Exome Enrichment and SOLiD Sequencing of Formalin Fixed Paraffin Embedded (FFPE) Prostate Cancer Tissue



Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Exome Sequencing of FFPE and Fresh Frozen Prostate Cancer Tissue
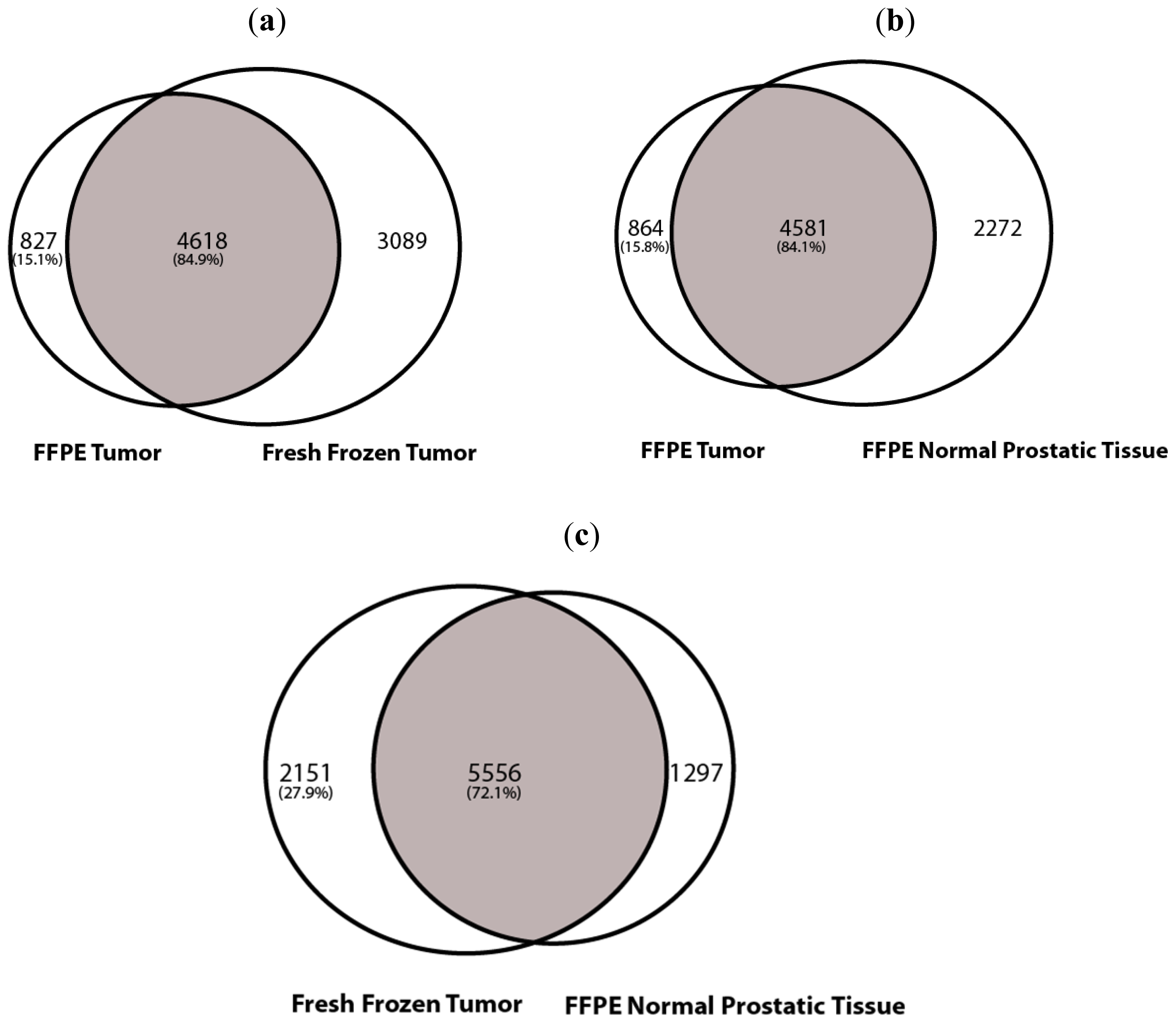
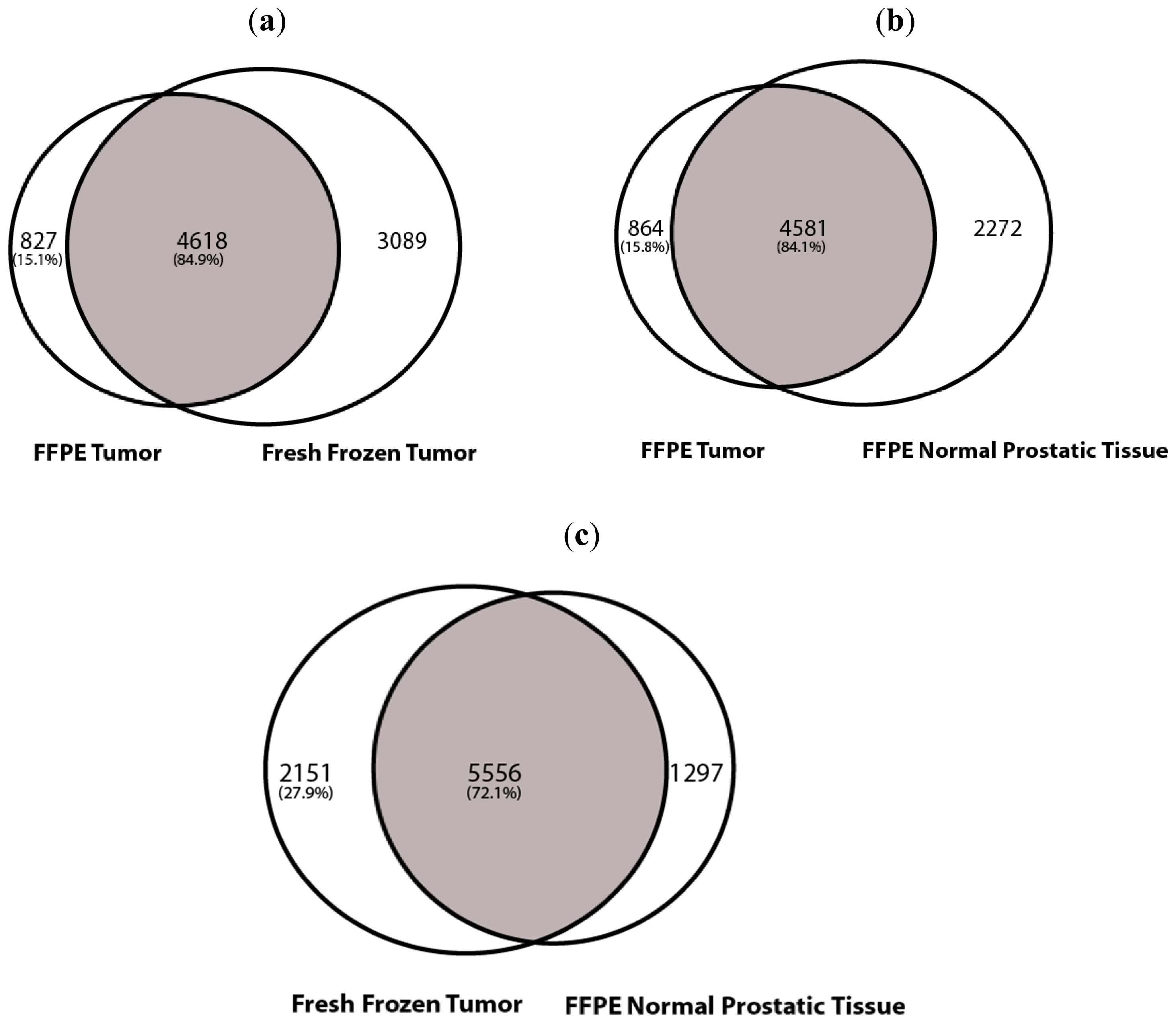
2.1.2. SNV Analysis
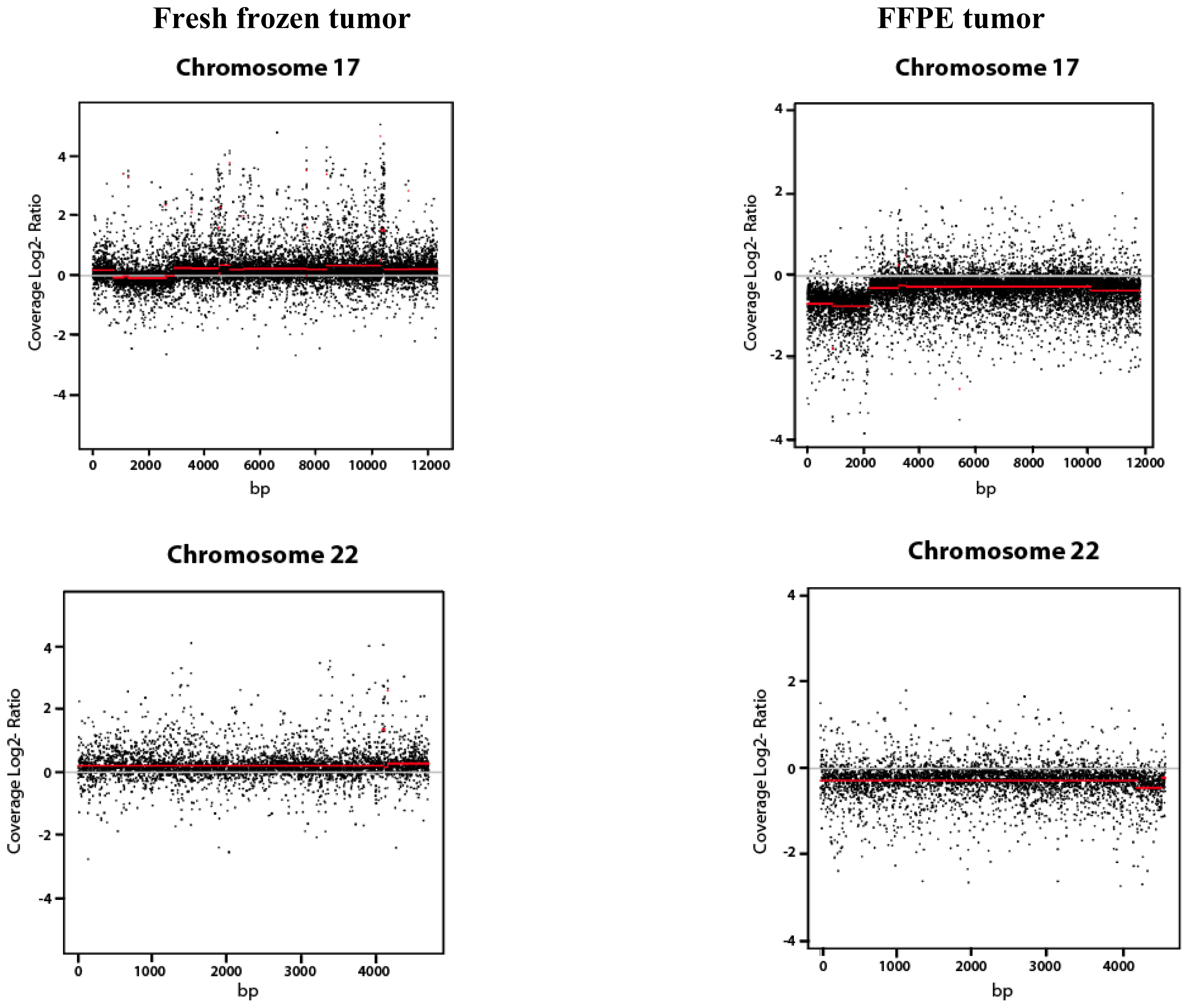
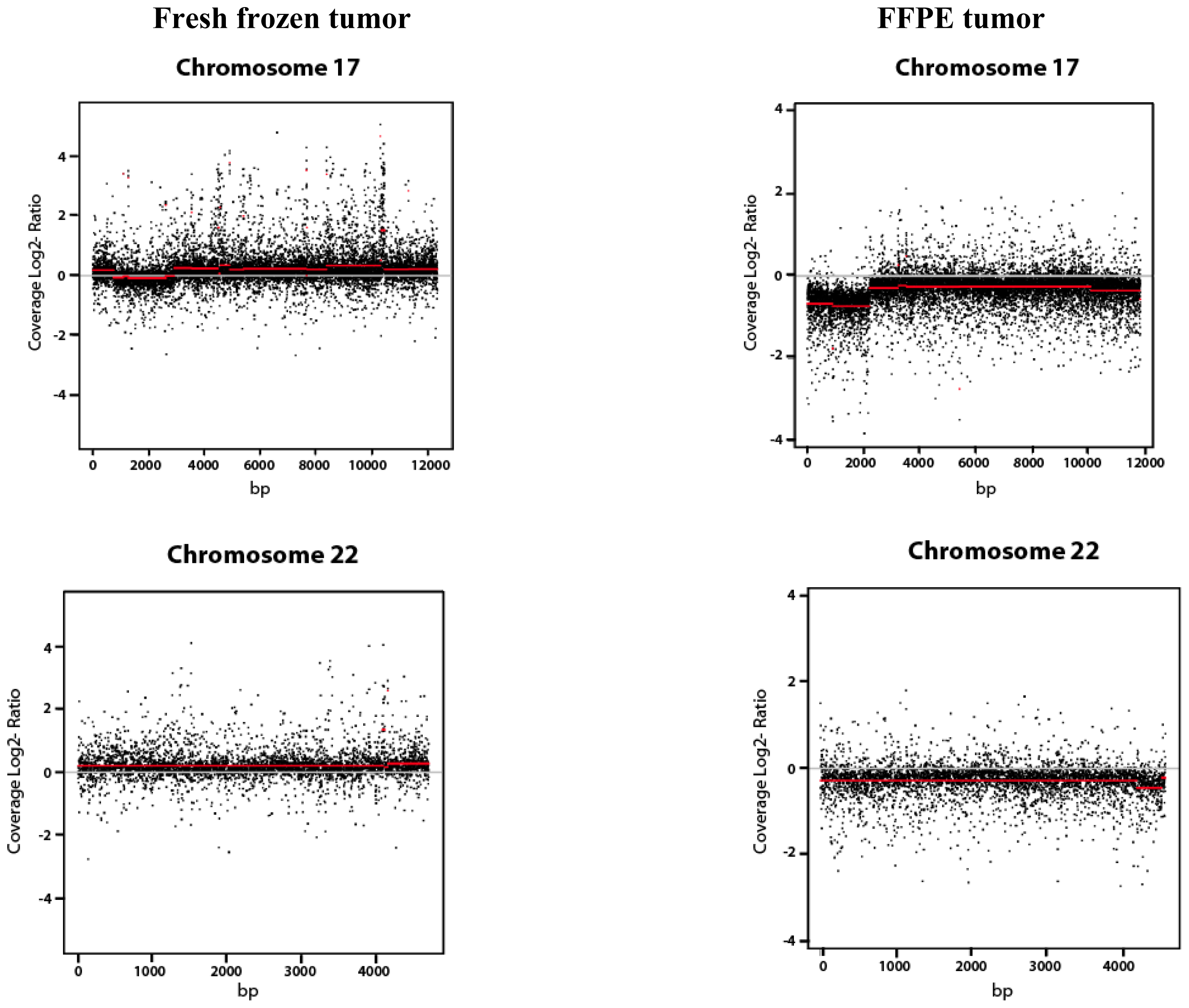
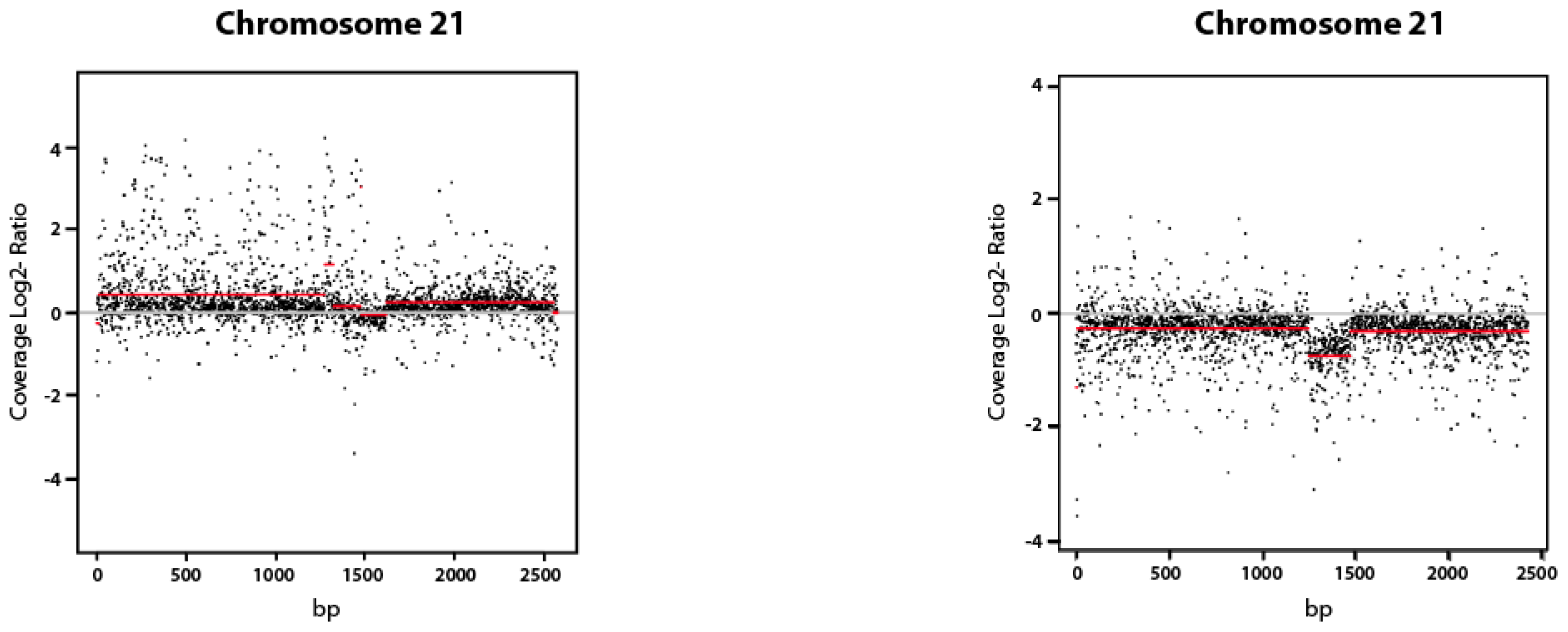
2.1.3. Copy Number Variation (CNV Analysis)
2.2. Discussion
3. Experimental Section
3.1. Tissue Storage
3.2. DNA Extraction and Preparation
3.3. Library Construction
3.4. Targeted Capture and Exome Sequencing
3.5. Bioinformatic Analyses
3.6. Determination of Copy Number Variations
4. Conclusions
Acknowledgments
References
