Phage Display-based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed against Human Pathogens


Abstract
:1. Introduction
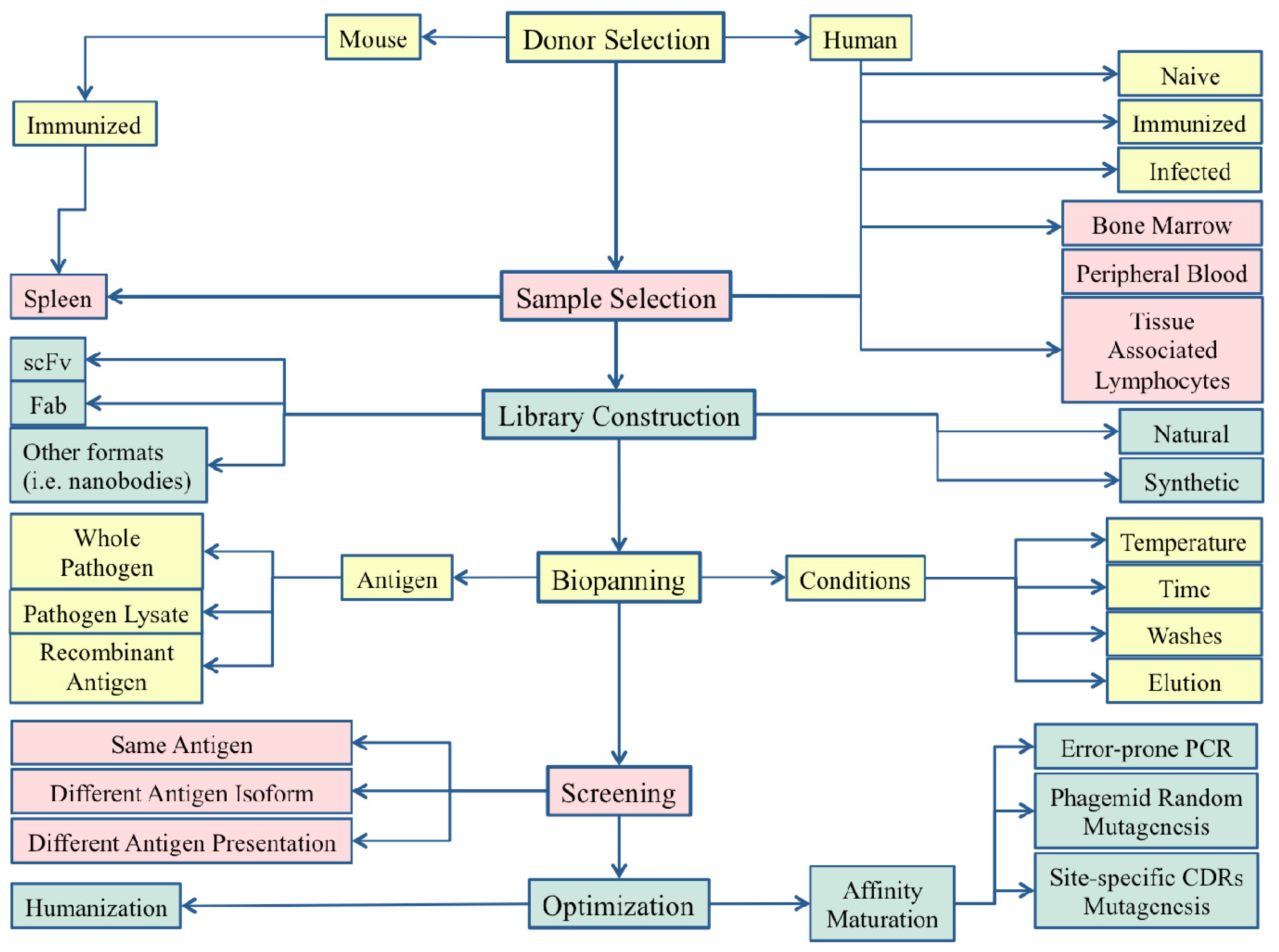
2. Phage Display: The Importance of the Correct Strategy
2.1. The Library Design and the Source of B-Cells
2.1.1. Fully Synthetic Library Design
2.1.2. Human Libraries from Bone Marrow and Peripheral Blood B-Cells
2.1.3. Libraries from Immunized Animals
2.2. Panning Condition Optimization and Target Antigen Presentation as Key Points for Phage Display Success
2.2.1. Importance of the Selection Conditions
2.2.2. Appropriate Selection of the Target Antigen and of the Correct Antigen Presentation
2.2.3. Cross-Selection Strategies
2.2.4. Selection Using “Exhaustive Panning” Strategy
2.3. Phage Display for Crucial Post-Selection Optimization of mAbs
2.3.1. Phage Display for mAb Optimization: The Example of Palivizumab and Motavizumab
2.3.2. Phage Display Optimization by Error-Prone Libraries Usage: The Example of Anthim
3. Conclusions
Acknowledgment
References
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Parmley, S.F; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar]
- Burioni, R; Plaisant, P; Delli Carri, V; Vannini, A; Spanu, T; Clementi, M; Fadda, G; Varaldo, P.E. An improved phage display vector for antibody repertoire cloning by construction of combinatorial libraries. Res. Virol 1997, 148, 161–164. [Google Scholar]
- Mancini, N; Carletti, S; Perotti, M; Canducci, F; Mammarella, M; Sampaolo, M; Burioni, R. Phage display for the production of human monoclonal antibodies against human pathogens. New Microbiol 2004, 27, 315–328. [Google Scholar]
- Burton, D.R; Barbas, C.F., III; Persson, M.A; Koenig, S; Chanock, R.M; Lerner, R.A. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. USA 1991, 88, 10134–10137. [Google Scholar]
- Williamson, R.A; Burioni, R; Sanna, P.P; Partridge, L.J; Barbas, C.F., III; Burton, D.R. Human monoclonal antibodies against a plethora of viral pathogens from single combinatorial libraries. Proc. Natl. Acad. Sci. USA 1993, 90, 4141–4145. [Google Scholar]
