Fasudil, a Rho-Kinase Inhibitor, Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice

Abstract
:1. Introduction
2. Results
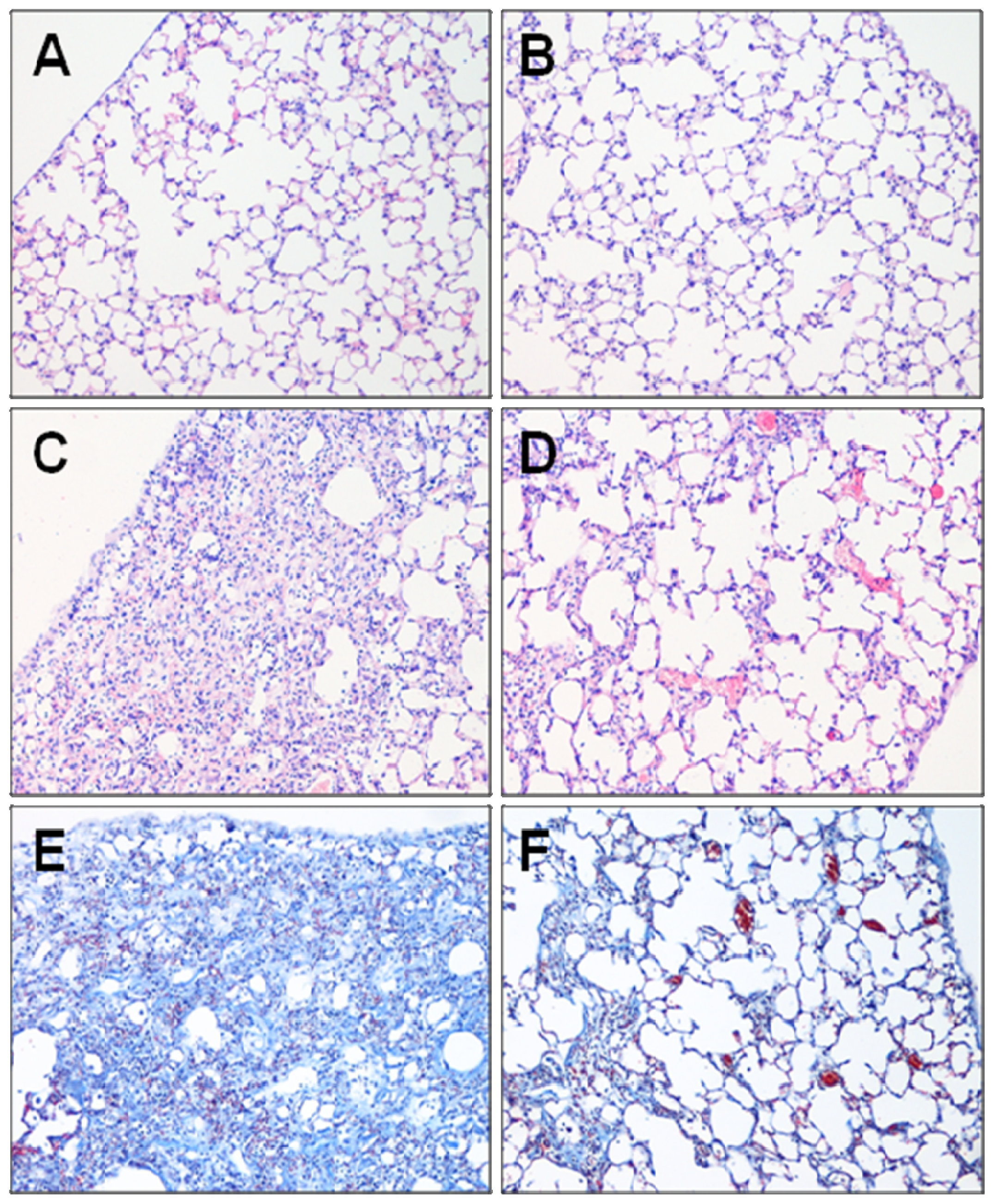
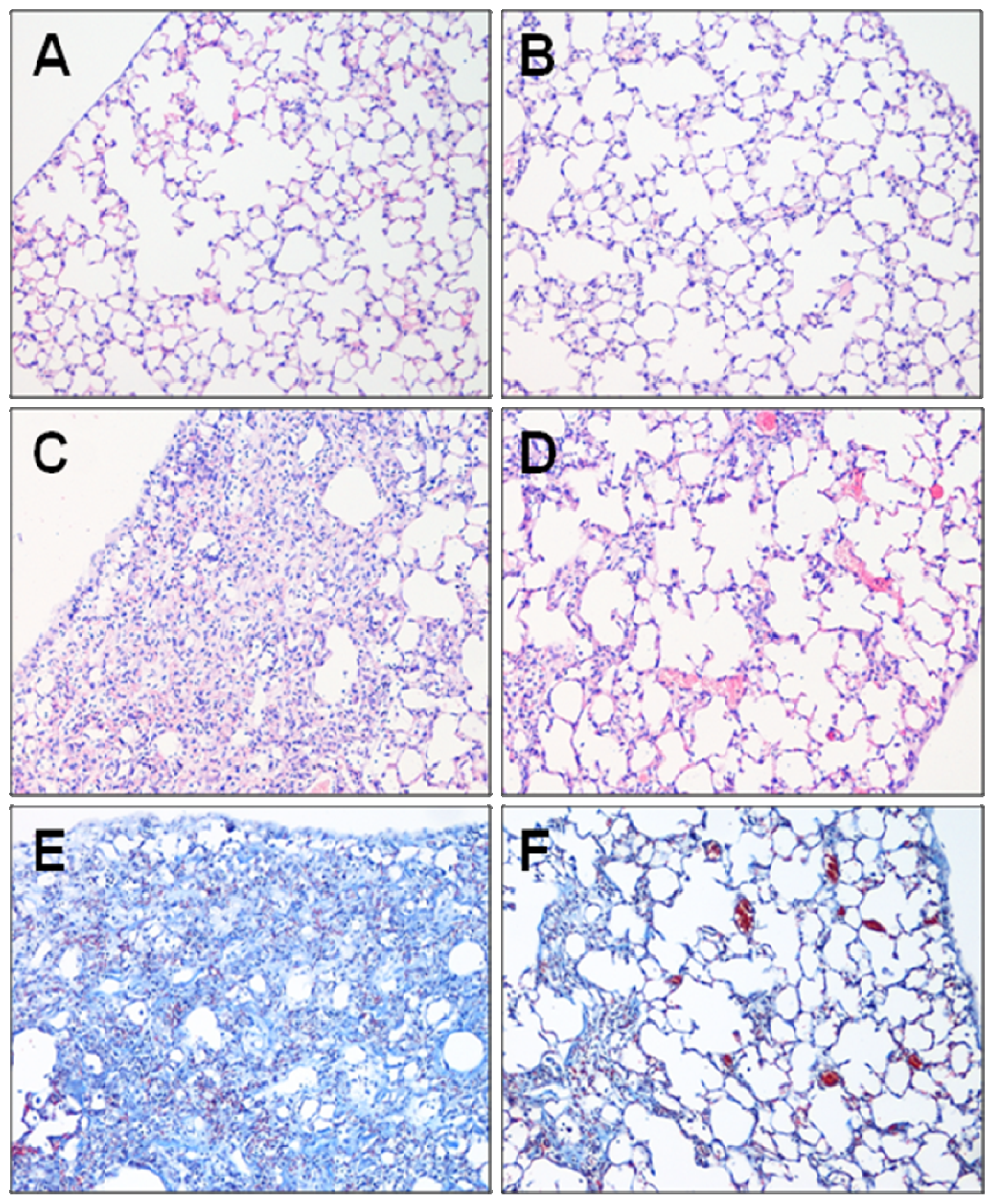
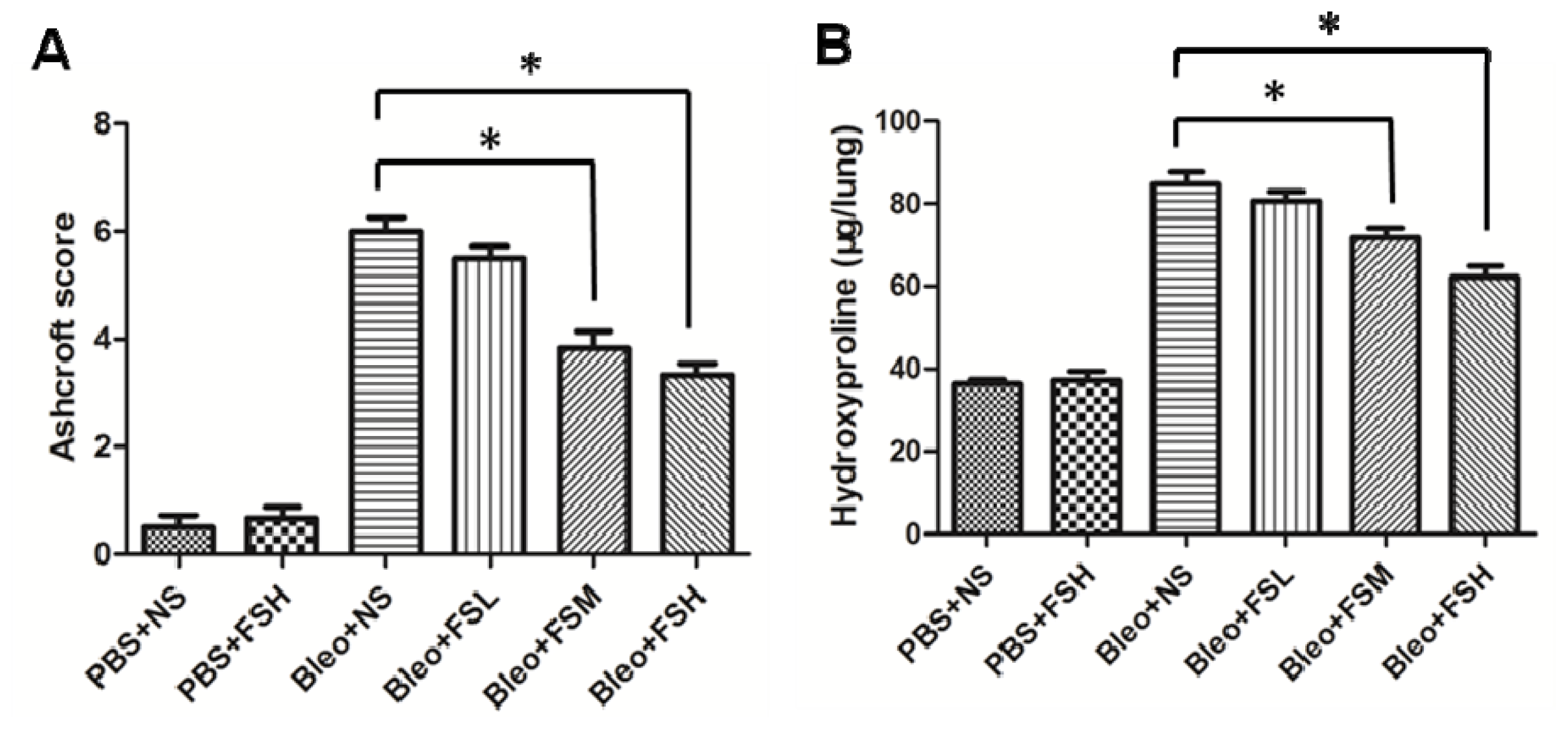
2.1. Effect of Fasudil on Histopathology
2.2. Effect of Fasudil on Hydroxyproline Level
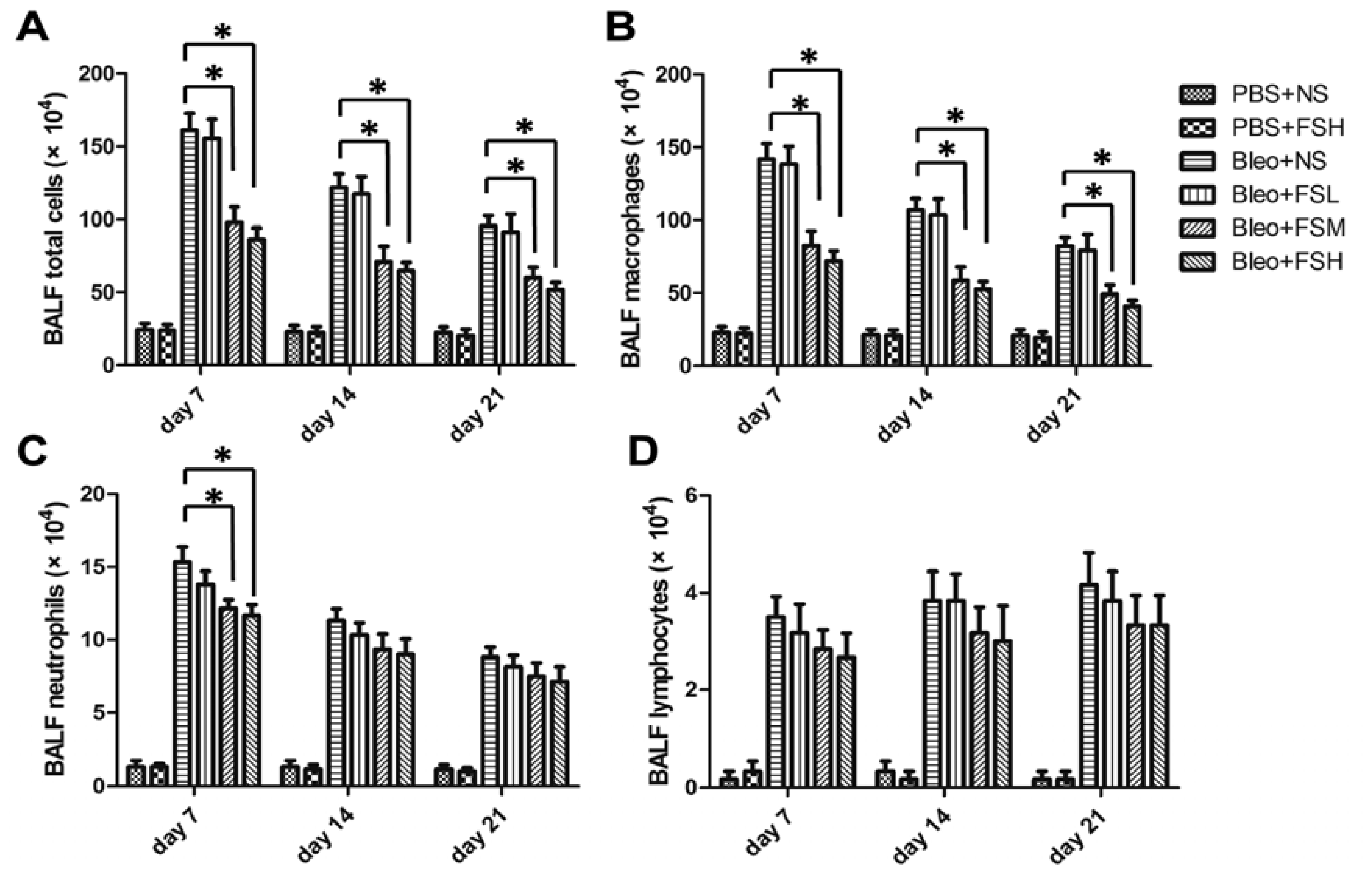
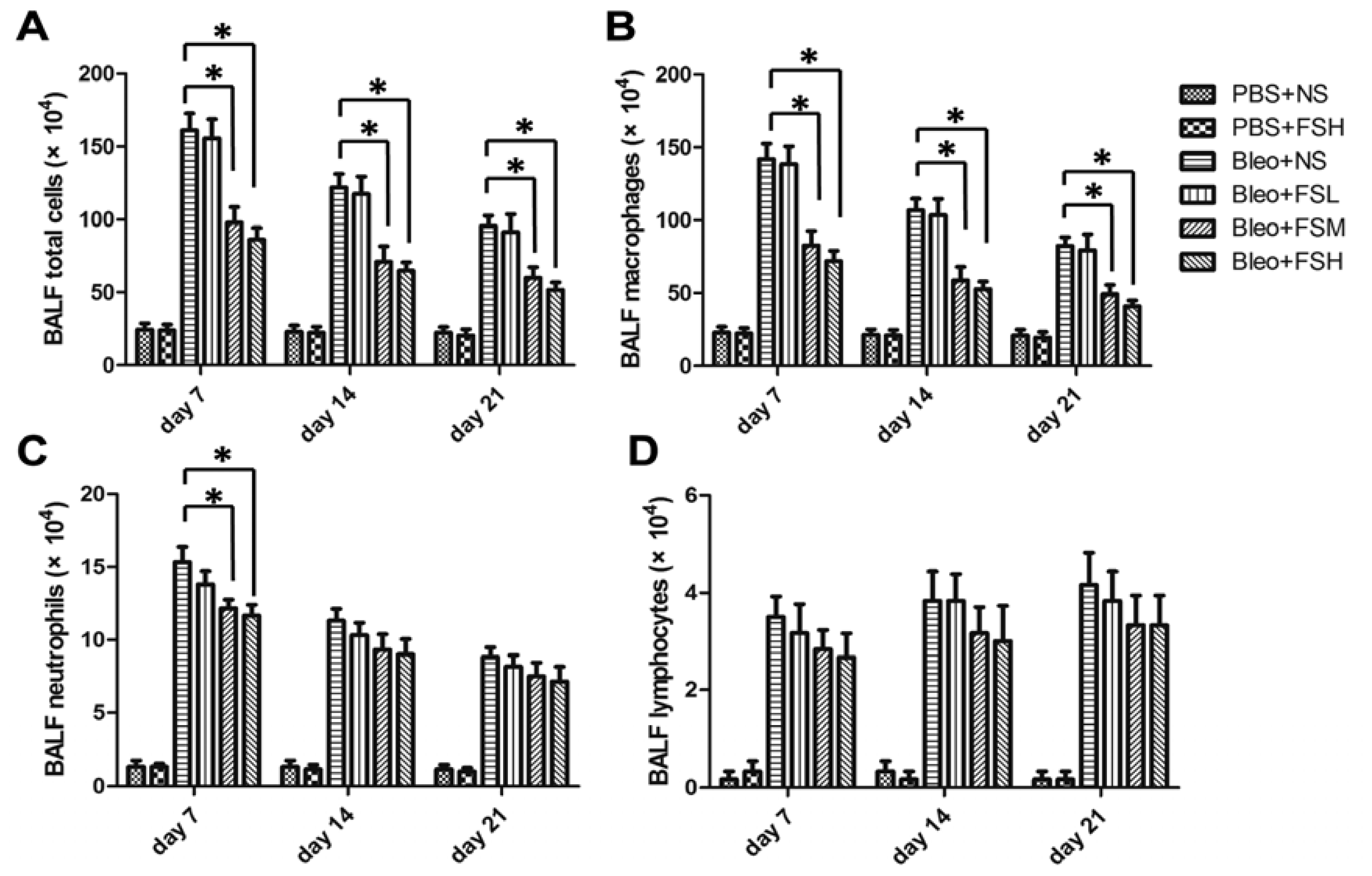
2.3. Effect of Fasudil on Infiltration of the Inflammatory Cells in Airways
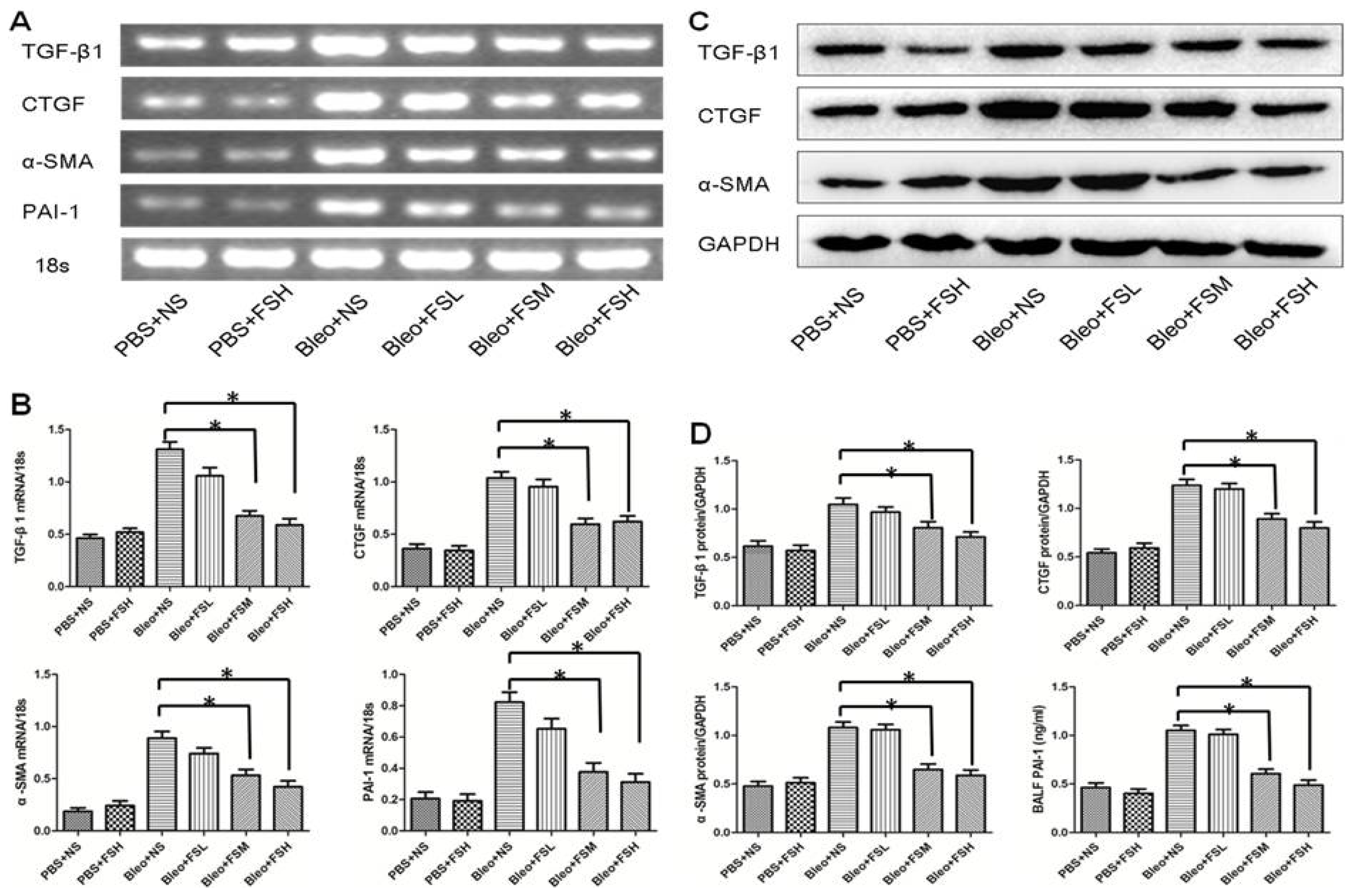
2.4. Effect of Fasudil on TGF-β1, CTGF, α-SMA, and PAI-1
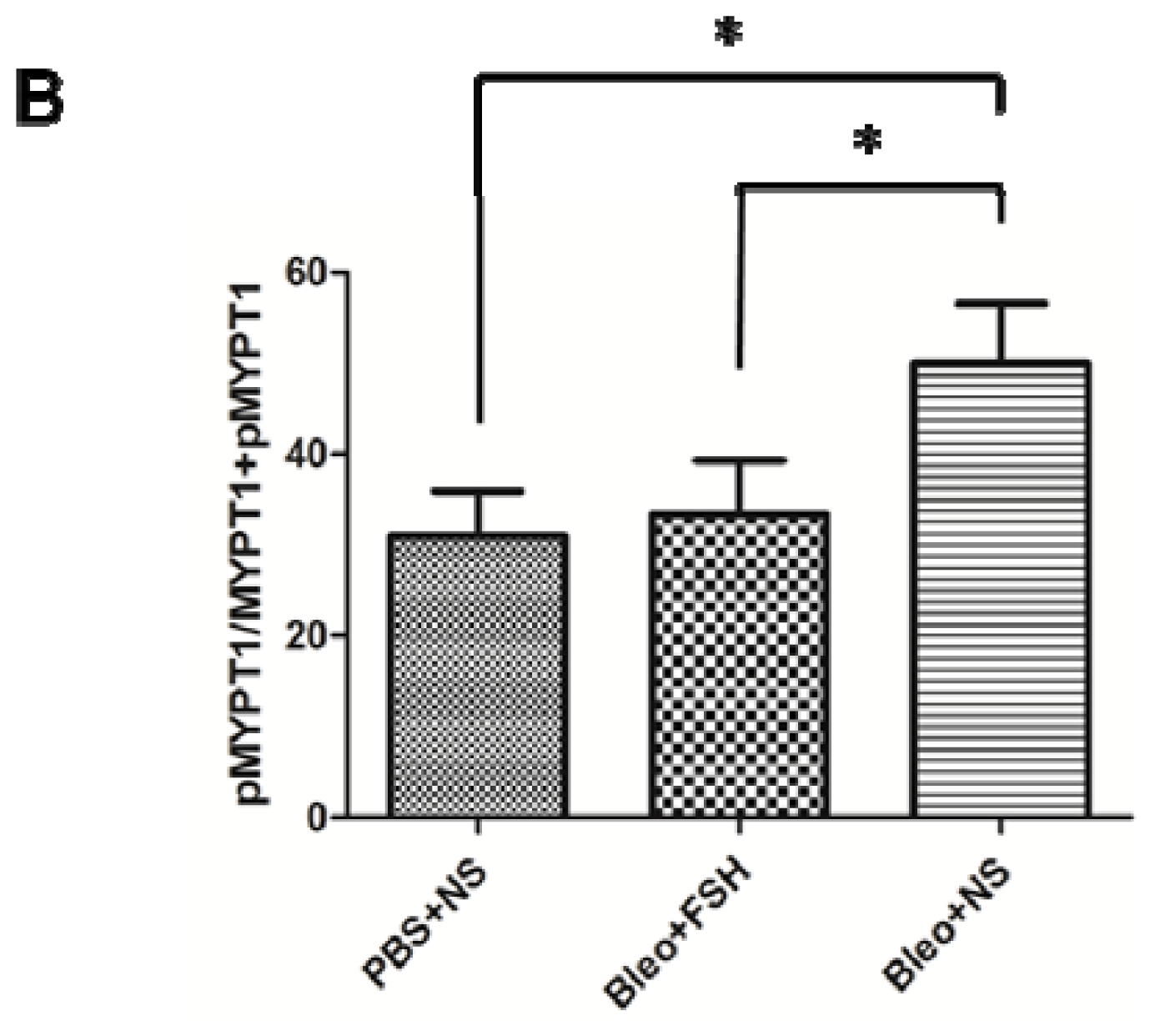
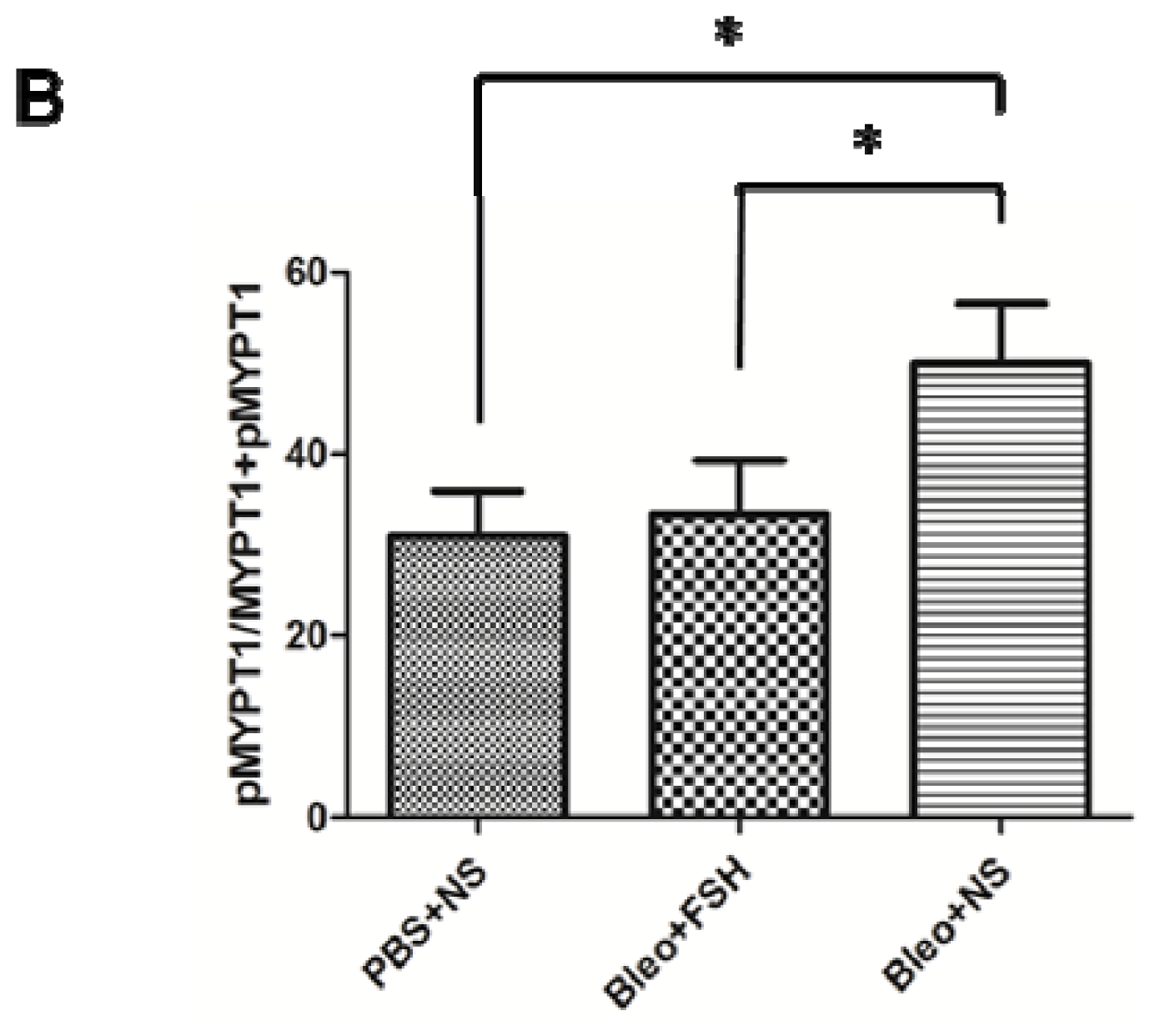
2.5. MYPT1 Phosphorylation Levels in Mouse Lungs
3. Discussion
4. Experimental Section
4.1. Study Design and Experimental Protocol
4.2. Histopathological Examination
4.3. Bronchoalveolar Lavage Fluid (BALF)
4.4. Enzyme-Linked Immunosorbent Assay
4.5. Hydroxyproline Assay
4.6. RT-PCR
4.7. Western Blot
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med 2011, 183, 788–824. [Google Scholar]
- Gross, T.J.; Hunninghake, G.W. Idiopathic pulmonary fibrosis. N. Engl. J. Med 2001, 345, 517–525. [Google Scholar]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med 2006, 174, 810–816. [Google Scholar]
- Shimokawa, H.; Takeshita, A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler Thromb Vasc. Biol 2005, 25, 1767–1775. [Google Scholar]
