The Neuroprotective Functions of Transforming Growth Factor Beta Proteins

Abstract
:1. Introduction
2. TGF-β in the Intact Brain
2.1. The Distribution of TGF-βs, Their Binding Proteins and Receptors in the Brain
2.2. The Physiological Functions of TGF-βs in the Brain
2.2.1. The Role of TGF-βs in Brain Development
2.2.2. TGF-βs and the Modulation of Synaptic Transmission
2.2.3. Proposed Neuroendocrine Functions of TGF-βs
3. TGF-β in Cerebral Ischemia
3.1. The Induction of TGF-βs in Response to Brain Ischemia
3.2. Cell Types Expressing TGF-βs Following Brain Ischemia
3.3. Effect of TGF-βs on Ischemic Brain Lesion
4. TGF-β in Traumatic CNS Injury
4.1. The Induction of TGF-βs in Response to Traumatic Injury
4.2. TGF-β Actions for Traumatic Injury
5. TGF-β in Multiple Sclerosis
5.1. The Expression Level of TGF-βs in EAE and MS
5.2. The Effects of TGF-βs in EAE and MS
5.3. Genetic Studies on the Involvement of TGF-βs in MS
5.4. TGF-β Subtypes in MS
6. TGF-β in Neurodegenerative Disorders
6.1. Alzheimer’s Disease
6.1.1. TGF-β Levels in AD and Its Animal Models
6.1.2. The Role of TGF-βs in AD Progression
6.1.3. TGF-β Signaling in AD
6.2. Parkinson’s Disease
6.2.1. TGF-β Levels and Their Alterations in PD
6.2.2. The Effect of TGF-β on the Survival of Dopaminergic Neurons in the Substantia Nigra
7. TGF-β in Neurodegeneration Resulted from Brain Infections
8. The Role of TGF-βs in Brain Tumors
8.1. Proliferative Actions of TGF-βs in Brain Tumors
8.2. The TGF-β System in the Treatment of Brain Tumors
9. The Neuroprotective Mechanisms of TGF-βs
9.1. Anti-Inflammatory Action
9.2. Scar Formation
9.3. Anti-Apoptotic Property
9.4. Protection against Excitotoxicity
9.5. Angiogenesis
9.6. Neuronal Regeneration
10. Conclusions
Acknowledgment
References
- Roberts, A.B.; Anzano, M.A.; Lamb, L.C.; Smith, J.M.; Sporn, M.B. New class of transforming growth factors potentiated by epidermal growth factor: Isolation from non-neoplastic tissues. Proc. Natl. Acad. Sci. USA 1981, 78, 5339–5343. [Google Scholar]
- Burt, D.W.; Law, A.S. Evolution of the transforming growth factor-beta superfamily. Prog. Growth Factor Res 1994, 5, 99–118. [Google Scholar]
- Lawrence, D.A. Transforming growth factor-beta: A general review. Eur. Cytokine Netw 1996, 7, 363–374. [Google Scholar]
- Clark, D.A.; Coker, R. Transforming growth factor-beta (TGF-beta). Int. J. Biochem. Cell. Biol 1998, 30, 293–298. [Google Scholar]
- Funkenstein, B.; Olekh, E.; Jakowlew, S.B. Identification of a novel transforming growth factor-beta (TGF-beta6) gene in fish: Regulation in skeletal muscle by nutritional state. BMC Mol. Biol 2010, 11, 37. [Google Scholar]
- Roberts, A.B. Molecular and cell biology of TGF-beta. Miner. Electrolyte. Metab 1998, 24, 111–119. [Google Scholar]
- Sinha, S.; Nevett, C.; Shuttleworth, C.A.; Kielty, C.M. Cellular and extracellular biology of the latent transforming growth factor-beta binding proteins. Matrix. Biol 1998, 17, 529–545. [Google Scholar]
- Oklu, R.; Hesketh, R. The latent transforming growth factor beta binding protein (LTBP) family. Biochem. J 2000, 352, 601–610. [Google Scholar]
- Khalil, N. TGF-beta: From latent to active. Microbes. Infect 1999, 1, 1255–1263. [Google Scholar]
- Konig, H.G.; Kogel, D.; Rami, A.; Prehn, J.H. TGF-β1 activates two distinct type I receptors in neurons: Implications for neuronal NF-κB signaling. J. Cell. Biol 2005, 168, 1077–1086. [Google Scholar]
- Gumienny, T.L.; Padgett, R.W. The other side of TGF-beta superfamily signal regulation: Thinking outside the cell. Trends Endocrinol. Metab 2002, 13, 295–299. [Google Scholar]
- Ten Dijke, P.; Hill, C.S. New insights into TGF-beta-Smad signalling. Trends Biochem. Sci 2004, 29, 265–273. [Google Scholar]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol 2007, 8, 970–982. [Google Scholar]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res 2012, 347, 11–20. [Google Scholar]
- Unsicker, K.; Flanders, K.C.; Cissel, D.S.; Lafyatis, R.; Sporn, M.B. Transforming growth factor beta isoforms in the adult rat central and peripheral nervous system. Neuroscience 1991, 44, 613–625. [Google Scholar]
- Vincze, C.; Pal, G.; Wappler, E.A.; Szabo, E.R.; Nagy, Z.G.; Lovas, G.; Dobolyi, A. Distribution of mRNAs encoding transforming growth factors-beta1, -2, and -3 in the intact rat brain and after experimentally induced focal ischemia. J. Comp. Neurol 2010, 518, 3752–3770. [Google Scholar]
- Komuta, Y.; Teng, X.; Yanagisawa, H.; Sango, K.; Kawamura, K.; Kawano, H. Expression of transforming growth factor-beta receptors in meningeal fibroblasts of the injured mouse brain. Cell. Mol. Neurobiol 2010, 30, 101–111. [Google Scholar]
- Bottner, M.; Krieglstein, K.; Unsicker, K. The transforming growth factor-betas: Structure, signaling, and roles in nervous system development and functions. J. Neurochem 2000, 75, 2227–2240. [Google Scholar]
- Dobolyi, A.; Palkovits, M. Expression of latent transforming growth factor beta binding proteins in the rat brain. J. Comp. Neurol 2008, 507, 1393–1408. [Google Scholar]
- Dobolyi, A. Transforming Growth Factor Beta in the Central Nervous System. In Neuroscience —Dealing with Frontiers; Contreras, C.M., Ed.; In Tech: Rijeka, Croatia, 2012; pp. 129–148. [Google Scholar]
- Bottner, M.; Unsicker, K.; Suter-Crazzolara, C. Expression of TGF-beta type II receptor mRNA in the CNS. Neuroreport 1996, 7, 2903–2907. [Google Scholar]
- Flanders, K.C.; Ludecke, G.; Engels, S.; Cissel, D.S.; Roberts, A.B.; Kondaiah, P.; Lafyatis, R.; Sporn, M.B.; Unsicker, K. Localization and actions of transforming growth factor-beta s in the embryonic nervous system. Development 1991, 113, 183–191. [Google Scholar]
