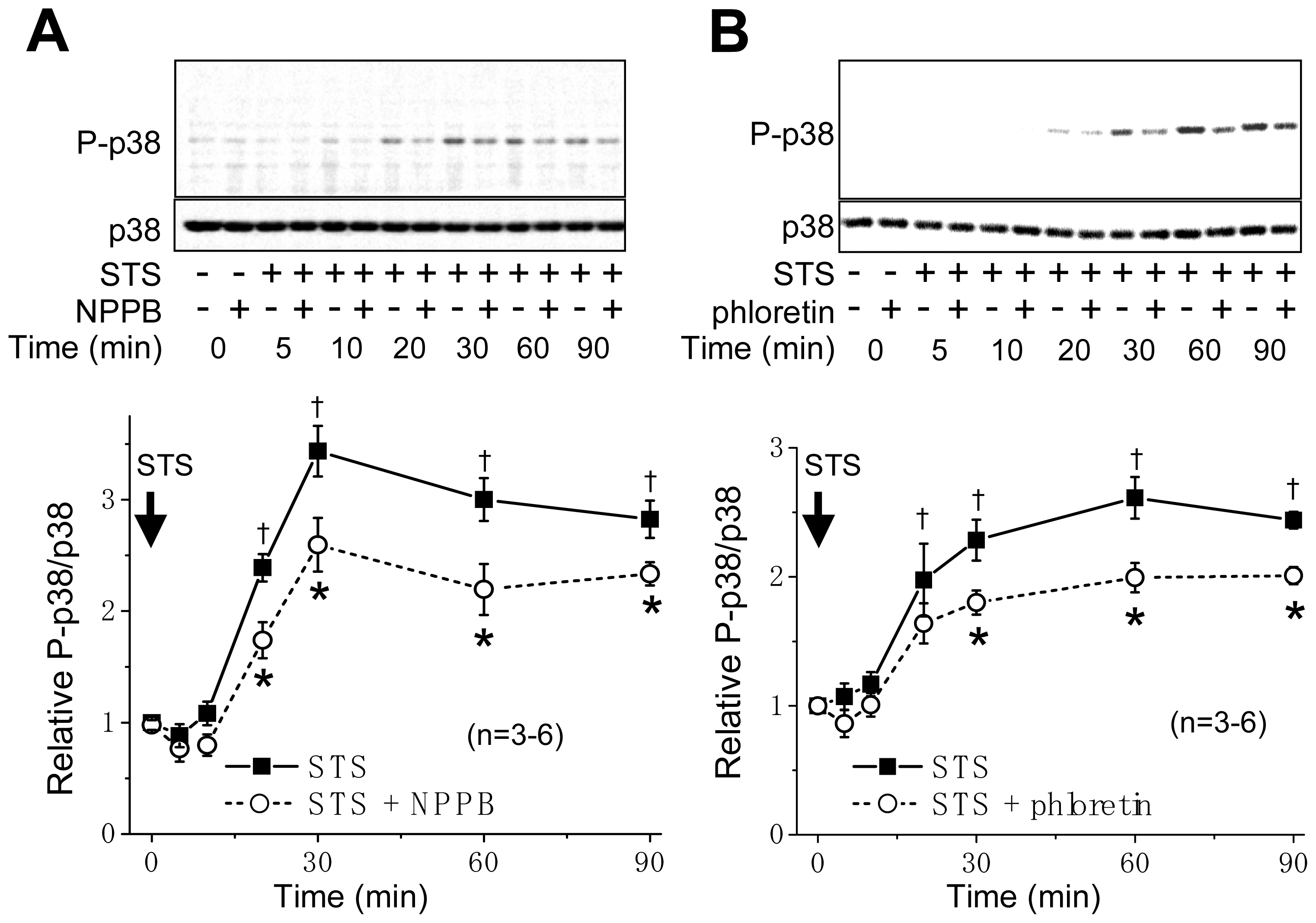
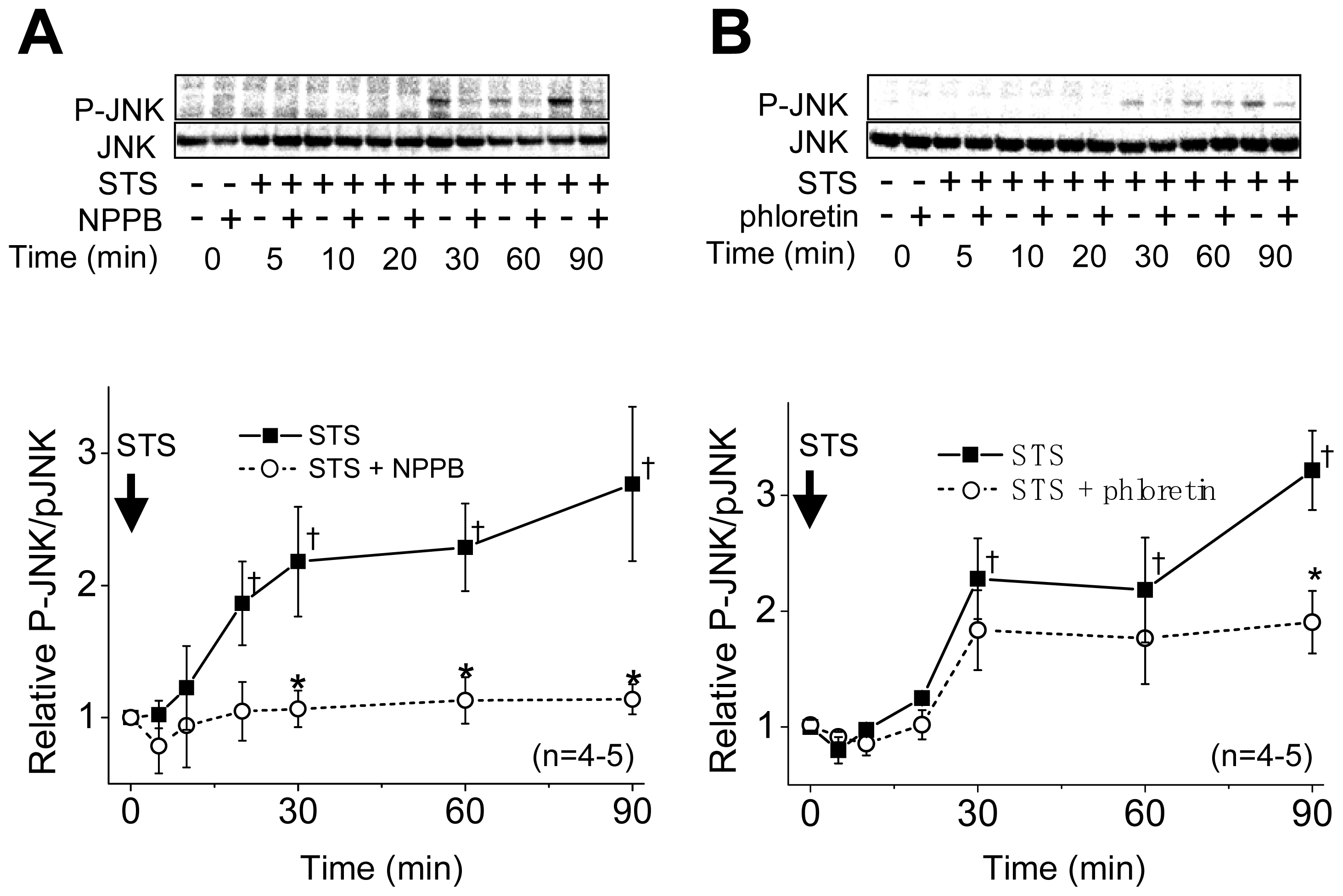
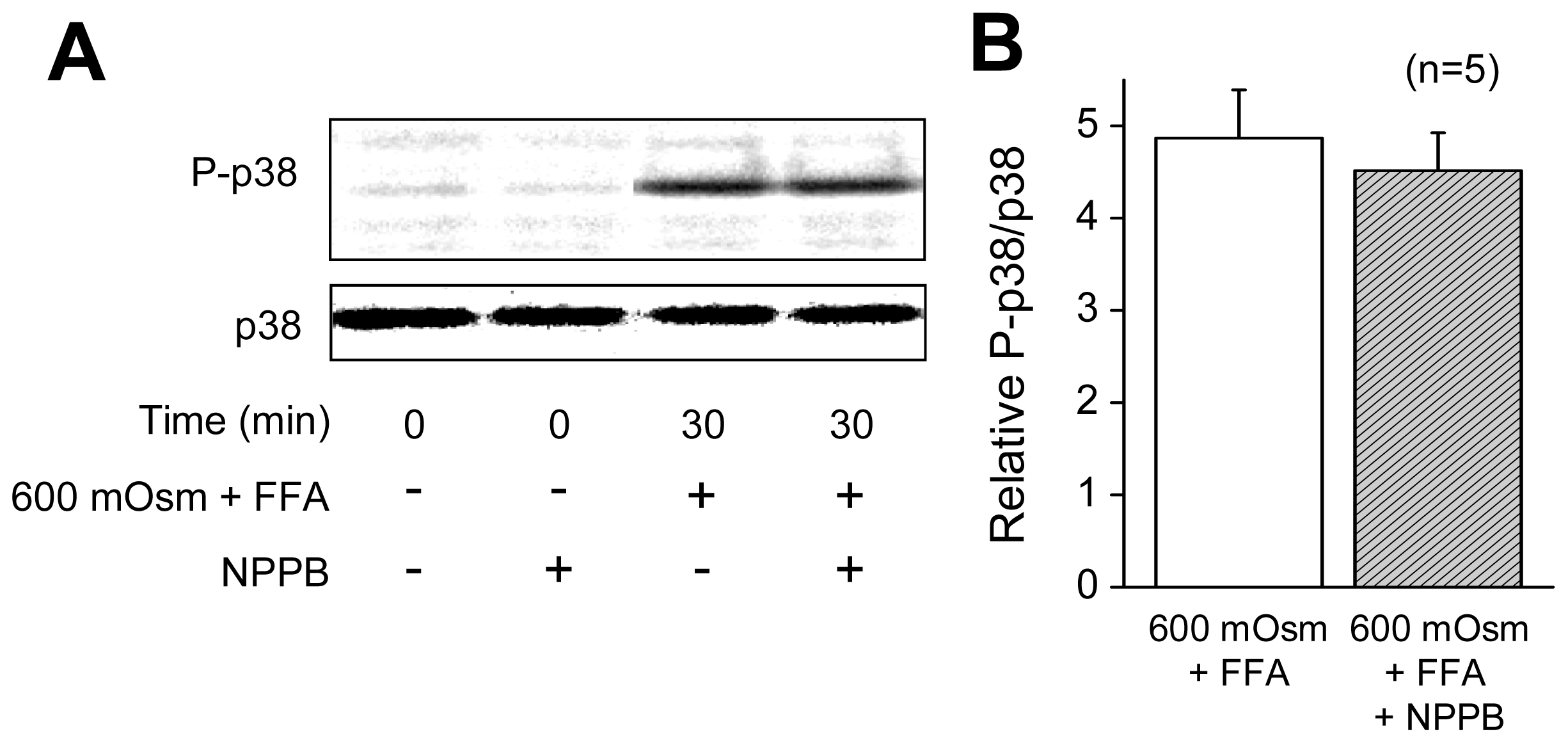
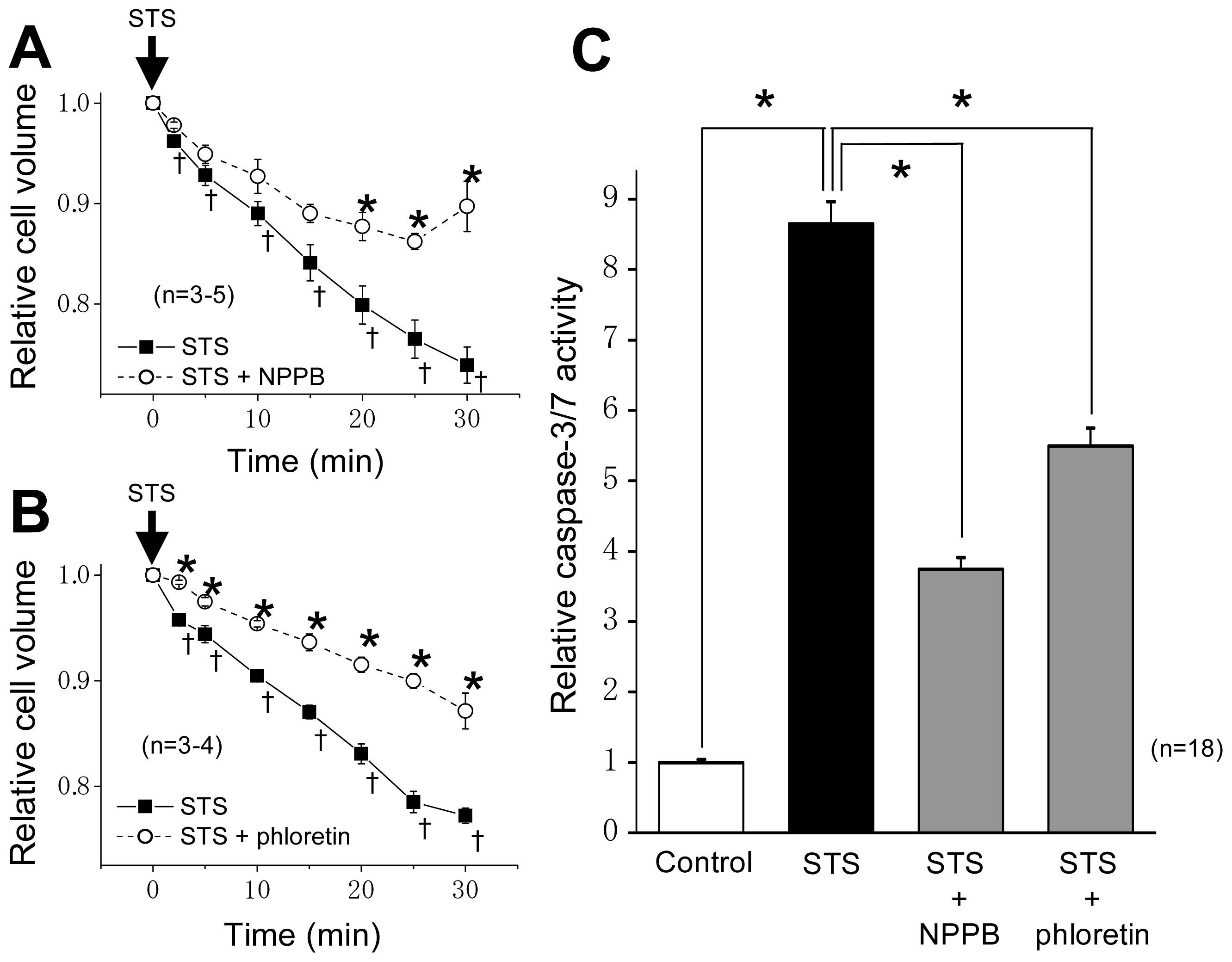
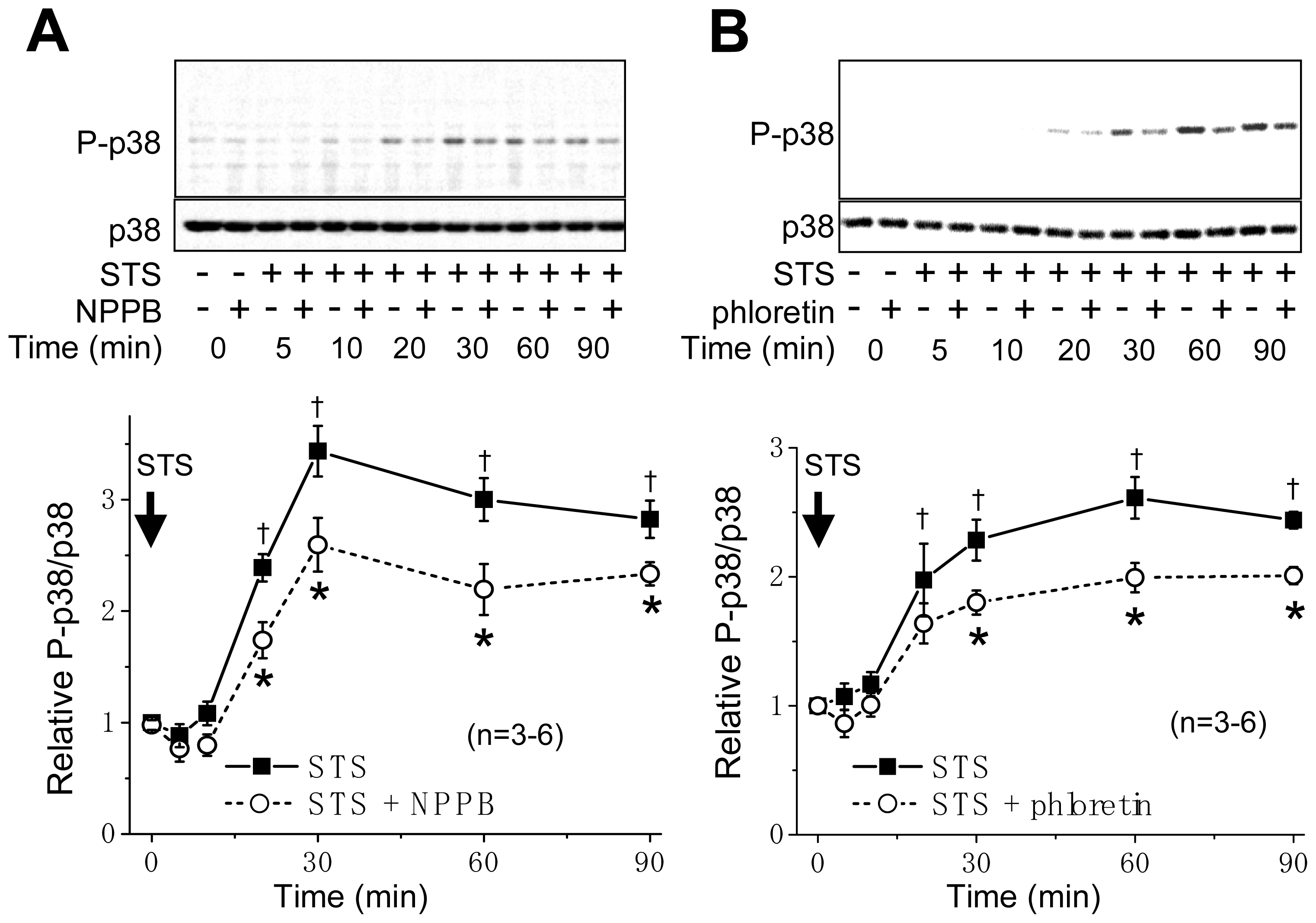
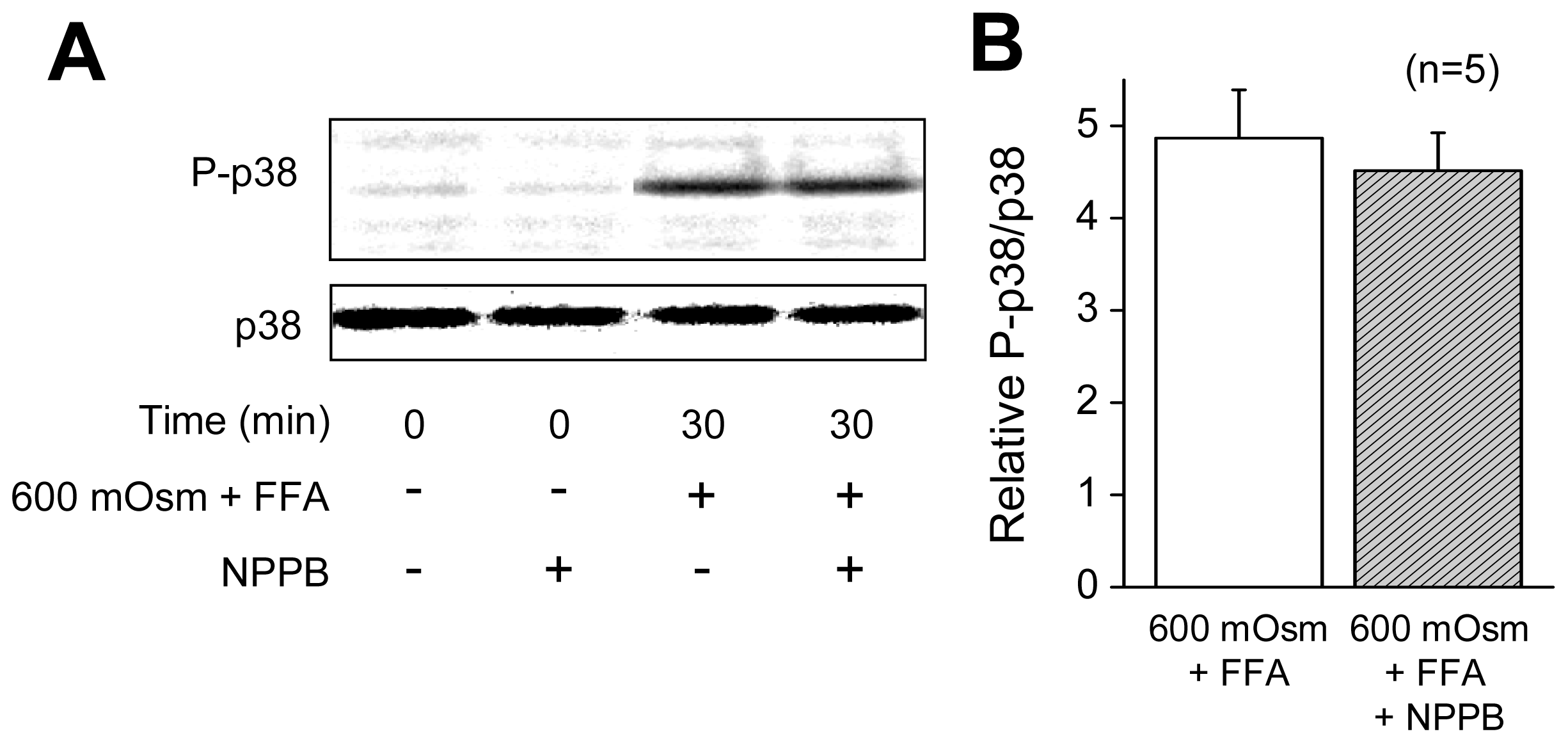
2.1. VSOR Blocker Sensitivity of STS-Induced AVD and Phosphorylation of p38 and JNK
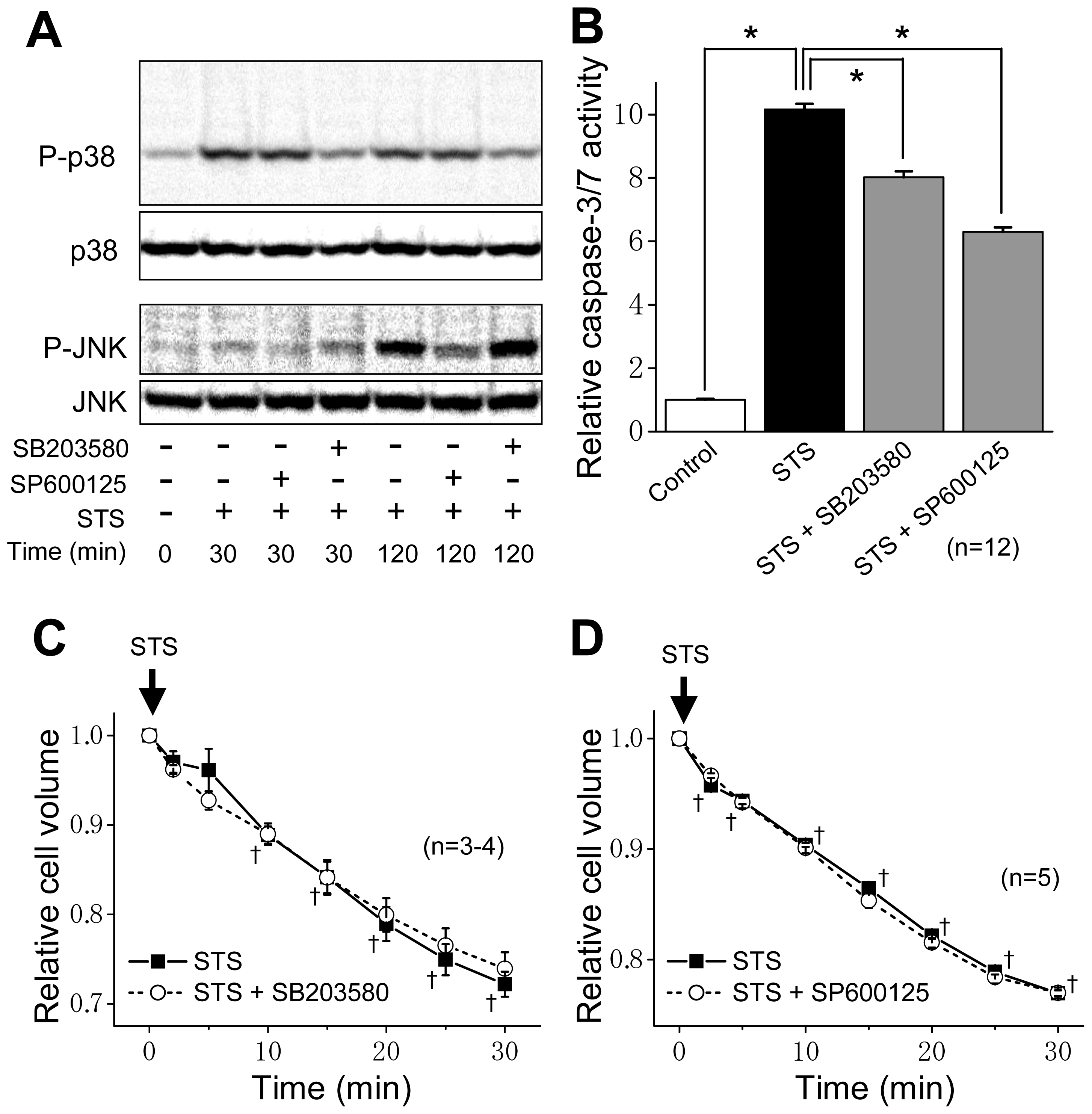
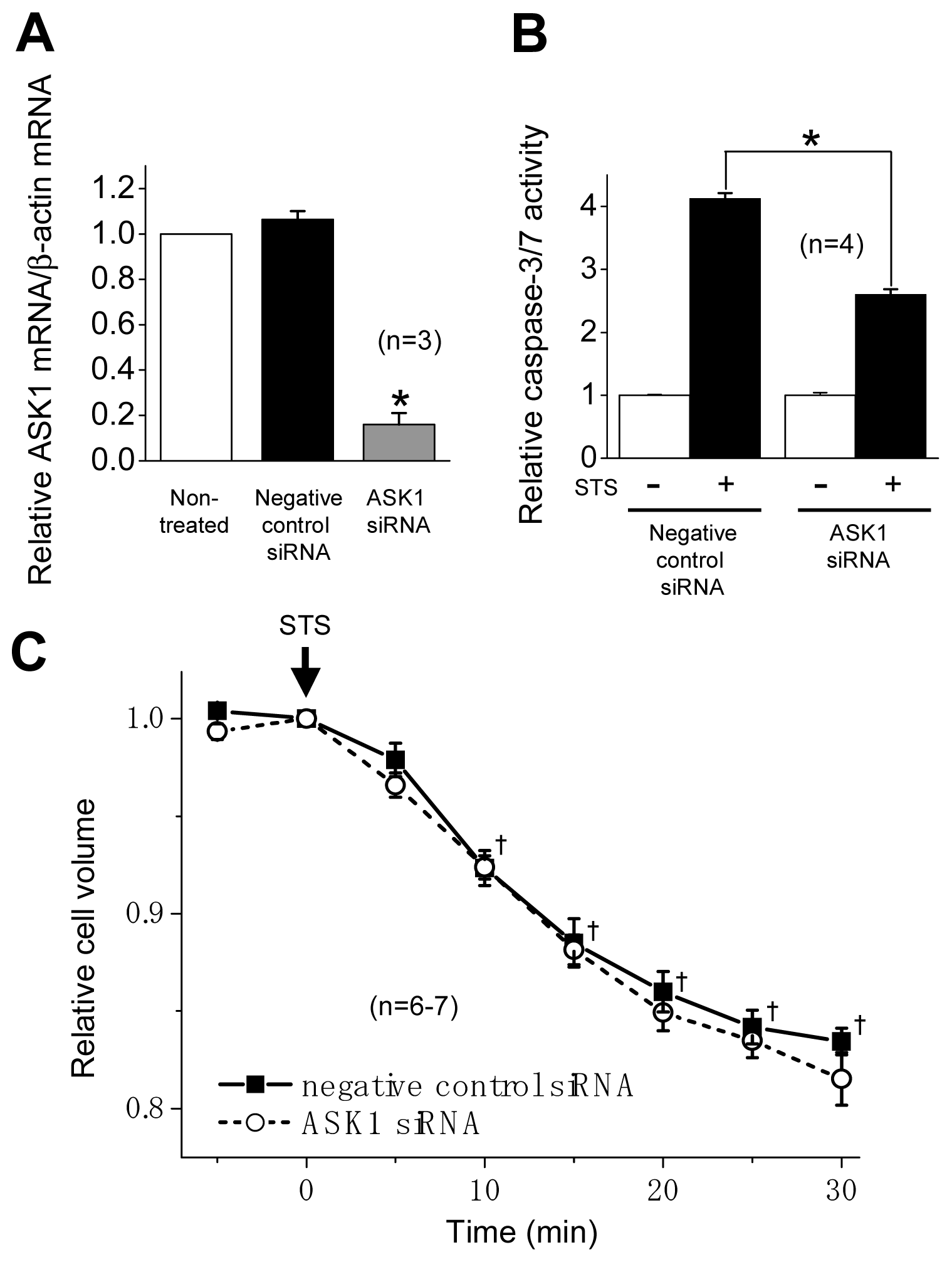
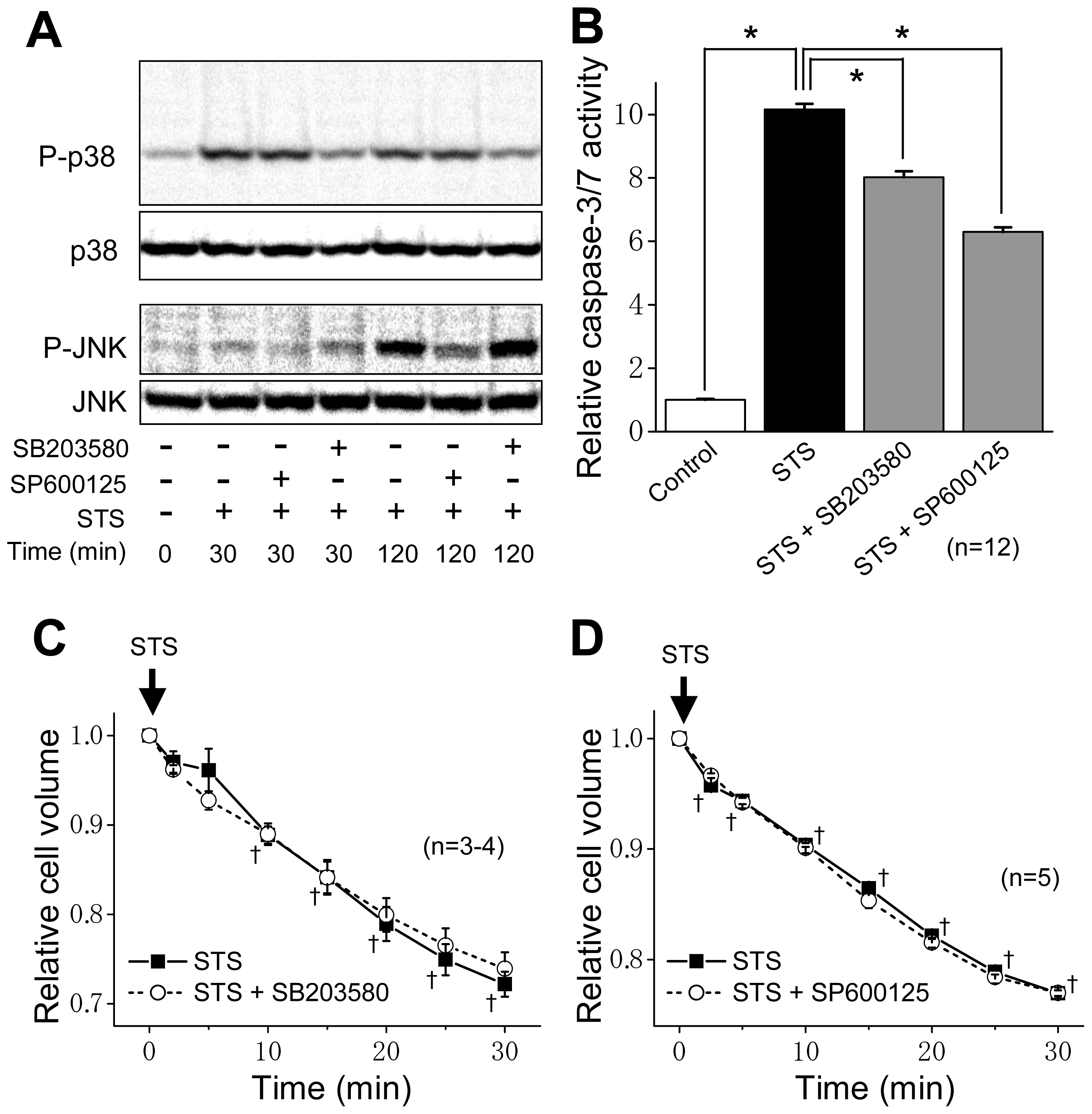
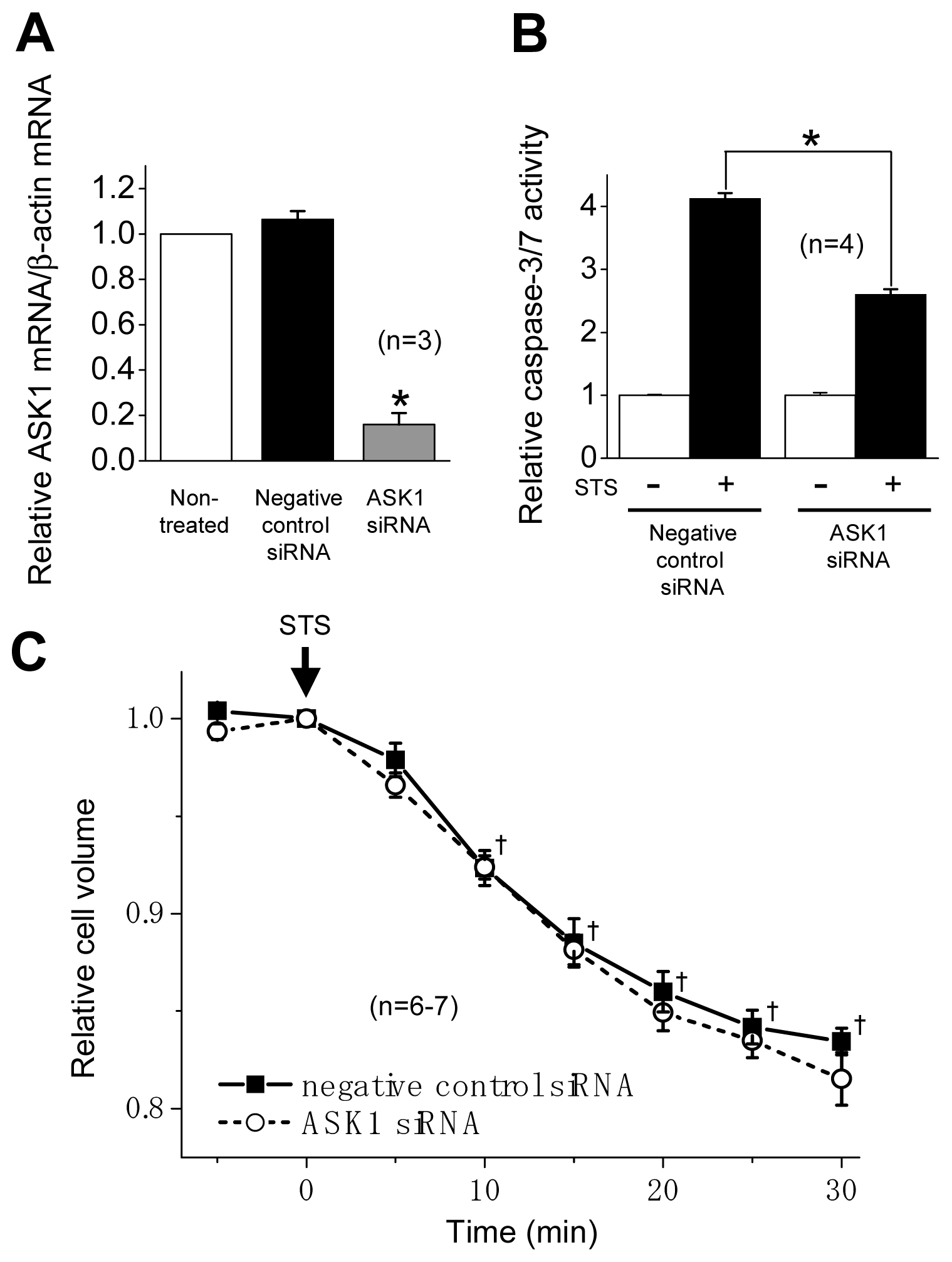
2.2. ASK1 Dependence of STS-Induced Caspase Activation but not of AVD
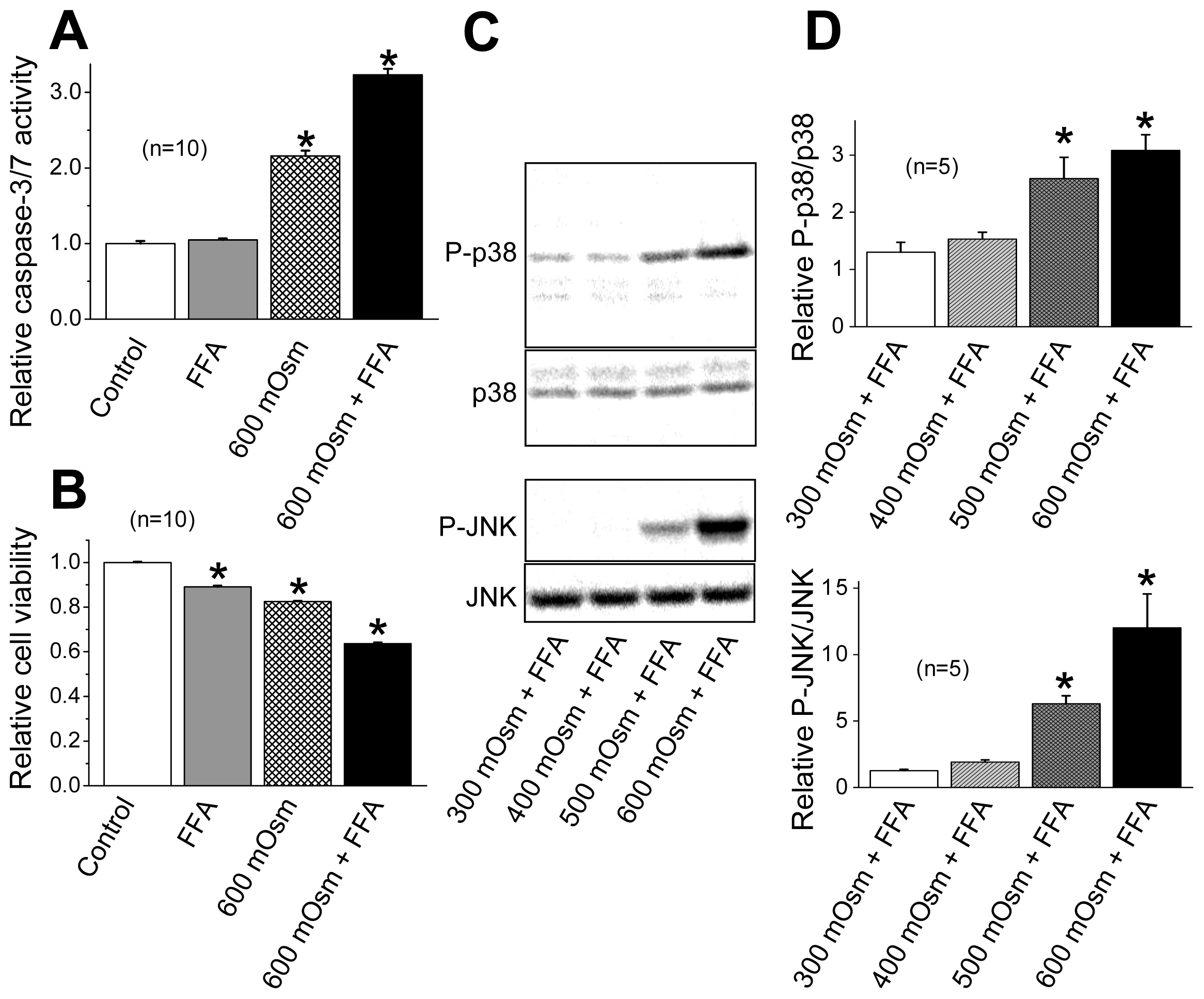
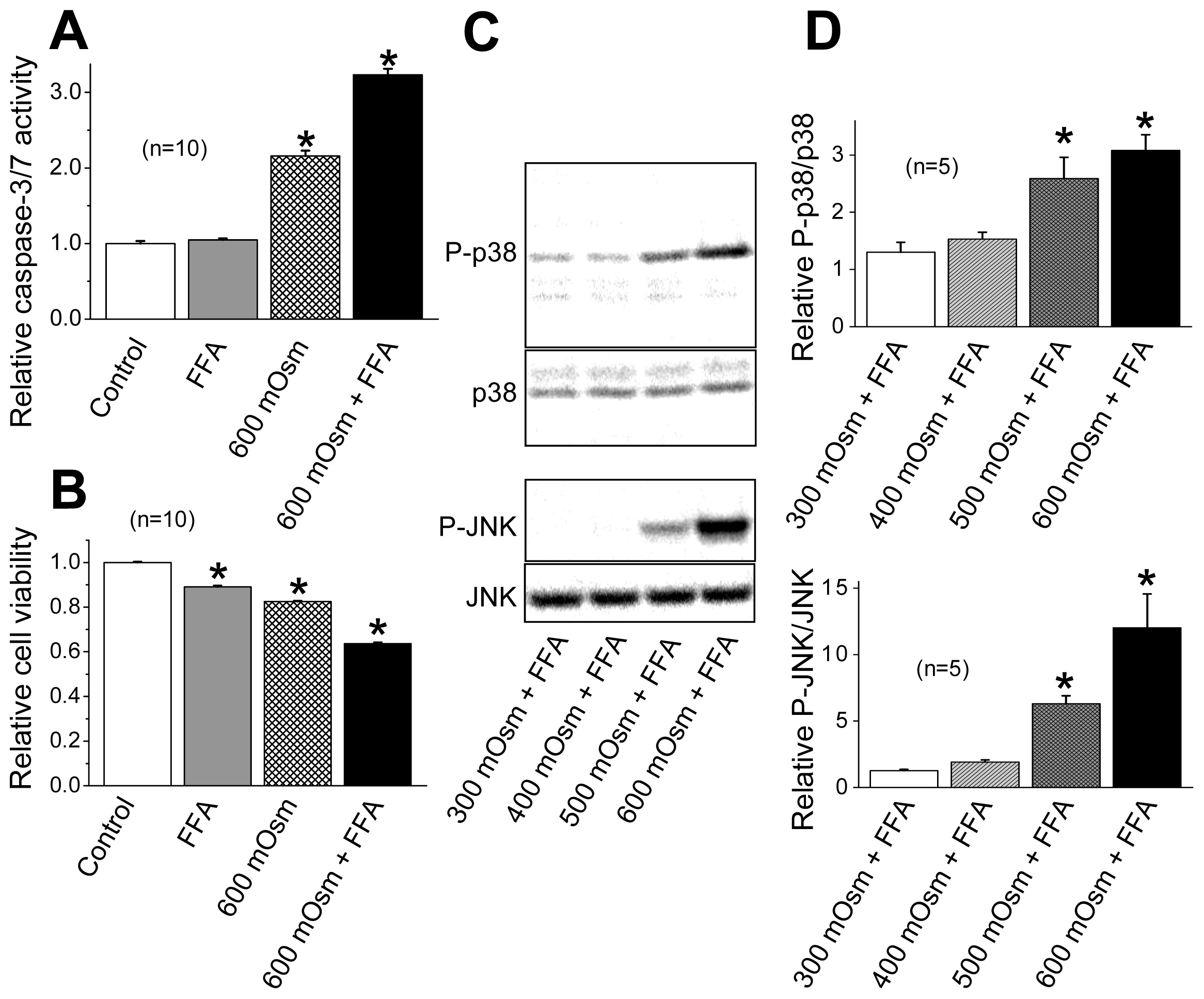
2.3. Induction of Phosphorylation of p38 and JNK by Sustained Cell Shrinkage
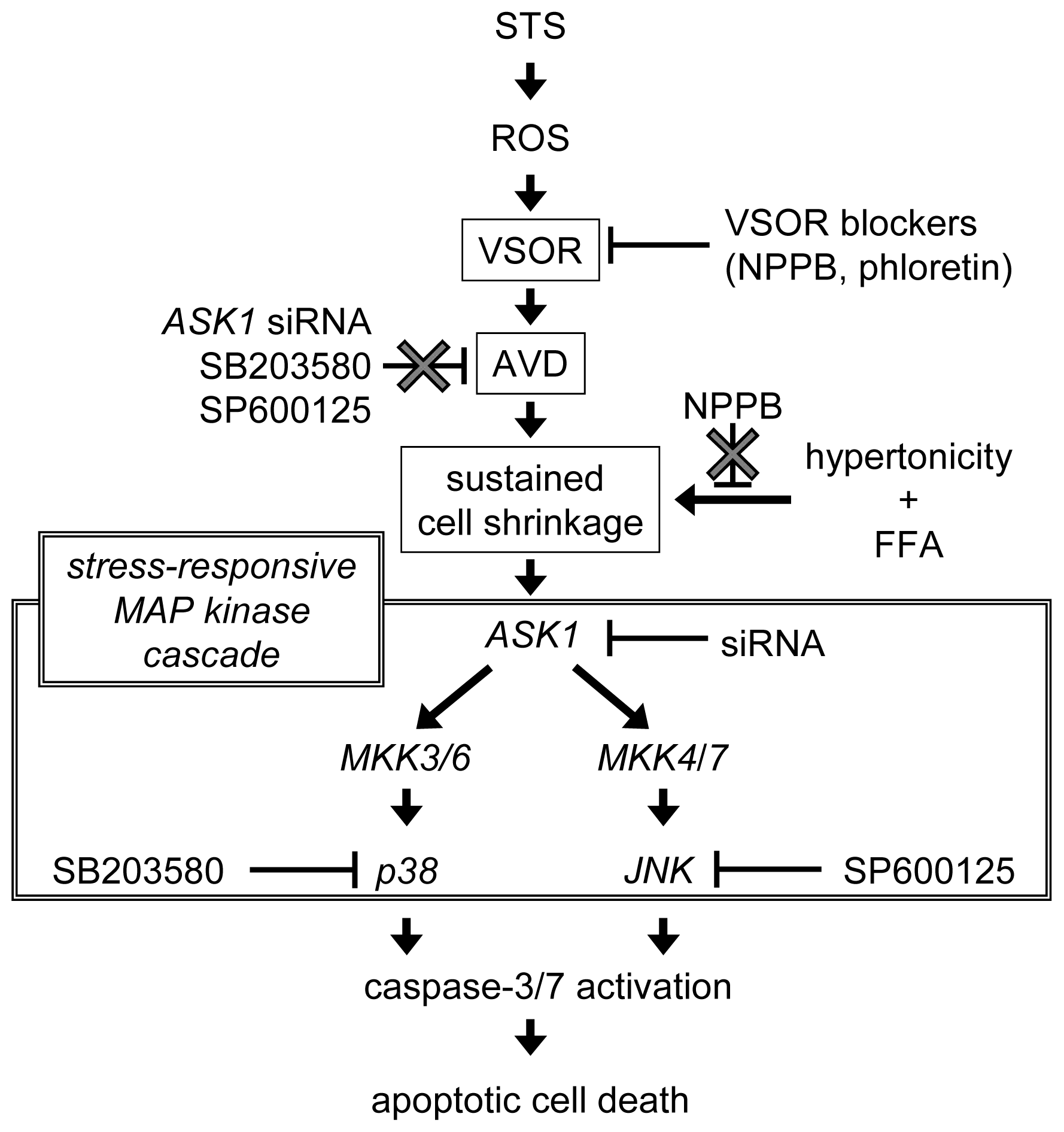
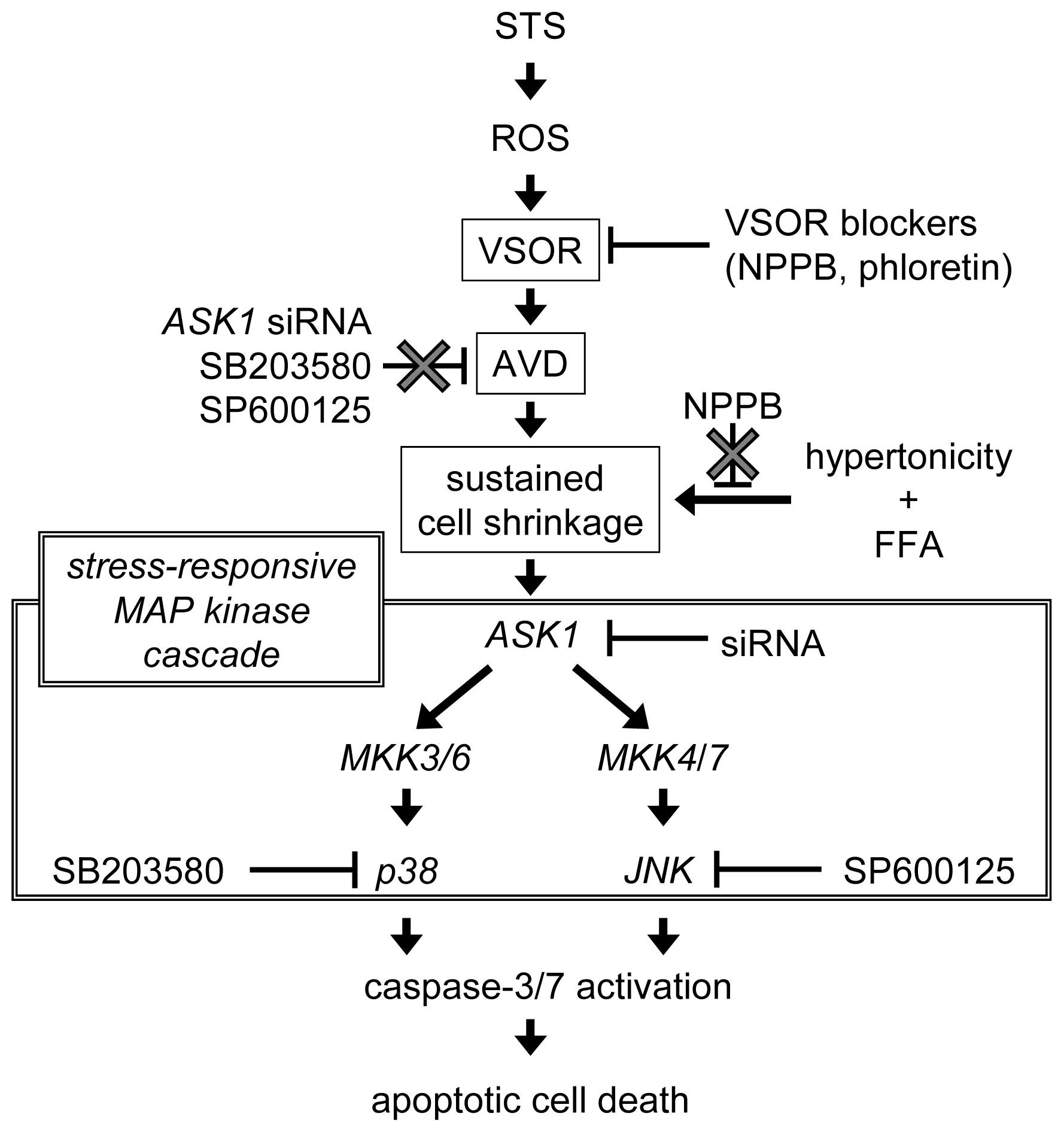
3. Discussion
4. Experimental Section
4.1. Chemicals and Antibodies
4.2. Cell Culture and Apoptosis Induction
4.3. Cell Viability Measurements
4.4. Caspase-3/7 Activity Measurements
4.5. Cell Volume Measurements
4.6. Western Blotting
4.7. RNA Interference
4.8. Quantitative RT-PCR
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar]
- Okada, Y.; Maeno, E.; Shimizu, T.; Dezaki, K.; Wang, J.; Morishima, S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J. Physiol 2001, 532, 3–16. [Google Scholar]
- Lang, F.; Busch, G.L.; Ritter, M.; Völkl, H.; Waldegger, S.; Gulbins, E.; Häussinger, D. Functional significance of cell volume regulatory mechanisms. Physiol. Rev 1998, 78, 247–306. [Google Scholar]