- Burioni, R; Plaisant, P; Manzin, A; Rosa, D; Delli Carri, V; Bugli, F; Solforosi, L; Abrignani, S; Varaldo, P.E; Fadda, G; et al. Dissection of human humoral immune response against hepatitis C virus E2 glycoprotein by repertoire cloning and generation of recombinant Fab fragments. Hepatology 1998, 28, 810–814. [Google Scholar]
- Zhang, M.Y; Xiao, X; Sidorov, I.A; Choudhry, V; Cham, F; Zhang, P.F; Bouma, P; Zwick, M; Choudhary, A; Montefiori, D.C; et al. Identification and characterization of a new cross-reactive human immunodeficiency virus type 1-neutralizing human monoclonal antibody. J Virol 2004, 78, 9233–9242. [Google Scholar]
- Mao, S; Gao, C; Lo, C.H; Wirsching, P; Wong, C.H; Janda, K.D. Phage-display library selection of high-affinity human single-chain antibodies to tumor-associated carbohydrate antigens sialyl Lewisx and Lewisx. Proc. Natl. Acad. Sci. USA 1999, 96, 6953–6958. [Google Scholar]
- Reiche, N; Jung, A; Brabletz, T; Vater, T; Kirchner, T; Faller, G. Generation and characterization of human monoclonal scFv antibodies against Helicobacter pylori antigens. Infect. Immun 2002, 70, 4158–4164. [Google Scholar]
- Kim, S.J; Jang, M.H; Stapleton, J.T; Yoon, S.O; Kim, K.S; Jeon, E.S; Hong, H.J. Neutralizing human monoclonal antibodies to hepatitis a virus recovered by phage display. Virology 2004, 318, 598–607. [Google Scholar]
- Chang, T.Y; Siegel, D.L. Isolation of an IgG anti-B from a human Fab-phage display library. Transfusion 2001, 41, 6–12. [Google Scholar]
- Roark, J.H; Bussel, J.B; Cines, D.B; Siegel, D.L. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood 2002, 100, 1388–1398. [Google Scholar]
- Clark, M.A; Hawkins, N.J; Papaioannou, A; Fiddes, R.J; Ward, R.L. Isolation of human anti-c-erbB-2 Fabs from a lymph node-derived phage display library. Clin. Exp. Immunol 1997, 109, 166–174. [Google Scholar]
- Roovers, R.C; van der Linden, E; de Bruine, A.P; Arends, J.W; Hoogenboom, H.R. Identification of colon tumour-associated antigens by phage antibody selections on primary colorectal carcinoma. Eur. J. Cancer 2001, 37, 542–549. [Google Scholar]