- Zhang, J.M.; Hoffmann, R.; Sieber-Blum, M. Mitogenic and anti-proliferative signals for neural crest cells and the neurogenic action of TGF-beta1. Dev. Dyn 1997, 208, 375–386. [Google Scholar]
- Aigner, L.; Bogdahn, U. TGF-beta in neural stem cells and in tumors of the central nervous system. Cell Tissue Res 2008, 331, 225–241. [Google Scholar]
- Vogel, T.; Ahrens, S.; Buttner, N.; Krieglstein, K. Transforming growth factor beta promotes neuronal cell fate of mouse cortical and hippocampal progenitors in vitro and in vivo: Identification of Nedd9 as an essential signaling component. Cereb. Cortex 2010, 20, 661–671. [Google Scholar]
- Mathieu, P.; Piantanida, A.P.; Pitossi, F. Chronic expression of transforming growth factor-beta enhances adult neurogenesis. Neuroimmunomodulation 2011, 17, 200–201. [Google Scholar]
- Battista, D.; Ferrari, C.C.; Gage, F.H.; Pitossi, F.J. Neurogenic niche modulation by activated microglia: Transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci 2006, 23, 83–93. [Google Scholar]
- Garcia-Campmany, L.; Marti, E. The TGFbeta intracellular effector Smad3 regulates neuronal differentiation and cell fate specification in the developing spinal cord. Development 2007, 134, 65–75. [Google Scholar]
- Gouin, A.; Bloch-Gallego, E.; Tanaka, H.; Rosenthal, A.; Henderson, C.E. Transforming growth factor-beta 3, glial cell line-derived neurotrophic factor, and fibroblast growth factor-2, act in different manners to promote motoneuron survival in vitro. J. Neurosci. Res 1996, 43, 454–464. [Google Scholar]
- Jiang, Y.; McLennan, I.S.; Koishi, K.; Hendry, I.A. Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience 2000, 97, 735–742. [Google Scholar]
- Jiang, Y.; Zhang, M.; Koishi, K.; McLennan, I.S. TGF-beta 2 attenuates the injury-induced death of mature motoneurons. J. Neurosci. Res 2000, 62, 809–813. [Google Scholar]
- Guenard, V.; Rosenbaum, T.; Gwynn, L.A.; Doetschman, T.; Ratner, N.; Wood, P.M. Effect of transforming growth factor-beta 1 and -beta 2 on Schwann cell proliferation on neurites. Glia 1995, 13, 309–318. [Google Scholar]
- Parkinson, D.B.; Dong, Z.; Bunting, H.; Whitfield, J.; Meier, C.; Marie, H.; Mirsky, R.; Jessen, K.R. Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: Examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J. Neurosci 2001, 21, 8572–8585. [Google Scholar]
- Feng, Z.; Ko, C.P. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J. Neurosci 2008, 28, 9599–9609. [Google Scholar]
- Heupel, K.; Sargsyan, V.; Plomp, J.J.; Rickmann, M.; Varoqueaux, F.; Zhang, W.; Krieglstein, K. Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural. Dev 2008, 3. [Google Scholar] [CrossRef]
- Fukushima, T.; Liu, R.Y.; Byrne, J.H. Transforming growth factor-beta2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 2007, 17, 5–9. [Google Scholar]
- Prevot, V.; Bouret, S.; Croix, D.; Takumi, T.; Jennes, L.; Mitchell, V.; Beauvillain, J.C. Evidence that members of the TGFbeta superfamily play a role in regulation of the GnRH neuroendocrine axis: Expression of a type I serine-threonine kinase receptor for TGRbeta and activin in GnRH neurones and hypothalamic areas of the female rat. J. Neuroendocrinol 2000, 12, 665–670. [Google Scholar]
- Bouret, S.; de Seranno, S.; Beauvillain, J.C.; Prevot, V. Transforming growth factor beta1 may directly influence gonadotropin-releasing hormone gene expression in the rat hypothalamus. Endocrinology 2004, 145, 1794–1801. [Google Scholar]
- Dhandapani, K.M.; Hadman, M.; de Sevilla, L.; Wade, M.F.; Mahesh, V.B.; Brann, D.W. Astrocyte protection of neurons: Role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway. J. Biol. Chem 2003, 278, 43329–43339. [Google Scholar]
- Sortino, M.A.; Chisari, M.; Merlo, S.; Vancheri, C.; Caruso, M.; Nicoletti, F.; Canonico, P.L.; Copani, A. Glia mediates the neuroprotective action of estradiol on beta-amyloid-induced neuronal death. Endocrinology 2004, 145, 5080–5086. [Google Scholar]
- Fevre-Montange, M.; Dumontel, C.; Chevallier, P.; Isnard, A.K.; Guigard, M.P.; Trouillas, J. Localization of transforming growth factors, TGFbeta1 and TGFbeta3, in hypothalamic magnocellular neurones and the neurohypophysis. J. Neuroendocrinol 2004, 16, 571–576. [Google Scholar]
- Beynon, A.L.; Thome, J.; Coogan, A.N. Age and time of day influences on the expression of transforming growth factor-beta and phosphorylated SMAD3 in the mouse suprachiasmatic and paraventricular nuclei. Neuroimmunomodulation 2009, 16, 392–399. [Google Scholar]
- Giacomini, D.; Paez-Pereda, M.; Stalla, J.; Stalla, G.K.; Arzt, E. Molecular interaction of BMP-4, TGF-beta, and estrogens in lactotrophs: Impact on the PRL promoter. Mol. Endocrinol 2009, 23, 1102–1114. [Google Scholar]
- Klempt, N.D.; Sirimanne, E.; Gunn, A.J.; Klempt, M.; Singh, K.; Williams, C.; Gluckman, P.D. Hypoxia-ischemia induces transforming growth factor beta 1 mRNA in the infant rat brain. Brain Res. Mol. Brain Res 1992, 13, 93–101. [Google Scholar]
- Lehrmann, E.; Kiefer, R.; Finsen, B.; Diemer, N.H.; Zimmer, J.; Hartung, H.P. Cytokines in cerebral ischemia: Expression of transforming growth factor beta-1 (TGF-beta 1) mRNA in the postischemic adult rat hippocampus. Exp. Neurol 1995, 131, 114–123. [Google Scholar]
- Knuckey, N.W.; Finch, P.; Palm, D.E.; Primiano, M.J.; Johanson, C.E.; Flanders, K.C.; Thompson, N.L. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res. Mol. Brain Res 1996, 40, 1–14. [Google Scholar]