- Okada, Y. Cell Volume Regulation. In The Molecular Mechanism and Volume Sensing Machinery; Elsevier: Amsterdam, The Netherlands, 1998; p. 214. [Google Scholar]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev 2009, 89, 193–277. [Google Scholar]
- Okada, Y. Volume expansion-sensing outward-rectifier Cl− channel: Fresh start to the molecular identity and volume sensor. Am. J. Physiol. Cell Physiol 1997, 273, C755–C789. [Google Scholar]
- Okada, Y. Cell volume-sensitive chloride channel: Phenotypic properties and molecular identity. Contrib. Nephrol 2006, 152, 9–24. [Google Scholar]
- Okada, Y.; Maeno, E.; Shimizu, T.; Manabe, K.; Mori, S.; Nabekura, T. Dual roles of plasmalemmal chloride channels in induction of cell death. Pflügers Archiv: Eur. J. Physiol 2004, 448, 287–295. [Google Scholar]
- Okada, Y.; Shimizu, T.; Maeno, E.; Tanabe, S.; Wang, X.; Takahashi, N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J. Membrane Biol 2006, 209, 21–29. [Google Scholar]
- Shimizu, T.; Numata, T.; Okada, Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl− channel. Proc. Natl. Acad. Sci. USA 2004, 101, 6770–6773. [Google Scholar]
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol 2009, 587, 2141–2149. [Google Scholar]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar]