- Niggli, V. Rho-kinase in human neutrophils: A role in signalling for myosin light chain phosphorylation and cell migration. FEBS Lett 1999, 445, 69–72. [Google Scholar]
- Shimizu, Y.; Dobashi, K.; Iizuka, K.; Horie, T.; Suzuki, K.; Tukagoshi, H.; Nakazawa, T.; Nakazato, Y.; Mori, M. Contribution of small GTPase Rho and its target protein ROCK in a murine model of lung fibrosis. Am. J. Respir. Crit. Care Med 2001, 163, 210–217. [Google Scholar]
- Bonniaud, P.; Margetts, P.J.; Ask, K.; Flanders, K.; Gauldie, J.; Kolb, M. TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J. Immunol 2005, 175, 5390–5395. [Google Scholar]
- Kothapalli, D.; Frazier, K.S.; Welply, A.; Segarini, P.R.; Grotendorst, G.R. Transforming growth factor beta induces anchorage-independent growth of NRK fibroblasts via a connective tissue growth factor-dependent signaling pathway. Cell Growth Differ 1997, 8, 61–68. [Google Scholar]
- Watts, K.L.; Spiteri, M.A. Connective tissue growth factor expression and induced by transforming growth factor-beta is abrogated by simvastatin via a Rho signaling mechanism. Am. J. Physiol. Lung Cell Mol. Physiol 2004, 287, L1323–L1332. [Google Scholar]
- Watts, K.L.; Cottrell, E.; Hoban, P.R.; Spiteri, M.A. RhoA signaling modulates cyclin D1 expression in human lung fibroblasts; implications for idiopathic pulmonary fibrosis. Respir. Res 2006, 7. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Zaiman, A.; Champion, H.C. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am. J. Physiol. Lung Cell Mol. Physiol 2008, 294, L24–L33. [Google Scholar]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med 2011, 17, 355–361. [Google Scholar]
- Bergeron, A.; Soler, P.; Kambouchner, M.; Loiseau, P.; Milleron, B.; Valeyre, D.; Hance, A.J.; Tazi, A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and il-10. Eur. Respir. J 2003, 22, 69–76. [Google Scholar]
- Kenyon, N.J.; Ward, R.W.; McGrew, G.; Last, J.A. TGF-β1 causes airway fibrosis and increased collagen I and III mRNA in mice. Thorax 2003, 8, 772–777. [Google Scholar]
- Golan-Gerstl, R.; Wallach-Dayan, S.B.; Amir, G.; Breuer, R. Epithelial cell apoptosis by fas ligand-positive myofibroblasts in lung fibrosis. Am. J. Respir. Cell Mol. Biol 2007, 36, 270–275. [Google Scholar]
- Senoo, T.; Hattori, N.; Tanimoto, T.; Furonaka, M.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Yokoyama, A.; Kohno, N. Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis. Thorax 2010, 65, 334–340. [Google Scholar]
- Goldstein, R.H.; Fine, A. Potential therapeutic initiatives for fibrogenic lung diseases. Chest 1995, 108, 848–855. [Google Scholar]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol 2008, 294, L152–L160. [Google Scholar]
- Singh, R.; Wang, B.; Shirvaikar, A.; Khan, S.; Kamat, S.; Schelling, J.R.; Konieczkowski, M.; Sedor, J.R. The IL-1 receptor and Rho directly associate to drive cell activation in inflammation. J. Clin. Invest 1999, 103, 1561–1570. [Google Scholar]