- Ata, K.A.; Lennmyr, F.; Funa, K.; Olsson, Y.; Terent, A. Expression of transforming growth factor-beta1, 2, 3 isoforms and type I and II receptors in acute focal cerebral ischemia: An immunohistochemical study in rat after transient and permanent occlusion of middle cerebral artery. Acta Neuropathol 1999, 97, 447–455. [Google Scholar]
- Zhu, Y.; Culmsee, C.; Roth-Eichhorn, S.; Krieglstein, J. Beta(2)-adrenoceptor stimulation enhances latent transforming growth factor-beta-binding protein-1 and transforming growth factor-beta1 expression in rat hippocampus after transient forebrain ischemia. Neuroscience 2001, 107, 593–602. [Google Scholar]
- Ali, C.; Docagne, F.; Nicole, O.; Lesne, S.; Toutain, J.; Young, A.; Chazalviel, L.; Divoux, D.; Caly, M.; Cabal, P.; et al. Increased expression of transforming growth factor-beta after cerebral ischemia in the baboon: An endogenous marker of neuronal stress? J. Cereb. Blood Flow Metab 2001, 21, 820–827. [Google Scholar]
- Doyle, K.P.; Cekanaviciute, E.; Mamer, L.E.; Buckwalter, M.S. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J. Neuroinflammation 2010, 7. [Google Scholar] [CrossRef]
- Krupinski, J.; Kumar, P.; Kumar, S.; Kaluza, J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke 1996, 27, 852–857. [Google Scholar]
- Wu, Z.; Hayashi, Y.; Zhang, J.; Nakanishi, H. Involvement of prostaglandin E2 released from leptomeningeal cells in increased expression of transforming growth factor-beta in glial cells and cortical neurons during systemic inflammation. J. Neurosci. Res 2007, 85, 184–192. [Google Scholar]
- Wu, Z.; Tokuda, Y.; Zhang, X.W.; Nakanishi, H. Age-dependent responses of glial cells and leptomeninges during systemic inflammation. Neurobiol. Dis 2008, 32, 543–551. [Google Scholar]
- Krohn, K. TGF-beta1-dependent differential expression of a rat homolog for latent TGF-beta binding protein in astrocytes and C6 glioma cells. Glia 1999, 25, 332–342. [Google Scholar]
- Flanders, K.C.; Ren, R.F.; Lippa, C.F. Transforming growth factor-betas in neurodegenerative disease. Prog. Neurobiol 1998, 54, 71–85. [Google Scholar]
- Morgan, T.E.; Nichols, N.R.; Pasinetti, G.M.; Finch, C.E. TGF-beta 1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp. Neurol 1993, 120, 291–301. [Google Scholar]
- Dhandapani, K.M.; Brann, D.W. Transforming growth factor-beta: A neuroprotective factor in cerebral ischemia. Cell Biochem. Biophys 2003, 39, 13–22. [Google Scholar]
- Battaglia, G.; Cannella, M.; Riozzi, B.; Orobello, S.; Maat-Schieman, M.L.; Aronica, E.; Busceti, C.L.; Ciarmiello, A.; Alberti, S.; Amico, E.; et al. Early defect of transforming growth factor beta1 formation in Huntington's disease. J. Cell. Mol. Med 2011, 15, 555–571. [Google Scholar]
- Lacmann, A.; Hess, D.; Gohla, G.; Roussa, E.; Krieglstein, K. Activity-dependent release of transforming growth factor-beta in a neuronal network in vitro. Neuroscience 2007, 150, 647–657. [Google Scholar]
- Lehrmann, E.; Kiefer, R.; Christensen, T.; Toyka, K.V.; Zimmer, J.; Diemer, N.H.; Hartung, H.P.; Finsen, B. Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia 1998, 24, 437–448. [Google Scholar]
- Gross, C.E.; Bednar, M.M.; Howard, D.B.; Sporn, M.B. Transforming growth factor-beta 1 reduces infarct size after experimental cerebral ischemia in a rabbit model. Stroke 1993, 24, 558–562. [Google Scholar]
- Henrich-Noack, P.; Prehn, J.H.; Krieglstein, J. Neuroprotective effects of TGF-beta 1. J. Neural. Transm. Suppl 1994, 43, 33–45. [Google Scholar]
- McNeill, H.; Williams, C.; Guan, J.; Dragunow, M.; Lawlor, P.; Sirimanne, E.; Nikolics, K.; Gluckman, P. Neuronal rescue with transforming growth factor-beta 1 after hypoxic-ischaemic brain injury. Neuroreport 1994, 5, 901–904. [Google Scholar]
- Pang, L.; Ye, W.; Che, X.M.; Roessler, B.J.; Betz, A.L.; Yang, G.Y. Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke 2001, 32, 544–552. [Google Scholar]
- Ma, M.; Ma, Y.; Yi, X.; Guo, R.; Zhu, W.; Fan, X.; Xu, G.; Frey, W.H., II; Liu, X. Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci 2008, 9, 117. [Google Scholar]
- Ruocco, A.; Nicole, O.; Docagne, F.; Ali, C.; Chazalviel, L.; Komesli, S.; Yablonsky, F.; Roussel, S.; MacKenzie, E.T.; Vivien, D.; Buisson, A. A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J. Cereb. Blood Flow Metab 1999, 19, 1345–1353. [Google Scholar]
- Boche, D.; Cunningham, C.; Gauldie, J.; Perry, V.H. Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J. Cereb. Blood Flow Metab 2003, 23, 1174–1182. [Google Scholar]
- Pera, J.; Zawadzka, M.; Kaminska, B.; Szczudlik, A. Influence of chemical and ischemic preconditioning on cytokine expression after focal brain ischemia. J. Neurosci. Res 2004, 78, 132–140. [Google Scholar]
- Lenzlinger, P.M.; Morganti-Kossmann, M.C.; Laurer, H.L.; McIntosh, T.K. The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol 2001, 24, 169–181. [Google Scholar]
- Helmy, A.; Carpenter, K.L.; Menon, D.K.; Pickard, J.D.; Hutchinson, P.J. The cytokine response to human traumatic brain injury: Temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab 2011, 31, 658–670. [Google Scholar]
- Lindholm, D.; Castren, E.; Kiefer, R.; Zafra, F.; Thoenen, H. Transforming growth factor-beta 1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. J. Cell Biol 1992, 117, 395–400. [Google Scholar]
- Wang, X.; Chen, W.; Liu, W.; Wu, J.; Shao, Y.; Zhang, X. The role of thrombospondin-1 and transforming growth factor-beta after spinal cord injury in the rat. J. Clin. Neurosci 2009, 16, 818–821. [Google Scholar]