- Wang, X.S.; Diener, K.; Jannuzzi, D.; Trollinger, D.; Tan, T.H.; Lichenstein, H.; Zukowski, M.; Yao, Z. Molecular cloning and characterization of a novel protein kinase with a catalytic domain homologous to mitogen-activated protein kinase kinase kinase. J. Biol. Chem 1996, 271, 31607–31611. [Google Scholar]
- Ichijo, H.; Nishida, E.; Irie, K.; Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar]
- Chen, Z.; Seimiya, H.; Naito, M.; Mashima, T.; Kizaki, A.; Dan, S.; Imaizumi, M.; Ichijo, H.; Miyazono, K.; Tsuruo, T. ASK1 mediates apoptotic cell death induced by genotoxic stress. Oncogene 1999, 18, 173–180. [Google Scholar]
- Kim, J.Y.; Yu, S.J.; Oh, H.J.; Lee, J.Y.; Kim, Y.; Sohn, J. Panaxydol induces apoptosis through an increased intracellular calcium level, activation of JNK and p38 MAPK and NADPH oxidase-dependent generation of reactive oxygen species. Apoptosis 2011, 16, 347–358. [Google Scholar]
- Subramanyam, M.; Takahashi, N.; Hasegawa, Y.; Mohri, T.; Okada, Y. Inhibition of a protein kinase Akt1 by ASK1 is involved in apoptotic inhibition of regulatory volume increase. J. Biol. Chem 2010, 285, 6109–6117. [Google Scholar]
- Matsuzawa, A.; Ichijo, H. Molecular mechanisms of the decision between life and death, regulation of apoptosis by apoptosis signal-regulating kinase 1. J. Biochem 2001, 130, 1–8. [Google Scholar]
- Kawasaki, H.; Morooka, T.; Shimohama, S.; Kimura, J.; Hirano, T.; Gotoh, Y.; Nishida, E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J. Biol. Chem 1997, 272, 18518–18521. [Google Scholar]