- Wu, B.P; Xiao, B; Wan, T.M; Zhang, Y.L; Zhang, Z.S; Zhou, D.Y; Lai, Z.S; Gao, C.F. Construction and selection of the natural immune Fab antibody phage display library from patients with colorectal cancer. World J. Gastroenterol 2001, 7, 811–815. [Google Scholar]
- Xu, M.Y; Xu, X.H; Chen, G.Z; Deng, X.L; Li, J; Yu, X.J; Chen, M.Z. Production of a human single-chain variable fragment antibody against esophageal carcinoma. World J. Gastroenterol 2004, 10, 2619–2623. [Google Scholar]
- Sixholo, J; van Wyngaardt, W; Mashau, C; Frischmuth, J; Du Plessis, D.H; Fehrsen, J. Improving the characteristics of a mycobacterial 16 kDa-specific chicken scFv. Biologicals 2011, 39, 110–116. [Google Scholar]
- De Genst, E; Saerens, D; Muyldermans, S; Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol 2006, 30, 187–198. [Google Scholar]
- Dooley, H; Flajnik, M.F. Antibody repertoire development in cartilaginous fish. Dev. Comp. Immunol 2006, 30, 43–56. [Google Scholar]
- Conrad, U; Scheller, J. Considerations on antibody-phage display methodology. Comb. Chem. High Throughput Screen 2005, 8, 117–126. [Google Scholar]
- Chan, C.E; Chan, A.H; Lim, A.P; Hanson, B.J. Comparison of the efficiency of antibody selection from semi-synthetic scFv and non-immune Fab phage display libraries against protein targets for rapid development of diagnostic immunoassays. J. Immunol. Methods 2011, 373, 79–88. [Google Scholar]
- Yu, R; Wang, S; Yu, Y.Z; Du, W.S; Yang, F; Yu, W.Y; Sun, Z.W. Neutralizing antibodies of botulinum neurotoxin serotype a screened from a fully synthetic human antibody phage display library. J. Biomol. Screen 2009, 14, 991–998. [Google Scholar]
- Tomlinson, I.M; Cox, J.P; Gherardi, E; Lesk, A.M; Chothia, C. The structural repertoire of the human V kappa domain. EMBO J 1995, 14, 4628–4638. [Google Scholar]
- Oliva, B; Bates, P.A; Querol, E; Aviles, F.X; Sternberg, M.J. Automated classification of antibody complementarity determining region 3 of the heavy chain (H3) loops into canonical forms and its application to protein structure prediction. J. Mol. Biol 1998, 279, 1193–1210. [Google Scholar]
- Clementi, N; de Marco, D; Mancini, N; Solforosi, L; Moreno, G.J; Gubareva, L.V; Mishin, V; di Pietro, A; Vicenzi, E; Siccardi, A.G; et al. PLoS One 2011, 6, e28001.