- Rimaniol, A.C.; Lekieffre, D.; Serrano, A.; Masson, A.; Benavides, J.; Zavala, F. Biphasic transforming growth factor-beta production flanking the pro-inflammatory cytokine response in cerebral trauma. Neuroreport 1995, 7, 133–136. [Google Scholar]
- Buss, A.; Pech, K.; Kakulas, B.A.; Martin, D.; Schoenen, J.; Noth, J.; Brook, G.A. TGF-beta 1 and TGF-beta 2 expression after traumatic human spinal cord injury. Spinal Cord 2008, 46, 364–371. [Google Scholar]
- Wyss-Coray, T.; Feng, L.; Masliah, E.; Ruppe, M.D.; Lee, H.S.; Toggas, S.M.; Rockenstein, E.M.; Mucke, L. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-β1. Am. J. Pathol 1995, 147, 53–67. [Google Scholar]
- Kawamoto, K.; Yagi, M.; Stover, T.; Kanzaki, S.; Raphael, Y. Hearing and hair cells are protected by adenoviral gene therapy with TGF-beta 1 and GDNF. Mol. Ther 2003, 7, 484–492. [Google Scholar]
- Hauser, S.L.; Oksenberg, J.R. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron 2006, 52, 61–76. [Google Scholar]
- Voumvourakis, K.I.; Antonelou, R.; Kitsos, D.K.; Stamboulis, E.; Tsiodras, S. TGF-beta/BMPs: Crucial crossroad in neural autoimmune disorders. Neurochem. Int 2011, 59, 542–550. [Google Scholar]
- Mirshafiey, A.; Mohsenzadegan, M. TGF-beta as a promising option in the treatment of multiple sclerosis. Neuropharmacology 2009, 56, 929–936. [Google Scholar]
- Mix, E.; Meyer-Rienecker, H.; Hartung, H.P.; Zettl, U.K. Animal models of multiple sclerosis—Potentials and limitations. Prog. Neurobiol 2010, 92, 386–404. [Google Scholar]
- Luo, J.; Ho, P.P.; Buckwalter, M.S.; Hsu, T.; Lee, L.Y.; Zhang, H.; Kim, D.K.; Kim, S.J.; Gambhir, S.S.; Steinman, L.; Wyss-Coray, T. Glia-dependent TGF-beta signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J. Clin. Invest 2007, 117, 3306–3315. [Google Scholar]
- Kiefer, R.; Schweitzer, T.; Jung, S.; Toyka, K.V.; Hartung, H.P. Sequential expression of transforming growth factor-beta1 by T-cells, macrophages, and microglia in rat spinal cord during autoimmune inflammation. J. Neuropathol. Exp. Neurol 1998, 57, 385–395. [Google Scholar]
- Beck, J.; Rondot, P.; Jullien, P.; Wietzerbin, J.; Lawrence, D.A. TGF-beta-like activity produced during regression of exacerbations in multiple sclerosis. Acta Neurol. Scand 1991, 84, 452–455. [Google Scholar]
- Soderstrom, M.; Hillert, J.; Link, J.; Navikas, V.; Fredrikson, S.; Link, H. Expression of IFN-gamma, IL-4, and TGF-beta in multiple sclerosis in relation to HLA-Dw2 phenotype and stage of disease. Mult. Scler 1995, 1, 173–180. [Google Scholar]
- Link, J.; Soderstrom, M.; Olsson, T.; Hojeberg, B.; Ljungdahl, A.; Link, H. Increased transforming growth factor-beta, interleukin-4, and interferon-gamma in multiple sclerosis. Ann. Neurol 1994, 36, 379–386. [Google Scholar]
- Rollnik, J.D.; Sindern, E.; Schweppe, C.; Malin, J.P. Biologically active TGF-beta 1 is increased in cerebrospinal fluid while it is reduced in serum in multiple sclerosis patients. Acta Neurol. Scand 1997, 96, 101–105. [Google Scholar]
- Meoli, E.M.; Oh, U.; Grant, C.W.; Jacobson, S. TGF-beta signaling is altered in the peripheral blood of subjects with multiple sclerosis. J. Neuroimmunol 2011, 230, 164–168. [Google Scholar]
- Olsson, T. Multiple sclerosis:cerebrospinal fluid. Ann. Neurol 1994, 36, S100–S102. [Google Scholar]
- Carrieri, P.B.; Provitera, V.; Bruno, R.; Perrella, M.; Tartaglia, G.; Busto, A.; Perrella, O. Possible role of transforming growth factor-beta in relapsing-remitting multiple sclerosis. Neurol. Res 1997, 19, 599–600. [Google Scholar]
- De Groot, C.J.; Montagne, L.; Barten, A.D.; Sminia, P.; van der Valk, P. Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J. Neuropathol. Exp. Neurol 1999, 58, 174–187. [Google Scholar]
- Kuruvilla, A.P.; Shah, R.; Hochwald, G.M.; Liggitt, H.D.; Palladino, M.A.; Thorbecke, G.J. Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 2918–2921. [Google Scholar]
- Link, J.; He, B.; Navikas, V.; Palasik, W.; Fredrikson, S.; Soderstrom, M.; Link, H. Transforming growth factor-beta 1 suppresses autoantigen-induced expression of pro-inflammatory cytokines but not of interleukin-10 in multiple sclerosis and myasthenia gravis. J. Neuroimmunol 1995, 58, 21–35. [Google Scholar]
- Diemel, L.T.; Jackson, S.J.; Cuzner, M.L. Role for TGF-beta1, FGF-2 and PDGF-AA in a myelination of CNS aggregate cultures enriched with macrophages. J. Neurosci. Res 2003, 74, 858–867. [Google Scholar]
- Ishikawa, M.; Jin, Y.; Guo, H.; Link, H.; Xiao, B.G. Nasal administration of transforming growth factor-beta1 induces dendritic cells and inhibits protracted-relapsing experimental allergic encephalomyelitis. Mult. Scler 1999, 5, 184–191. [Google Scholar]
- Jin, Y.X.; Xu, L.Y.; Guo, H.; Ishikawa, M.; Link, H.; Xiao, B.G. TGF-beta1 inhibits protracted-relapsing experimental autoimmune encephalomyelitis by activating dendritic cells. J. Autoimmun 2000, 14, 213–220. [Google Scholar]
- Weinberg, A.D.; Whitham, R.; Swain, S.L.; Morrison, W.J.; Wyrick, G.; Hoy, C.; Vandenbark, A.A.; Offner, H. Transforming growth factor-beta enhances the in vivo effector function and memory phenotype of antigen-specific T helper cells in experimental autoimmune encephalomyelitis. J. Immunol 1992, 148, 2109–2117. [Google Scholar]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med 2002, 8, 1115–1121. [Google Scholar]