- Montaner, S.; Perona, R.; Saniger, L.; Lacal, J.C. Multiple signalling pathways lead to the activation of the nuclear factor kappaB by the Rho family of GTPases. J. Biol. Chem 1998, 273, 12779–12785. [Google Scholar]
- Higashi, M.; Shimokawa, H.; Hattori, T.; Hiroki, J.; Mukai, Y.; Morikawa, K.; Ichiki, T.; Takahashi, S.; Takeshita, A. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: Effect on endothelial NAD(P)H oxidase system. Circ. Res 2003, 93, 767–775. [Google Scholar]
- Radeff, J.M.; Nagy, Z.; Stern, P.H. Rho and Rho kinase are involved in parathyroid hormone stimulated protein kinase C alpha translocation and IL-6 promoter activity in osteoblastic cells. J. Bone Miner. Res 2004, 19, 1882–1891. [Google Scholar]
- Takemoto, M.; Sun, J.; Hiroki, J.; Shimokawa, H.; Liao, J.K. Rho-kinasemediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation 2002, 106, 57–62. [Google Scholar]
- Funakoshi, Y.; Ichiki, T.; Shimokawa, H.; Egashira, K.; Takeda, K.; Kaibuchi, K.; Takeya, M.; Yoshimura, T.; Takeshita, A. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension 2001, 38, 100–104. [Google Scholar]
- Hattori, T.; Shimokawa, H.; Higashi, M.; Hiroki, J.; Mukai, Y.; Kaibuchi, K.; Takeshita, A. Long-term treatment with a specific Rho-kinase inhibitor suppresses cardiac allograft vasculopathy in mice. Circ. Res 2004, 94, 46–52. [Google Scholar]