- Burioni, R; Canducci, F; Mancini, N; Clementi, N; Sassi, M; de Marco, D; Diotti, R.A; Saita, D; Sampaolo, M; Sautto, G; et al. Monoclonal antibodies isolated from human B cells neutralize a broad range of H1 subtype influenza A viruses including swine-origin Influenza virus (S-OIV). Virology 2010, 399, 144–152. [Google Scholar]
- Burioni, R; Canducci, F; Mancini, N; Clementi, N; Sassi, M; de Marco, D; Saita, D; Diotti, R.A; Sautto, G; Sampaolo, M; et al. Molecular cloning of the first human monoclonal antibodies neutralizing with high potency swine-origin influenza A pandemic virus (S-OIV). New Microbiol 2009, 32, 319–324. [Google Scholar]
- De Marco, D.C.N; Mancini, N; Solforosi, L; Moreno, G.J; Sun, X; Tumpey, T.M; Gubareva, L.V; Mishin, V; Clementi, M; Burioni, R. A non-VH1-69 heterosubtypic neutralizing human monoclonal antibody protects mice against H1N1 and H5N1 viruses. PLoS One 2012, 7, e34415. [Google Scholar]
- Burioni, R; Mancini, N; de Marco, D; Clementi, N; Perotti, M; Nitti, G; Sassi, M; Canducci, F; Shvela, K; Bagnarelli, P; et al. Anti-HIV-1 response elicited in rabbits by anti-idiotype monoclonal antibodies mimicking the CD4-binding site. PLoS One 2008, 3, e3423. [Google Scholar]
- Nabel, G.J; Fauci, A.S. Induction of unnatural immunity: Prospects for a broadly protective universal influenza vaccine. Nat. Med 2010, 16, 1389–1391. [Google Scholar]
- Monto, A.S; Ansaldi, F; Aspinall, R; McElhaney, J.E; Montano, L.F; Nichol, K.L; Puig-Barbera, J; Schmitt, J; Stephenson, I. Influenza control in the 21st century: Optimizing protection of older adults. Vaccine 2009, 27, 5043–5053. [Google Scholar]
- Ansaldi, F; Canepa, P; Parodi, V; Bacilieri, S; Orsi, A; Compagnino, F; Icardi, G; Durando, P. Adjuvanted seasonal influenza vaccines and perpetual viral metamorphosis: The importance of cross-protection. Vaccine 2009, 27, 3345–3348. [Google Scholar]
- Lambert, L.C; Fauci, A.S. Influenza vaccines for the future. N. Engl. J. Med 2010, 363, 2036–2044. [Google Scholar]
- Stanekova, Z; Vareckova, E. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol. J 2010, 7, 351. [Google Scholar]
- Steel, J. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. mBio 2010, 1. [Google Scholar] [CrossRef]
- Desogus, A; Burioni, R; Ingianni, A; Bugli, F; Pompei, R; Fadda, G. Production and characterization of a human recombinant monoclonal Fab fragment specific for influenza A viruses. Clin. Diagn. Lab. Immunol 2003, 10, 680–685. [Google Scholar]
- Karlsson Hedestam, G.B; Fouchier, R.A; Phogat, S; Burton, D.R; Sodroski, J; Wyatt, R.T. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat. Rev. Microbiol 2008, 6, 143–155. [Google Scholar]
- Gamblin, S.J; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem 2010, 285, 28403–28409. [Google Scholar]
- Mancini, N; Solforosi, L; Clementi, N; de Marco, D; Clementi, M; Burioni, R. A potential role for monoclonal antibodies in prophylactic. Antivir. Res 2011, 92, 15–26. [Google Scholar]
- Kashyap, A.K; Steel, J; Rubrum, A; Estelles, A; Briante, R; Ilyushina, N.A; Xu, L; Swale, R.E; Faynboym, A.M; Foreman, P.K.; et al. Protection from the 2009 H1N1 pandemic influenza by an antibody from combinatorial survivor-based libraries. PLoS Pathog 2010, 6, e1000990. [Google Scholar]
- Jayasekera, J.P; Moseman, E.A; Carroll, M.C. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol 2007, 81, 3487–3494. [Google Scholar]
- Throsby, M; van den Brink, E; Jongeneelen, M; Poon, L.L; Alard, P; Cornelissen, L; Bakker, A; Cox, F; van Deventer, E; Guan, Y; et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 2008, 3, e3942. [Google Scholar]
- Goodchild, S.A; Dooley, H; Schoepp, R.J; Flajnik, M; Lonsdale, S.G. Isolation and characterisation of Ebolavirus-specific recombinant antibody fragments from murine and shark immune libraries. Mol. Immunol 2011, 48, 2027–2037. [Google Scholar]