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One 2010, 5, e15661. [Google Scholar]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet 2010, 11, 31–46. [Google Scholar]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [Green Version]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar]
- Ding, L.; Wendl, M.C.; Koboldt, D.C.; Mardis, E.R. Analysis of next-generation genomic data in cancer: Accomplishments and challenges. Hum. Mol. Genet 2010, 19, R188–R196. [Google Scholar]
- Schafer, S.C.; Pfnur, M.; Yerly, S.; Fandel, T.M.; Jichlinski, P.; Lehr, H.A. Cryopreservation of prostate cancer tissue during routine processing of fresh unfixed prostatectomy specimen: Demonstration and validation of a new technique. Prostate 2009, 69, 191–197. [Google Scholar]
- Coombs, N.J.; Gough, A.C.; Primrose, J.N. Optimisation of DNA and RNA extraction from archival formalin-fixed tissue. Nucleic Acids Res 1999, 27, e12. [Google Scholar]
- Talaulikar, D.; Gray, J.X.; Shadbolt, B.; McNiven, M.; Dahlstrom, J.E. A comparative study of the quality of DNA obtained from fresh frozen and formalin-fixed decalcified paraffin-embedded bone marrow trephine biopsy specimens using two different methods. J. Clin. Pathol 2008, 61, 119–123. [Google Scholar]
- Schweiger, M.R.; Kerick, M.; Timmermann, B.; Isau, M. The power of NGS technologies to delineate the genome organization in cancer: From mutations to structural variations and epigenetic alterations. Cancer Metastasis Rev 2011, 30, 199–210. [Google Scholar]
- Kerick, M.; Isau, M.; Timmermann, B.; Sultmann, H.; Herwig, R.; Krobitsch, S.; Schaefer, G.; Verdorfer, I.; Bartsch, G.; Klocker, H.; et al. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genomics 2011, 4, 68. [Google Scholar]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet 2010, 11, 685–696. [Google Scholar]