- Zhang, Y.; Zhang, J.; Navrazhina, K.; Argaw, A.T.; Zameer, A.; Gurfein, B.T.; Brosnan, C.F.; John, G.R. TGFbeta1 induces Jagged1 expression in astrocytes via ALK5 and Smad3 and regulates the balance between oligodendrocyte progenitor proliferation and differentiation. Glia 2010, 58, 964–974. [Google Scholar]
- Green, A.J.; Barcellos, L.F.; Rimmler, J.B.; Garcia, M.E.; Caillier, S.; Lincoln, R.R.; Bucher, P.; Pericak-Vance, M.A.; Haines, J.L.; Hauser, S.L.; Oksenberg, J.R. Sequence variation in the transforming growth factor-beta1 (TGFB1) gene and multiple sclerosis susceptibility. J. Neuroimmunol 2001, 116, 116–124. [Google Scholar]
- Weinshenker, B.G.; Hebrink, D.; Kantarci, O.H.; Schaefer-Klein, J.; Atkinson, E.; Schaid, D.; McMurray, C.M. Genetic variation in the transforming growth factor beta1 gene in multiple sclerosis. J. Neuroimmunol 2001, 120, 138–145. [Google Scholar]
- Izad, M.; Vodjgani, M.; Niknam, M.H.; Amirzargar, A.; Shahbeigi, S.; Heidari, A.B.; Keramatipour, M. Cytokines genes polymorphisms and risk of multiple sclerosis. Am. J. Med. Sci 2010, 339, 327–331. [Google Scholar]
- Peress, N.S.; Perillo, E.; Seidman, R.J. Glial transforming growth factor (TGF)-beta isotypes in multiple sclerosis: Differential glial expression of TGF-beta 1, 2 and 3 isotypes in multiple sclerosis. J. Neuroimmunol 1996, 71, 115–123. [Google Scholar]
- Goris, A.; Williams-Gray, C.H.; Foltynie, T.; Brown, J.; Maranian, M.; Walton, A.; Compston, D.A.; Barker, R.A.; Sawcer, S.J. Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson’s disease. J. Neurol 2007, 254, 846–848. [Google Scholar]
- Keidel, M.; Stude, P. Brain Lesions. In Encyclopedia of the Human Brain; Ramachandran, V.S., Ed.; Academic Press: New York, NY, USA, 2002; Volume 1, pp. 529–544. [Google Scholar]
- Peskind, E.R. Neurobiology of Alzheimer’s disease. J. Clin. Psychiatry 1996, 57, 5–8. [Google Scholar]
- Caraci, F.; Battaglia, G.; Bruno, V.; Bosco, P.; Carbonaro, V.; Giuffrida, M.L.; Drago, F.; Sortino, M.A.; Nicoletti, F.; Copani, A. TGF-beta1 pathway as a new target for neuroprotection in Alzheimer’s disease. CNS Neurosci. Ther 2011, 17, 237–249. [Google Scholar]
- Wirths, O.; Breyhan, H.; Marcello, A.; Cotel, M.C.; Bruck, W.; Bayer, T.A. Inflammatory changes are tightly associated with neurodegeneration in the brain and spinal cord of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 747–757. [Google Scholar]
- Lippa, C.F.; Smith, T.W.; Flanders, K.C. Transforming growth factor-beta: Neuronal and glial expression in CNS degenerative diseases. Neurodegeneration 1995, 4, 425–432. [Google Scholar]
- Morimoto, K.; Horio, J.; Satoh, H.; Sue, L.; Beach, T.; Arita, S.; Tooyama, I.; Konishi, Y. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients compared to the brains of non-demented patients with and without increasing AD pathology. J. Alzheimers Dis 2011, 25, 59–76. [Google Scholar]
- Lifshitz, V.; Weiss, R.; Benromano, T.; Kfir, E.; Blumenfeld-Katzir, T.; Tempel-Brami, C.; Assaf, Y.; Xia, W.; Wyss-Coray, T.; Weiner, H.L.; Frenkel, D. Immunotherapy of cerebrovascular amyloidosis in a transgenic mouse model. Neurobiol. Aging 2012, 33, 432.e1–432.e13. [Google Scholar]
- Uribe-San Martin, R.; Herrera-Molina, R.; Olavarria, L.; Ramirez, G.; von Bernhardi, R. Reduction of beta-amyloid-induced neurotoxicity on hippocampal cell cultures by moderate acidosis is mediated by transforming growth factor beta. Neuroscience 2009, 158, 1338–1347. [Google Scholar]
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Invest 2006, 116, 3060–3069. [Google Scholar]
- Rezai-Zadeh, K.; Gate, D.; Gowing, G.; Town, T. How to get from here to there: Macrophage recruitment in Alzheimer’s disease. Curr. Alzheimer. Res 2011, 8, 156–163. [Google Scholar]
- El Khoury, J.; Luster, A.D. Mechanisms of microglia accumulation in Alzheimer’s disease: Therapeutic implications. Trends Pharmacol. Sci 2008, 29, 626–632. [Google Scholar]
- Weiss, R.; Lifshitz, V.; Frenkel, D. TGF-beta1 affects endothelial cell interaction with macrophages and T cells leading to the development of cerebrovascular amyloidosis. Brain Behav. Immun 2011, 25, 1017–1024. [Google Scholar]
- Wyss-Coray, T.; Lin, C.; von Euw, D.; Masliah, E.; Mucke, L.; Lacombe, P. Alzheimer’s disease-like cerebrovascular pathology in transforming growth factor-beta 1 transgenic mice and functional metabolic correlates. Ann. N. Y. Acad. Sci 2000, 903, 317–323. [Google Scholar]
- Nicolakakis, N.; Aboulkassim, T.; Aliaga, A.; Tong, X.K.; Rosa-Neto, P.; Hamel, E. Intact memory in TGF-beta1 transgenic mice featuring chronic cerebrovascular deficit: Recovery with pioglitazone. J. Cereb. Blood Flow Metab 2011, 31, 200–211. [Google Scholar]
- Ongali, B.; Nicolakakis, N.; Lecrux, C.; Aboulkassim, T.; Rosa-Neto, P.; Papadopoulos, P.; Tong, X.K.; Hamel, E. Transgenic mice overexpressing APP and transforming growth factor-beta1 feature cognitive and vascular hallmarks of Alzheimer's disease. Am. J. Pathol 2011, 177, 3071–3080. [Google Scholar]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med 2001, 7, 612–618. [Google Scholar]
- Caraci, F.; Bosco, P.; Signorelli, M.; Spada, R.S.; Cosentino, F.I.; Toscano, G.; Bonforte, C.; Muratore, S.; Prestianni, G.; Panerai, S.; et al. The CC genotype of transforming growth factor-beta1 increases the risk of late-onset Alzheimer’s disease and is associated with AD-related depression. Eur. Neuropsychopharmacol 2012, 22, 281–289. [Google Scholar]
- Haas, A.; Liraz, O.; Michaelson, D.M. The Effects of Apolipoproteins E3 and E4 on the transforming growth factor-beta system in targeted replacement Mice. Neurodegener. Dis 2012, 10, 41–45. [Google Scholar]