- Gerstenbruch, S; Brooks, C.L; Kosma, P; Brade, L; Mackenzie, C.R; Evans, S.V; Brade, H; Muller-Loennies, S. Analysis of cross-reactive and specific anti-carbohydrate antibodies against lipopolysaccharide from Chlamydophila psittaci. Glycobiology 2010, 20, 461–472. [Google Scholar]
- Sowa, K.M; Cavanagh, D.R; Creasey, A.M; Raats, J; McBride, J; Sauerwein, R; Roeffen, W.F; Arnot, D.E. Isolation of a monoclonal antibody from a malaria patient-derived phage display library recognising the Block 2 region of Plasmodium falciparum merozoite surface protein-1. Mol. Biochem. Parasitol 2001, 112, 143–147. [Google Scholar]
- Knapp, B; Hundt, E; Enders, B; Kupper, H.A. Protection of Aotus monkeys from malaria infection by immunization with recombinant hybrid proteins. Infect. Immun 1992, 60, 2397–2401. [Google Scholar]
- Perera, K.L; Handunnetti, S.M; Holm, I; Longacre, S; Mendis, K. Baculovirus merozoite surface protein 1 C-terminal recombinant antigens are highly protective in a natural primate model for human Plasmodium vivax malaria. Infect. Immun 1998, 66, 1500–1506. [Google Scholar]
- Diggs, C.L; Ballou, W.R; Miller, L.H. The major merozoite surface protein as a malaria vaccine target. Parasitol. Today 1993, 9, 300–302. [Google Scholar]
- Matsumoto, S; Yukitake, H; Kanbara, H; Yamada, T. Long-lasting protective immunity against rodent malaria parasite infection at the blood stage by recombinant BCG secreting merozoite surface protein-1. Vaccine 1999, 18, 832–834. [Google Scholar]
- Burghaus, P.A; Wellde, B.T; Hall, T; Richards, R.L; Egan, A.F; Riley, E.M; Ballou, W.R; Holder, A.A. Immunization of Aotus nancymai with recombinant C-terminus of Plasmodium falciparum merozoite surface protein 1 in liposomes and alum adjuvant does not induce protection against a challenge infection. Infect. Immun 1996, 64, 3614–3619. [Google Scholar]
- Chang, S.P; Case, S.E; Gosnell, W.L; Hashimoto, A; Kramer, K.J; Tam, L.Q; Hashiro, C.Q; Nikaido, C.M; Gibson, H.L; Lee-Ng, C.T; et al. A recombinant baculovirus 42-kilodalton C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 protects Aotus monkeys against malaria. Infect. Immun 1996, 64, 253–261. [Google Scholar]
- Cavanagh, D.R; Elhassan, I.M; Roper, C; Robinson, V.J; Giha, H; Holder, A.A; Hviid, L; Theander, T.G; Arnot, D.E; McBride, J.S. A longitudinal study of type-specific antibody responses to Plasmodium falciparum merozoite surface protein-1 in an area of unstable malaria in Sudan. J. Immunol 1998, 161, 347–359. [Google Scholar]
- Miller, L.H; Roberts, T; Shahabuddin, M; McCutchan, T.F. Analysis of sequence diversity in the Plasmodium falciparum merozoite surface protein-1 (MSP-1). Mol. Biochem. Parasitol 1993, 59, 1–14. [Google Scholar]
- Conway, D.J; Cavanagh, D.R; Tanabe, K; Roper, C; Mikes, Z.S; Sakihama, N; Bojang, K.A; Oduola, A.M; Kremsner, P.G; Arnot, D.E; et al. A principal target of human immunity to malaria identified by molecular population genetic and immunological analyses. Nat. Med 2000, 6, 689–692. [Google Scholar]
- Choo, Q.L; Kuo, G; Weiner, A.J; Overby, L.R; Bradley, D.W; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar]
- Arichi, T; Saito, T; Major, M.E; Belyakov, I.M; Shirai, M; Engelhard, V.H; Feinstone, S.M; Berzofsky, J.A. Retraction. Proc. Natl. Acad. Sci. USA 2001, 98, 6901–6904. [Google Scholar]
- Weiner, A.J; Brauer, M.J; Rosenblatt, J; Richman, K.H; Tung, J; Crawford, K; Bonino, F; Saracco, G; Choo, Q.L; Houghton, M; et al. Variable and hypervariable domains are found in the regions of HCV corresponding to the flavivirus envelope and NS1 proteins and the pestivirus envelope glycoproteins. Virology 1991, 180, 842–848. [Google Scholar]