- Steinberg, K.P.; Hudson, L.D.; Goodman, R.B.; Hough, C.L.; Lanken, P.N.; Hyzy, R.; Thompson, B.T.; Ancukiewicz, M. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N. Engl. J. Med 2006, 354, 1671–1684. [Google Scholar]
- Khalil, N.; O’Connor, R.N.; Flanders, K.C.; Unruh, H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: An immunohistochemical study. Am. J. Respir. Cell Mol. Biol 1996, 14, 131–138. [Google Scholar]
- Border, W.A.; Noble, N.A. Transforming growth factor-β in tissue fibrosis. N. Engl. J. Med 1994, 331, 1286–1292. [Google Scholar]
- Duncan, M.R.; Frazier, K.S.; Abramson, S.; Williams, S.; Klapper, H.; Huang, X.; Grotendorst, G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: Down-regulation by cAMP. FASEB. J 1999, 13, 1774–1786. [Google Scholar]
- Giri, S.N.; Hyde, D.M.; Hollinger, M.A. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax 1993, 48, 959–966. [Google Scholar]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest 1997, 100, 768–776. [Google Scholar]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 1998, 275, L365–L371. [Google Scholar]
- Liu, M.; Gu, M.; Wu, Y.; Zhu, P.; Zhang, W.; Yin, C.; Zhang, W.J. Therapeutic effect of Y-27632 on chronic allograft nephropathy in rats. J. Surg. Res 2009, 157, e117–e127. [Google Scholar]
- Wu, G.; Tu, Y.; Jia, R. The influence of fasudil on the epithelial-mesenchymal transdifferentiation of renal tubular epithelial cells from diabetic rats. Biomed. Pharmacother 2010, 64, 124–129. [Google Scholar]