- Yue, T.L.; Wang, C.; Gu, J.L.; Ma, X.L.; Kumar, S.; Lee, J.C.; Feuerstein, G.Z.; Thomas, H.; Maleeff, B.; Ohlstein, E.H. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ. Res 2000, 86, 692–699. [Google Scholar]
- Krumschnabel, G.; Maehr, T.; Nawaz, M.; Schwarzbaum, P.J.; Manzl, C. Staurosporine-induced cell death in salmonid cells: The role of apoptotic volume decrease, ion fluxes and MAP kinase signaling. Apoptosis 2007, 12, 1755–1768. [Google Scholar]
- Wang, X.; Martindale, J.L.; Liu, Y.; Holbrook, N.J. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J 1998, 333, 291–300. [Google Scholar]
- Okada, Y.; Maeno, E. Apoptosis, cell volume regulation and volume-regulatory chloride channels. Comp. Biochem. Physiol. Part A 2001, 130, 377–383. [Google Scholar]
- Kubo, M.; Okada, Y. Volume-regulatory Cl− channel currents in cultured human epithelial cells. J. Physiol 1992, 456, 351–371. [Google Scholar]
- Fan, H.-T.; Morishima, S.; Kida, H.; Okada, Y. Phloretin differentially inhibits volume-sensitive and cAMP-activated, but not Ca-activated, Cl− channels. Br. J. Pharmacol 2001, 133, 1096–1106. [Google Scholar]
- Cuenda, A.; Rouse, J.; Doza, Y.N.; Meier, R.; Cohen, P.; Gallagher, T.F.; Young, P.R.; Lee, J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 1995, 364, 229–233. [Google Scholar]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar]