- Bukh, J; Purcell, R.H; Miller, R.H. At least 12 genotypes of hepatitis C virus predicted by sequence analysis of the putative E1 gene of isolates collected worldwide. Proc. Natl. Acad. Sci. USA 1993, 90, 8234–8238. [Google Scholar]
- Prabhu, R; Khalap, N; Burioni, R; Clementi, M; Garry, R.F; Dash, S. Inhibition of hepatitis C virus nonstructural protein, helicase activity, and viral replication by a recombinant human antibody clone. Am. J. Pathol 2004, 165, 1163–1173. [Google Scholar]
- Plaisant, P; Burioni, R; Manzin, A; Solforosi, L; Candela, M; Gabrielli, A; Fadda, G; Clementi, M. Human monoclonal recombinant Fabs specific for HCV antigens obtained by repertoire cloning in phage display combinatorial vectors. Res. Virol 1997, 148, 165–169. [Google Scholar]
- Barbas, C.F., III; Kang, A.S.; Lerner, R.A.; Benkovic, S.J. Assembly of combinatorial antibody libraries on phage surfaces: The gene III site. Proc. Natl. Acad. Sci. USA 1991, 88, 7978–7982. [Google Scholar]
- Mancini, N; Diotti, R.A; Perotti, M; Sautto, G; Clementi, N; Nitti, G; Patel, A.H; Ball, J.K; Clementi, M; Burioni, R. Hepatitis C virus (HCV) infection may elicit neutralizing antibodies targeting epitopes conserved in all viral genotypes. PLoS One 2009, 4, e8254. [Google Scholar]
- Perotti, M; Ghidoli, N; Altara, R; Diotti, R.A; Clementi, N; de Marco, D; Sassi, M; Clementi, M; Burioni, R; Mancini, N. Hepatitis C virus (HCV)-driven stimulation of subfamily-restricted natural IgM antibodies in mixed cryoglobulinemia. Autoimmun. Rev 2008, 7, 468–472. [Google Scholar]
- Burioni, R; Perotti, M; Mancini, N; Clementi, M. Perspectives for the utilization of neutralizing human monoclonal antibodies as anti-HCV drugs. J. Hepatol 2008, 49, 299–300. [Google Scholar]
- Mancini, N; Carletti, S; Perotti, M; Romano, L; Craxi, R.D; Craxi, A; Zanetti, A.R; Clementi, M; Burioni, R. Modulation of epitope-specific anti-hepatitis C virus E2 (anti-HCV/E2) antibodies by anti-viral treatment. J. Med. Virol 2006, 78, 1304–1311. [Google Scholar]
- Mancini, N; Canducci, F; Carletti, S; Berardinelli, E; Serafini, G; Grieco, A; Perotti, M; Malcangi, G; Danieli, M.G; Varaldo, P.E; et al. Heterogeneity of the humoral anti-HCV/E2 response in persistently infected patients as demonstrated by divergent patterns of inhibition of the binding of anti-HCV/E2 human monoclonal antibodies. J. Biol. Regul. Homeost. Agents 2003, 17, 183–187. [Google Scholar]
- Burioni, R; Matsuura, Y; Mancini, N; Tani, H; Miyamura, T; Varaldo, P.E; Clementi, M. Diverging effects of human recombinant anti-hepatitis C virus (HCV) antibody fragments derived from a single patient on the infectivity of a vesicular stomatitis virus/HCV pseudotype. J. Virol 2002, 76, 11775–11779. [Google Scholar]
- Allander, T; Drakenberg, K; Beyene, A; Rosa, D; Abrignani, S; Houghton, M; Widell, A; Grillner, L; Persson, M.A. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen. Virol 2000, 81, 2451–2459. [Google Scholar]
- Johansson, D.X; Voisset, C; Tarr, A.W; Aung, M; Ball, J.K; Dubuisson, J; Persson, M.A. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc. Natl. Acad. Sci. USA 2007, 104, 16269–16274. [Google Scholar]
- Giang, E; Dorner, M; Prentoe, J.C; Dreux, M; Evans, M.J; Bukh, J; Rice, C.M; Ploss, A; Burton, D.R; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6205–6210. [Google Scholar]
- Ditzel, H.J. Rescue of a broader range of antibody specificities using an epitope-masking strategy. Methods Mol. Biol 2002, 178, 179–186. [Google Scholar]
- Tsui, P; Tornetta, M.A; Ames, R.S; Silverman, C; Porter, T; Weston, C; Griego, S; Sweet, R.W. Progressive epitope-blocked panning of a phage library for isolation of human RSV antibodies. J. Immunol. Methods 2002, 263, 123–132. [Google Scholar]
- Sblattero, D; Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat. Biotechnol 2000, 18, 75–80. [Google Scholar]