- Lee, C.G.; Cho, S.; Homer, R.J.; Elias, J.A. Genetic control of transforming growth factor-beta1-induced emphysema and fibrosis in the murine lung. Proc. Am. Thorac. Soc 2006, 3, 476–477. [Google Scholar]
- Cutroneo, K.R.; White, S.L.; Phan, S.H.; Ehrlich, H.P. Therapies for bleomycin induced lung fibrosis through regulation of TGF-beta1 induced collagen gene expression. J. Cell Physiol 2007, 211, 585–589. [Google Scholar]
- Idell, S.; James, K.K.; Levin, E.G.; Schwartz, B.S.; Manchanda, N.; Maunder, R.J.; Martin, T.R.; McLarty, J.; Fair, D.S. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J. Clin. Invest 1989, 84, 695–705. [Google Scholar]
- Bertozzi, P.; Astedt, B.; Zenzius, L.; Lynch, K.; LeMaire, F.; Zapol, W.; Chapman, H.A., Jr. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N. Engl. J. Med 1990, 322, 890–897. [Google Scholar]
- Chapman, H.A.; Allen, C.L.; Stone, O.L. Abnormalities in pathways of alveolar fibrin turnover among patients with interstitial lung disease. Am. Rev. Respir. Dis 1986, 133, 437–443. [Google Scholar]
- Eitzman, D.T.; McCoy, R.D.; Zheng, X.; Fay, W.P.; Shen, T.; Ginsburg, D.; Simon, R.H. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J. Clin. Invest 1996, 97, 232–237. [Google Scholar]