- Cui, H.; Shao, C.; Liu, Q.; Yu, W.; Fang, J.; Yu, W.; Ali, A.; Ding, K. Heparanase enhances nerve-growth-factor-induced PC12 cell neuritogenesis via the p38 MAPK pathway. Biochem. J 2011, 440, 273–282. [Google Scholar]
- Zhang, D.; Guo, M.; Zhang, W.; Lu, X.Y. Adiponectin stimulates proliferation of adult hippocampal neural stem/progenitor cells through activation of p38 mitogen-activated protein kinase (p38MAPK)/glycogen synthase kinase 3β (GSK-3β)/β-catenin signaling cascade. J. Biol. Chem 2011, 286, 44913–44920. [Google Scholar]
- Polimeni, M.; Valente, E.; Aldieri, E.; Khadjavi, A.; Giribaldi, G.; Prato, M. Haemozoin induces early cytokine-mediated lysozyme release from human monocytes through p38 MAPK- and NF-kappaB- dependent mechanisms. PLoS One 2012, 7, e39497. [Google Scholar]
- Kang, S.; Chemaly, E.R.; Hajjar, R.J.; Lebeche, D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J. Biol. Chem 2011, 286, 18465–18473. [Google Scholar]
- Alrashdan, Y.A.; Alkhouri, H.; Chen, E.; Lalor, D.J.; Poniris, M.; Henness, S.; Brightling, C.E.; Burgess, J.K.; Armour, C.L.; Ammit, A.J.; et al. Asthmatic airway smooth muscle CXCL10 production: Mitogen-activated protein kinase JNK involvement. Am. J. Physiol. Lung. Cell. Mol. Physiol 2012, 302, L1118–L1127. [Google Scholar]
- Joetham, A.; Ohnishi, H.; Okamoto, M.; Takeda, K.; Schedel, M.; Domenico, J.; Dakhama, A.; Gelfand, E.W. Loss of T regulatory cell suppression following signaling through glucocorticoid-induced tumor necrosis receptor (GITR) is dependent on c-Jun N-terminal kinase activation. J. Biol. Chem 2012, 287, 17100–17108. [Google Scholar]