- Lee, H.G.; Ueda, M.; Zhu, X.; Perry, G.; Smith, M.A. Ectopic expression of phospho-Smad2 in Alzheimer’s disease: Uncoupling of the transforming growth factor-beta pathway? J. Neurosci. Res 2006, 84, 1856–1861. [Google Scholar]
- Ueberham, U.; Ueberham, E.; Gruschka, H.; Arendt, T. Altered subcellular location of phosphorylated Smads in Alzheimer’s disease. Eur. J. Neurosci 2006, 24, 2327–2334. [Google Scholar]
- Ueberham, U.; Hilbrich, I.; Ueberham, E.; Rohn, S.; Glockner, P.; Dietrich, K.; Bruckner, M.K.; Arendt, T. Transcriptional control of cell cycle-dependent kinase 4 by Smad proteins-implications for Alzheimer’s disease. Neurobiol. Aging 2012, (in press). [Google Scholar]
- Caraci, F.; Spampinato, S.; Sortino, M.A.; Bosco, P.; Battaglia, G.; Bruno, V.; Drago, F.; Nicoletti, F.; Copani, A. Dysfunction of TGF-beta1 signaling in Alzheimer’s disease: Perspectives for neuroprotection. Cell Tissue Res 2012, 347, 291–301. [Google Scholar]
- Harris, M.K.; Shneyder, N.; Borazanci, A.; Korniychuk, E.; Kelley, R.E.; Minagar, A. Movement disorders. Med. Clin. North Am 2009, 93, 371–388. [Google Scholar]
- Krieglstein, K.; Unsicker, K. Transforming growth factor-beta promotes survival of midbrain dopaminergic neurons and protects them against N-methyl-4-phenylpyridinium ion toxicity. Neuroscience 1994, 63, 1189–1196. [Google Scholar]
- Schober, A.; Peterziel, H.; von Bartheld, C.S.; Simon, H.; Krieglstein, K.; Unsicker, K. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-beta for its neuroprotective action. Neurobiol. Dis 2007, 25, 378–391. [Google Scholar]
- Krieglstein, K.; Suter-Crazzolara, C.; Fischer, W.H.; Unsicker, K. TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. Embo. J 1995, 14, 736–742. [Google Scholar]
- Krieglstein, K.; Suter-Crazzolara, C.; Unsicker, K. Development of mesencephalic dopaminergic neurons and the transforming growth factor-beta superfamily. J. Neural. Transm. Suppl 1995, 46, 209–216. [Google Scholar]
- Farkas, L.M.; Dunker, N.; Roussa, E.; Unsicker, K.; Krieglstein, K. Transforming growth factor-beta(s) are essential for the development of midbrain dopaminergic neurons in vitro and in vivo. J. Neurosci 2003, 23, 5178–5186. [Google Scholar]
- Krieglstein, K.; Henheik, P.; Farkas, L.; Jaszai, J.; Galter, D.; Krohn, K.; Unsicker, K. Glial cell line-derived neurotrophic factor requires transforming growth factor-beta for exerting its full neurotrophic potential on peripheral and CNS neurons. J. Neurosci 1998, 18, 9822–9834. [Google Scholar]
- Poulsen, K.T.; Armanini, M.P.; Klein, R.D.; Hynes, M.A.; Phillips, H.S.; Rosenthal, A. TGF beta 2 and TGF beta 3 are potent survival factors for midbrain dopaminergic neurons. Neuron 1994, 13, 1245–1252. [Google Scholar]
- Roussa, E.; Oehlke, O.; Rahhal, B.; Heermann, S.; Heidrich, S.; Wiehle, M.; Krieglstein, K. Transforming growth factor beta cooperates with persephin for dopaminergic phenotype induction. Stem Cells 2008, 26, 1683–1694. [Google Scholar]
- Roussa, E.; von Bohlen und Halbach, O.; Krieglstein, K. TGF-beta in dopamine neuron development, maintenance and neuroprotection. Adv. Exp. Med. Biol 2009, 651, 81–90. [Google Scholar]
- Zhang, J.; Pho, V.; Bonasera, S.J.; Holtzman, J.; Tang, A.T.; Hellmuth, J.; Tang, S.; Janak, P.H.; Tecott, L.H.; Huang, E.J. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat. Neurosci 2007, 10, 77–86. [Google Scholar]
- Polazzi, E.; Altamira, L.E.; Eleuteri, S.; Barbaro, R.; Casadio, C.; Contestabile, A.; Monti, B. Neuroprotection of microglial conditioned medium on 6-hydroxydopamine-induced neuronal death: Role of transforming growth factor beta-2. J. Neurochem 2009, 110, 545–556. [Google Scholar]
- Rahhal, B.; Heermann, S.; Ferdinand, A.; Rosenbusch, J.; Rickmann, M.; Krieglstein, K. In vivo requirement of TGF-beta/GDNF cooperativity in mouse development: Focus on the neurotrophic hypothesis. Int. J. Dev. Neurosci 2009, 27, 97–102. [Google Scholar]
- Tapia-Gonzalez, S.; Giraldez-Perez, R.M.; Cuartero, M.I.; Casarejos, M.J.; Mena, M.A.; Wang, X.F.; Sanchez-Capelo, A. Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol. Neurodegener 2011, 6, 72. [Google Scholar]
- Brundin, P.; Karlsson, J.; Emgard, M.; Schierle, G.S.; Hansson, O.; Petersen, A.; Castilho, R.F. Improving the survival of grafted dopaminergic neurons: A review over current approaches. Cell Transplant 2000, 9, 179–195. [Google Scholar]
- Mantel, P.Y.; Schmidt-Weber, C.B. Transforming growth factor-beta: Recent advances on its role in immune tolerance. Methods Mol. Biol 2011, 677, 303–338. [Google Scholar]
- Nishino, Y.; Ooishi, R.; Kurokawa, S.; Fujino, K.; Murakami, M.; Madarame, H.; Hashimoto, O.; Sugiyama, K.; Funaba, M. Gene expression of the TGF-beta family in rat brain infected with Borna disease virus. Microbes Infect 2009, 11, 737–743. [Google Scholar]
- Stitz, L.; Planz, O.; Bilzer, T.; Frei, K.; Fontana, A. Transforming growth factor-beta modulates T cell-mediated encephalitis caused by Borna disease virus. Pathogenic importance of CD8+ cells and suppression of antibody formation. J. Immunol 1991, 147, 3581–3586. [Google Scholar]
- Baloul, L.; Lafon, M. Apoptosis and rabies virus neuroinvasion. Biochimie 2003, 85, 777–788. [Google Scholar]
- Kossmann, T.; Morganti-Kossmann, M.C.; Orenstein, J.M.; Britt, W.J.; Wahl, S.M.; Smith, P.D. Cytomegalovirus production by infected astrocytes correlates with transforming growth factor-beta release. J. Infect. Dis 2003, 187, 534–541. [Google Scholar]