- Nishikimi, T.; Koshikawa, S.; Ishikawa, Y.; Akimoto, K.; Inaba, C.; Ishimura, K.; Ono, H.; Matsuoka, H. Inhibition of Rho-kinase attenuates nephrosclerosis and improves survival in salt-loaded spontaneously hypertensive stroke-prone rats. J. Hypertens 2007, 25, 1053–1063. [Google Scholar]
- Ishimaru, K.; Ueno, H.; Kagitani, S.; Takabayashi, D.; Takata, M.; Inoue, H. Fasudil attenuates myocardial fibrosis in association with inhibition of monocyte/macrophage infiltration in the heart of DOCA/salt hypertensive rats. J. Cardiovasc. Pharmacol 2007, 50, 187–194. [Google Scholar]
- Vicari, R.M.; Chaitman, B.; Keefe, D.; Smith, W.B.; Chrysant, S.G.; Tonkon, M.J.; Bittar, N.; Weiss, R.J.; Morales-Ballejo, H.; Thadani, U. Efficacy and safety of fasudil in patients with stable angina: A double-blind, placebo-controlled, phase 2 trial. J. Am. Coll. Cardiol 2005, 46, 1803–1811. [Google Scholar]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol 1988, 41, 467–470. [Google Scholar]
- Nie, L.; Xiang, R.; Zhou, W.; Lu, B.; Cheng, D.; Gao, J. Attenuation of acute lung inflammation induced by cigarette smoke in CXCR3 knockout mice. Respir. Res 2008, 9, 82. [Google Scholar]
- Moore, B.B.; Paine, R., 3rd; Christensen, P.J.; Moore, T.A.; Sitterding, S.; Ngan, R.; Wilke, C.A.; Kuziel, W.A.; Toews, G.B. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J. Immunol 2001, 167, 4368–4377. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | S/AS | Primer Sequence (5′ to 3′) | Products (bp) |
|---|---|---|---|