- Krumschnabel, G.; Maehr, T.; Nawaz, M.; Schwarzbaum, P.; Manzl, C. Staurosporine-induced cell death in salmonid cells: The role of apoptotic volume decrease, ion fluxes and MAP kinase signaling. Apoptosis 2007, 12, 1755–1768. [Google Scholar]
- Wehner, F.; Shimizu, T.; Sabirov, R.; Okada, Y. Hypertonic activation of a non-selective cation conductance in HeLa cells and its contribution to cell volume regulation. FEBS Lett 2003, 551, 20–24. [Google Scholar]
- Shimizu, T.; Wehner, F.; Okada, Y. Inhibition of hypertonicity-induced cation channels sensitizes HeLa cells to shrinkage-induced apoptosis. Cell Physiol. Biochem 2006, 18, 295–302. [Google Scholar]
- Nielsen, M.B.; Christensen, S.T.; Hoffmann, E.K. Effects of osmotic stress on the activity of MAPKs and PDGFR-β-mediated signal transduction in NIH-3T3 fibroblasts. Am. J. Physiol. Cell Physiol 2008, 294, C1046–C1055. [Google Scholar]
- Juo, P.; Kuo, C.J.; Reynolds, S.E.; Konz, R.F.; Raingeaud, J.; Davis, R.J.; Biemann, H.P.; Blenis, J. Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol. Cell. Biol 1997, 17, 24–35. [Google Scholar]
- Kim, H.W.; Cho, K.J.; Lee, S.K.; Kim, G.W. Apoptosis signal-regulating kinase 1 (Ask1) targeted small interfering RNA on ischemic neuronal cell death. Brain Res 2011, 1412, 73–78. [Google Scholar]