- Thie, H; Voedisch, B; Dubel, S; Hust, M; Schirrmann, T. Affinity maturation by phage display. Methods Mol. Biol 2009, 525, 309–322. [Google Scholar]
- Low, N.M; Holliger, P.H; Winter, G. Mimicking somatic hypermutation: Affinity maturation of antibodies displayed on bacteriophage using a bacterial mutator strain. J. Mol. Biol 1996, 260, 359–368. [Google Scholar]
- Irving, R.A; Kortt, A.A; Hudson, P.J. Affinity maturation of recombinant antibodies using E. coli mutator cells. Immunotechnology 1996, 2, 127–143. [Google Scholar]
- Fujii, R; Kitaoka, M; Hayashi, K. One-step random mutagenesis by error-prone rolling circle amplification. Nucleic Acids Res 2004, 32, e145. [Google Scholar]
- Martineau, P. Error-prone polymerase chain reaction for modification of scFvs. Methods Mol. Biol 2002, 178, 287–294. [Google Scholar]
- Tindall, K.R; Kunkel, T.A. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry 1988, 27, 6008–6013. [Google Scholar]
- Stott, E.J; Taylor, G; Ball, L.A; Anderson, K; Young, K.K; King, A.M; Wertz, G.W. Immune and histopathological responses in animals vaccinated with recombinant vaccinia viruses that express individual genes of human respiratory syncytial virus. J. Virol 1987, 61, 3855–3861. [Google Scholar]
- Connors, M; Collins, P.L; Firestone, C.Y; Murphy, B.R. Respiratory syncytial virus (RSV) F, G, M2 (22K), and N proteins each induce resistance to RSV challenge, but resistance induced by M2 and N proteins is relatively short-lived. J. Virol 1991, 65, 1634–1637. [Google Scholar]
- Lamb, R.A. Paramyxovirus fusion: A hypothesis for changes. Virology 1993, 197, 1–11. [Google Scholar]
- Lamb, R.A; Jardetzky, T.S. Structural basis of viral invasion: Lessons from paramyxovirus F. Curr. Opin. Struct. Biol 2007, 17, 427–436. [Google Scholar]
- Magro, M; Mas, V; Chappell, K; Vazquez, M; Cano, O; Luque, D; Terron, M.C; Melero, J.A; Palomo, C. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc. Natl. Acad. Sci. USA 2012, 109, 3089–3094. [Google Scholar]
- Beeler, J.A; van Wyke Coelingh, K. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: Effect of mutation upon fusion function. J. Virol 1989, 63, 2941–2950. [Google Scholar]
- Kabat, E.A; Wu, T.T. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J. Immunol 1991, 147, 1709–1719. [Google Scholar]
- Hieter, P.A; Maizel, J.V., Jr; Leder, P. Evolution of human immunoglobulin kappa J region genes. J. Biol. Chem 1982, 257, 1516–1522. [Google Scholar]
- Wu, H; Pfarr, D.S; Tang, Y; An, L.L; Patel, N.K; Watkins, J.D; Huse, W.D; Kiener, P.A; Young, J.F. Ultra-potent antibodies against respiratory syncytial virus: Effects of binding kinetics and binding valence on viral neutralization. J. Mol. Biol 2005, 350, 126–144. [Google Scholar]
- Johnson, S; Oliver, C; Prince, G.A; Hemming, V.G; Pfarr, D.S; Wang, S.C; Dormitzer, M; O’Grady, J; Koenig, S; Tamura, J.K; et al. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 1997, 176, 1215–1224. [Google Scholar]
- Maynard, J.A; Maassen, C.B; Leppla, S.H; Brasky, K; Patterson, J.L; Iverson, B.L; Georgiou, G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat. Biotechnol 2002, 20, 597–601. [Google Scholar]
- Little, S.F; Novak, J.M; Lowe, J.R; Leppla, S.H; Singh, Y; Klimpel, K.R; Lidgerding, B.C; Friedlander, A.M. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology 1996, 142, 707–715. [Google Scholar]
- Little, S.F; Leppla, S.H; Cora, E. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect. Immun 1988, 56, 1807–1813. [Google Scholar]
- Krebber, A; Bornhauser, S; Burmester, J; Honegger, A; Willuda, J; Bosshard, H.R; Pluckthun, A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J. Immunol. Methods 1997, 201, 35–55. [Google Scholar]
- Fromant, M; Blanquet, S; Plateau, P. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal. Biochem 1995, 224, 347–353. [Google Scholar]