- Enam, S.; Sweet, T.M.; Amini, S.; Khalili, K.; del Valle, L. Evidence for involvement of transforming growth factor beta1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch. Pathol. Lab. Med 2004, 128, 282–291. [Google Scholar]
- Cupp, C.; Taylor, J.P.; Khalili, K.; Amini, S. Evidence for stimulation of the transforming growth factor beta 1 promoter by HIV-1 Tat in cells derived from CNS. Oncogene 1993, 8, 2231–2236. [Google Scholar]
- Sawaya, B.E.; Thatikunta, P.; Denisova, L.; Brady, J.; Khalili, K.; Amini, S. Regulation of TNFalpha and TGFbeta-1 gene transcription by HIV-1 Tat in CNS cells. J. Neuroimmunol 1998, 87, 33–42. [Google Scholar]
- Masliah, E.; Ge, N.; Achim, C.L.; Wiley, C.A. Cytokine receptor alterations during HIV infection in the human central nervous system. Brain Res 1994, 663, 1–6. [Google Scholar]
- Johnson, M.D.; Kim, P.; Tourtellotte, W.; Federspiel, C.F. Transforming growth factor beta and monocyte chemotactic protein-1 are elevated in cerebrospinal fluid of immunocompromised patients with HIV-1 infection. J. Neuro. AIDS 2004, 2, 33–43. [Google Scholar]
- da Cunha, A.; Jefferson, J.J.; Tyor, W.R.; Glass, J.D.; Jannotta, F.S.; Cottrell, J.R.; Resau, J.H. Transforming growth factor-beta1 in adult human microglia and its stimulated production by interleukin-1. J. Interferon. Cytokine. Res 1997, 17, 655–664. [Google Scholar]
- Vitkovic, L. Neuropathogenesis of HIV-1 infection: Interactions between interleukin-1 and transforming growth factor-beta 1. Mol. Psychiatry 1997, 2, 111–112. [Google Scholar]
- Perrella, O.; Carreiri, P.B.; Perrella, A.; Sbreglia, C.; Gorga, F.; Guarnaccia, D.; Tarantino, G. Transforming growth factor beta-1 and interferon-alpha in the AIDS dementia complex (ADC): Possible relationship with cerebral viral load? Eur. Cytokine Netw 2001, 12, 51–55. [Google Scholar]
- Ossege, L.M.; Voss, B.; Wiethege, T.; Sindern, E.; Malin, J.P. Detection of transforming growth factor beta 1 mRNA in cerebrospinal fluid cells of patients with meningitis by non-radioactive in situ hybridization. J. Neurol 1994, 242, 14–19. [Google Scholar]
- Matsumura, S.; Shibakusa, T.; Fujikawa, T.; Yamada, H.; Inoue, K.; Fushiki, T. Increase in transforming growth factor-beta in the brain during infection is related to fever, not depression of spontaneous motor activity. Neuroscience 2007, 144, 1133–1140. [Google Scholar]
- Fontana, A.; Constam, D.B.; Frei, K.; Malipiero, U.; Pfister, H.W. Modulation of the immune response by transforming growth factor beta. Int. Arch. Allergy Immunol 1992, 99, 1–7. [Google Scholar]
- Malipiero, U.; Koedel, U.; Pfister, W.; Fontana, A. Bacterial meningitis: The role of transforming growth factor-Beta in innate immunity and secondary brain damage. Neurodegener. Dis 2007, 4, 43–50. [Google Scholar]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech 2001, 52, 401–410. [Google Scholar]
- Rich, J.N.; Zhang, M.; Datto, M.B.; Bigner, D.D.; Wang, X.F. Transforming growth factor-beta-mediated p15(INK4B) induction and growth inhibition in astrocytes is SMAD3-dependent and a pathway prominently altered in human glioma cell lines. J. Biol. Chem 1999, 274, 35053–35058. [Google Scholar]
- Sasaki, A.; Naganuma, H.; Satoh, E.; Kawataki, T.; Amagasaki, K.; Nukui, H. Participation of thrombospondin-1 in the activation of latent transforming growth factor-beta in malignant glioma cells. Neurol. Med. Chir. (Tokyo) 2001, 41, 253–258, discussion 258–259. [Google Scholar]
- Jennings, M.T.; Pietenpol, J.A. The role of transforming growth factor beta in glioma progression. J. Neurooncol 1998, 36, 123–140. [Google Scholar]
- Zhang, L.; Sato, E.; Amagasaki, K.; Nakao, A.; Naganuma, H. Participation of an abnormality in the transforming growth factor-beta signaling pathway in resistance of malignant glioma cells to growth inhibition induced by that factor. J. Neurosurg 2006, 105, 119–128. [Google Scholar]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar]
- Hau, P.; Jachimczak, P.; Schlaier, J.; Bogdahn, U. TGF-beta2 signaling in high-grade gliomas. Curr. Pharm. Biotechnol 2011, 12, 2150–2157. [Google Scholar]
- Wick, W.; Naumann, U.; Weller, M. Transforming growth factor-beta: A molecular target for the future therapy of glioblastoma. Curr. Pharm. Des 2006, 12, 341–349. [Google Scholar]
- Naumann, U.; Maass, P.; Gleske, A.K.; Aulwurm, S.; Weller, M.; Eisele, G. Glioma gene therapy with soluble transforming growth factor-beta receptors II and III. Int. J. Oncol 2008, 33, 759–765. [Google Scholar]
- Ueda, R.; Fujita, M.; Zhu, X.; Sasaki, K.; Kastenhuber, E.R.; Kohanbash, G.; McDonald, H.A.; Harper, J.; Lonning, S.; Okada, H. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin. Cancer Res 2009, 15, 6551–6559. [Google Scholar]
- Kawano, H.; Kimura-Kuroda, J.; Komuta, Y.; Yoshioka, N.; Li, H.P.; Kawamura, K.; Li, Y.; Raisman, G. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res 2012, 349, 169–180. [Google Scholar]
- Hunt, N.H.; Grau, G.E. Cytokines: Accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol 2003, 24, 491–499. [Google Scholar]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem 2007, 14, 1189–1197. [Google Scholar]
- Kiefer, R.; Streit, W.J.; Toyka, K.V.; Kreutzberg, G.W.; Hartung, H.P. Transforming growth factor-beta 1: A lesion-associated cytokine of the nervous system. Int. J. Dev. Neurosci 1995, 13, 331–339. [Google Scholar]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar]
- Wyss-Coray, T.; Borrow, P.; Brooker, M.J.; Mucke, L. Astroglial overproduction of TGF-beta 1 enhances inflammatory central nervous system disease in transgenic mice. J. Neuroimmunol 1997, 77, 45–50. [Google Scholar]