- Friis, M.B.; Friborg, C.R.; Schneider, L.; Nielsen, M.B.; Lambert, I.H.; Christensen, S.T.; Hoffmann, E.K. Cell shrinkage as a signal to apoptosis in NIH 3T3 fibroblasts. J. Physiol 2005, 567, 427–443. [Google Scholar]
- Redman, P.T.; He, K.; Hartnett, K.A.; Jefferson, B.S.; Hu, L.; Rosenberg, P.A.; Levitan, E.S.; Aizenman, E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. USA 2007, 104, 3568–3573. [Google Scholar]
- Bossy-Wetzel, E.; Talantova, M.V.; Lee, W.D.; Schölzke, M.N.; Harrop, A.; Mathews, E.; Götz, T.; Han, J.; Ellisman, M.H.; Perkins, G.A.; et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 2004, 41, 351–365. [Google Scholar]
- Gao, J.; Wu, D.; Guo, T.B.; Ruan, Q.; Li, T.; Lu, Z.; Xu, M.; Dai, W.; Lu, L. K+ channel activity and redox status are differentially required for JNK activation by UV and reactive oxygen species. Exp. Cell Res 2004, 297, 461–471. [Google Scholar]
- Heimlich, G.; Cidlowski, J.A. Selective role of intracellular chloride in the regulation of the intrinsic but not extrinsic pathway of apoptosis in Jurkat T-cells. J. Biol. Chem 2006, 281, 2232–2241. [Google Scholar]
- Dezaki, K.; Maeno, E.; Sato, K.; Akita, T.; Okada, Y. Early-phase occurrence of K+ and Cl− efflux in addition to Ca2+ mobilization is a prerequisite to apoptosis in HeLa cells. Apoptosis 2012, 17, 821–831. [Google Scholar]
- Liu, H.-T.; Akita, T.; Shimizu, T.; Sabirov, R.Z.; Okada, Y. Bradykinin-induced astrocyte-neuron signaling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J. Physiol 2009, 587, 2197–2209. [Google Scholar]