| TGFβ1 | S | GACCGCAACAACGCCATCT | 306 |
| AS | GCCCTGTATTCCGTCTCCTT | ||
| CTGF | S | CTTCTGCGATTTCGGCTCC | 352 |
| AS | GGCTCGCATCATAGTTGGGT | ||
| α-SMA | S | CTGCCGAGCGTGAGATTGT | 485 |
| AS | CTTCGTCGTATTCCTGTTTGCT | ||
| PAI-1 | S | CAGATGTCTTCAGCCCTTGC | 444 |
| AS | GAAGTCCACCTGTTTCACCAT | ||
| 18s | S | AGCAGGACTGGGAGACTACG | 223 |
| AS | AGCAGGCTCTGGTGGGTGAT |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, C.; Huang, H.; Liu, J.; Wang, Y.; Lu, Z.; Xu, Z. Fasudil, a Rho-Kinase Inhibitor, Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. Int. J. Mol. Sci. 2012, 13, 8293-8307. https://doi.org/10.3390/ijms13078293
Jiang C, Huang H, Liu J, Wang Y, Lu Z, Xu Z. Fasudil, a Rho-Kinase Inhibitor, Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. International Journal of Molecular Sciences. 2012; 13(7):8293-8307. https://doi.org/10.3390/ijms13078293
Chicago/Turabian StyleJiang, Chunguo, Hui Huang, Jia Liu, Yanxun Wang, Zhiwei Lu, and Zuojun Xu. 2012. "Fasudil, a Rho-Kinase Inhibitor, Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice" International Journal of Molecular Sciences 13, no. 7: 8293-8307. https://doi.org/10.3390/ijms13078293
APA StyleJiang, C., Huang, H., Liu, J., Wang, Y., Lu, Z., & Xu, Z. (2012). Fasudil, a Rho-Kinase Inhibitor, Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. International Journal of Molecular Sciences, 13(7), 8293-8307. https://doi.org/10.3390/ijms13078293