- Stemmer, W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. [Google Scholar]
- Ter Meulen, J. Monoclonal antibodies in infectious diseases: Clinical pipeline in 2011. Infect. Dis. Clin. N. Am 2011, 25, 789–802. [Google Scholar]

{kind=link}
| Antibody library origin | PROs | CONs | Donor | B-cell source | PROs | CONs |
|---|---|---|---|---|---|---|
| Humans | Selection of mAbs potentially useful for human administration | Difficulties to obtain immunological reagents (i.e., immunized humans) | Vaccinated donors/convalescent patients | Peripheral blood | Sample easy to obtain | Limited library extension |
| Bone marrow | Library extension | Difficulties related to bone marrow sampling | ||||
| Animals | Useful for diagnostic tools development or research usage; Possibility to immunize with synthetic molecules | Humanization or chimerization of selected mAbs required before human administration | Infected/immunized animals | Spleen/ Peripheral blood/Bone marrow | Easy sampling | |
| Synthetic | Library in silico design; No immunization or infection and tissue sampling required; Selective pathogen targeting | Possible limitations in library extension; Possible Ab misfolding and possible drawbacks for mAb production |
| mAb Name | Origin | Ig Class or Ab Format | Molecular Target | Major Indication | Development Status |
|---|---|---|---|---|---|
| Edobacumab | Mus Musculus | IgM | Lipid A (LPS) | Septic Shock | Phase III |
| Nebacumab | Homo sapiens | IgM kappa | Lipid A (LPS) | Septic Shock | Phase I |
| Panobacumab | Homo sapiens | IgM kappa | P. aeruginosa serotype IATS O11 | Nosocomial pneumonia caused by serotype 011 positive P.aeruginosa | Phase I |
| KB001 | Human (from Mus musculus | Fab | P. aeruginosa PcrV | P. aeruginosa infection | Phase I/II |
| Felvizumab | Human (from Mus musculus ) | IgG1 | RSV Glycoprotein F | RSV infection | Phase III |
| Motavizumab (Numax®) | Human (from Mus musculus ) | IgG1 kappa | RSV Glycoprotein F | RSV infection | Phase II |
| Palivizumab (Synagis®) | Human (from Mus musculus ) | IgG1 kappa | RSV Glycoprotein F | RSV infection | Phase M |
| Sevirumab (Protovir™) | Human (from Mus musculus ) | IgG1 kappa | HCMV gB glycoprotein gH envelope glycoprotein | HCMV infection | Phase III |
| Suvizumab | Human (from Mus musculus ) | IgG1 | HIV-1 IIIB gp120 V3 loop | HIV infection | Phase I |
| Tuvirumab | Homo sapiens | chronic HBV infection | Phase II | ||
| Efungumab (Mycograb®) | Phage display Human Antibody | Human scFv | Fungal HSP90 | Fungal diseases | Phase III |
| Aurograb® | Phage display Human Antibody | Human scFv | Staph ABC transporter GrfA | MRSA, to be used with vancomycin | Phase III |
| Raxibacumab (Abthrax®) | Phage display Human Antibody | Human IgG | B. anthracis PA toxin | Anthrax biodefense | Phase III |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Clementi, N.; Mancini, N.; Solforosi, L.; Castelli, M.; Clementi, M.; Burioni, R. Phage Display-based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed against Human Pathogens. Int. J. Mol. Sci. 2012, 13, 8273-8292. https://doi.org/10.3390/ijms13078273
Clementi N, Mancini N, Solforosi L, Castelli M, Clementi M, Burioni R. Phage Display-based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed against Human Pathogens. International Journal of Molecular Sciences. 2012; 13(7):8273-8292. https://doi.org/10.3390/ijms13078273
Chicago/Turabian StyleClementi, Nicola, Nicasio Mancini, Laura Solforosi, Matteo Castelli, Massimo Clementi, and Roberto Burioni. 2012. "Phage Display-based Strategies for Cloning and Optimization of Monoclonal Antibodies Directed against Human Pathogens" International Journal of Molecular Sciences 13, no. 7: 8273-8292. https://doi.org/10.3390/ijms13078273