- Akita, T.; Okada, Y. Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J. Physiol 2011, 589, 3909–3927. [Google Scholar]
- Akita, T.; Fedorovich, S.V.; Okada, Y. Ca2+ nanodomain-mediated component of swelling-induced volume-sensitive outwardly rectifying anion current triggered by autocrine action of ATP in mouse astrocytes. Cell Physiol. Biochem 2011, 28, 1181–1190. [Google Scholar]
- Hazama, A.; Okada, Y. Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. J. Physiol 1988, 402, 687–702. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hasegawa, Y.; Shimizu, T.; Takahashi, N.; Okada, Y. The Apoptotic Volume Decrease Is an Upstream Event of MAP Kinase Activation during Staurosporine-Induced Apoptosis in HeLa Cells. Int. J. Mol. Sci. 2012, 13, 9363-9379. https://doi.org/10.3390/ijms13079363
Hasegawa Y, Shimizu T, Takahashi N, Okada Y. The Apoptotic Volume Decrease Is an Upstream Event of MAP Kinase Activation during Staurosporine-Induced Apoptosis in HeLa Cells. International Journal of Molecular Sciences. 2012; 13(7):9363-9379. https://doi.org/10.3390/ijms13079363
Chicago/Turabian StyleHasegawa, Yuichi, Takahiro Shimizu, Nobuyuki Takahashi, and Yasunobu Okada. 2012. "The Apoptotic Volume Decrease Is an Upstream Event of MAP Kinase Activation during Staurosporine-Induced Apoptosis in HeLa Cells" International Journal of Molecular Sciences 13, no. 7: 9363-9379. https://doi.org/10.3390/ijms13079363