- Schweiger, M.R.; Kerick, M.; Timmermann, B.; Albrecht, M.W.; Borodina, T.; Parkhomchuk, D.; Zatloukal, K.; Lehrach, H. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis. PLoS One 2009, 4, e5548. [Google Scholar]
- Homer, N.; Merriman, B.; Nelson, S.F. BFAST: An alignment tool for large scale genome resequencing. PLoS One 2009, 4, e7767. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010, 20, 1297–1303. [Google Scholar]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011, 43, 491–498. [Google Scholar]
- Cingolani, P. snpEff: Variant effect prediction. Available online: http://snpeff.sourceforge.net accessed on 15 February 2012.


{kind=link}
{kind=link}
{kind=link}
| Parameters | FFPE non-tumor | FFPE tumor | Fresh-frozen tumor |
|---|---|---|---|
| Reads | 100,675,212 | 99,910,447 | 113,195,686 |
| Rejected reads | 9,011,246 | 10,280,465 | 7,809,343 |
| Valid reads | 91,663,966 | 89,629,982 | 105,386,343 |
| Non-uniquely mapped reads | 47,691,531 | 47,113,995 | 54,345,991 |
| Uniquely mapped reads | 43,972,435 | 42,515,987 | 51,040,352 |
| On-target reads (within ± 150 bp) | 36,016,954 | 33,363,890 | 42,077,913 |
| On-target reads in% | 81.91% | 78.47% | 82.44% |
| All Intervals/Exons | 165,481 | 165,481 | 165,481 |
| Targeted exons | 162,490 | 162,699 | 163,027 |
| Targeted exons in% | 98.19% | 98.32% | 98.52% |
| Non-targeted exons | 2,991 | 2,782 | 2,454 |
| Total number of non synonymous SNVs | 6,853 | 5,445 | 7,707 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Menon, R.; Deng, M.; Boehm, D.; Braun, M.; Fend, F.; Boehm, D.; Biskup, S.; Perner, S. Exome Enrichment and SOLiD Sequencing of Formalin Fixed Paraffin Embedded (FFPE) Prostate Cancer Tissue. Int. J. Mol. Sci. 2012, 13, 8933-8942. https://doi.org/10.3390/ijms13078933
Menon R, Deng M, Boehm D, Braun M, Fend F, Boehm D, Biskup S, Perner S. Exome Enrichment and SOLiD Sequencing of Formalin Fixed Paraffin Embedded (FFPE) Prostate Cancer Tissue. International Journal of Molecular Sciences. 2012; 13(7):8933-8942. https://doi.org/10.3390/ijms13078933
Chicago/Turabian StyleMenon, Roopika, Mario Deng, Diana Boehm, Martin Braun, Falko Fend, Detlef Boehm, Saskia Biskup, and Sven Perner. 2012. "Exome Enrichment and SOLiD Sequencing of Formalin Fixed Paraffin Embedded (FFPE) Prostate Cancer Tissue" International Journal of Molecular Sciences 13, no. 7: 8933-8942. https://doi.org/10.3390/ijms13078933
APA StyleMenon, R., Deng, M., Boehm, D., Braun, M., Fend, F., Boehm, D., Biskup, S., & Perner, S. (2012). Exome Enrichment and SOLiD Sequencing of Formalin Fixed Paraffin Embedded (FFPE) Prostate Cancer Tissue. International Journal of Molecular Sciences, 13(7), 8933-8942. https://doi.org/10.3390/ijms13078933