- Stoll, G.; Schroeter, M.; Jander, S.; Siebert, H.; Wollrath, A.; Kleinschnitz, C.; Bruck, W. Lesion-associated expression of transforming growth factor-beta-2 in the rat nervous system: Evidence for down-regulating the phagocytic activity of microglia and macrophages. Brain Pathol 2004, 14, 51–58. [Google Scholar]
- Yu, P.; Wang, H.; Katagiri, Y.; Geller, H.M. An in vitro model of reactive astrogliosis and its effect on neuronal growth. Method. Mol. Biol 2012, 814, 327–340. [Google Scholar]
- Johns, L.D.; Babcock, G.; Green, D.; Freedman, M.; Sriram, S.; Ransohoff, R.M. Transforming growth factor-beta 1 differentially regulates proliferation and MHC class-II antigen expression in forebrain and brainstem astrocyte primary cultures. Brain Res 1992, 585, 229–236. [Google Scholar]
- Cullen, D.K.; Simon, C.M.; LaPlaca, M.C. Strain rate-dependent induction of reactive astrogliosis and cell death in three-dimensional neuronal-astrocytic co-cultures. Brain Res 2007, 1158, 103–115. [Google Scholar]
- Logan, A.; Baird, A.; Berry, M. Decorin attenuates gliotic scar formation in the rat cerebral hemisphere. Exp. Neurol 1999, 159, 504–510. [Google Scholar]
- Moon, L.D.; Fawcett, J.W. Reduction in CNS scar formation without concomitant increase in axon regeneration following treatment of adult rat brain with a combination of antibodies to TGFbeta1 and beta2. Eur. J. Neurosci 2001, 14, 1667–1677. [Google Scholar]
- Yoshioka, N.; Kimura-Kuroda, J.; Saito, T.; Kawamura, K.; Hisanaga, S.; Kawano, H. Small molecule inhibitor of type I transforming growth factor-beta receptor kinase ameliorates the inhibitory milieu in injured brain and promotes regeneration of nigrostriatal dopaminergic axons. J. Neurosci. Res 2011, 89, 381–393. [Google Scholar]
- Lagord, C.; Berry, M.; Logan, A. Expression of TGFbeta 2 but not TGFbeta 1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol. Cell. Neurosci 2002, 20, 69–92. [Google Scholar]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci 2010, 30, 5843–5854. [Google Scholar]
- Moreels, M.; Vandenabeele, F.; Dumont, D.; Robben, J.; Lambrichts, I. Alpha-smooth muscle actin (alpha-SMA) and nestin expression in reactive astrocytes in multiple sclerosis lesions: Potential regulatory role of transforming growth factor-beta 1 (TGF-beta1). Neuropathol. Appl. Neurobiol 2008, 34, 532–546. [Google Scholar]
- Schwab, J.M.; Beschorner, R.; Nguyen, T.D.; Meyermann, R.; Schluesener, H.J. Differential cellular accumulation of connective tissue growth factor defines a subset of reactive astrocytes, invading fibroblasts, and endothelial cells following central nervous system injury in rats and humans. J. Neurotrauma 2001, 18, 377–388. [Google Scholar]
- Yun, S.J.; Kim, M.O.; Kim, S.O.; Park, J.; Kwon, Y.K.; Kim, I.S.; Lee, E.H. Induction of TGF-beta-inducible gene-h3 (betaig-h3) by TGF-beta1 in astrocytes: Implications for astrocyte response to brain injury. Brain Res. Mol. Brain Res 2002, 107, 57–64. [Google Scholar]
- Law, A.K.; Gupta, D.; Levy, S.; Wallace, D.C.; McKeon, R.J.; Buck, C.R. TGF-beta1 induction of the adenine nucleotide translocator 1 in astrocytes occurs through Smads and Sp1 transcription factors. BMC Neurosci 2004, 5, 1. [Google Scholar]
- Chopp, M.; Li, Y. Apoptosis in focal cerebral ischemia. Acta Neurochir. Suppl 1996, 66, 21–26. [Google Scholar]
- Deigner, H.P.; Haberkorn, U.; Kinscherf, R. Apoptosis modulators in the therapy of neurodegenerative diseases. Expert Opin. Investig. Drugs 2000, 9, 747–764. [Google Scholar]
- Namura, S.; Zhu, J.; Fink, K.; Endres, M.; Srinivasan, A.; Tomaselli, K.J.; Yuan, J.; Moskowitz, M.A. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci 1998, 18, 3659–3668. [Google Scholar]
- Buisson, A.; Lesne, S.; Docagne, F.; Ali, C.; Nicole, O.; MacKenzie, E.T.; Vivien, D. Transforming growth factor-beta and ischemic brain injury. Cell. Mol. Neurobiol 2003, 23, 539–550. [Google Scholar]
- Zhu, Y.; Ahlemeyer, B.; Bauerbach, E.; Krieglstein, J. TGF-beta1 inhibits caspase-3 activation and neuronal apoptosis in rat hippocampal cultures. Neurochem. Int 2001, 38, 227–235. [Google Scholar]
- Bye, N.; Zieba, M.; Wreford, N.G.; Nichols, N.R. Resistance of the dentate gyrus to induced apoptosis during ageing is associated with increases in transforming growth factor-beta1 messenger RNA. Neuroscience 2001, 105, 853–862. [Google Scholar]
- Zhu, Y.; Yang, G.Y.; Ahlemeyer, B.; Pang, L.; Che, X.M.; Culmsee, C.; Klumpp, S.; Krieglstein, J. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J. Neurosci 2002, 22, 3898–3909. [Google Scholar]
- Ferrer, I. Apoptosis: Future targets for neuroprotective strategies. Cerebrovasc. Dis 2006, 21, 9–20. [Google Scholar]
- Kim, E.S.; Kim, R.S.; Ren, R.F.; Hawver, D.B.; Flanders, K.C. Transforming growth factor-beta inhibits apoptosis induced by beta-amyloid peptide fragment 25–35 in cultured neuronal cells. Brain Res. Mol. Brain Res 1998, 62, 122–130. [Google Scholar]
- Caraci, F.; Battaglia, G.; Busceti, C.; Biagioni, F.; Mastroiacovo, F.; Bosco, P.; Drago, F.; Nicoletti, F.; Sortino, M.A.; Copani, A. TGF-beta 1 protects against Abeta-neurotoxicity via the phosphatidylinositol-3-kinase pathway. Neurobiol. Dis 2008, 30, 234–242. [Google Scholar]
- Hicks, S.D.; Miller, M.W. Effects of ethanol on transforming growth factor Beta1-dependent and-independent mechanisms of neural stem cell apoptosis. Exp. Neurol 2011, 229, 372–380. [Google Scholar]
- Marushige, K.; Marushige, Y. Induction of apoptosis by transforming growth factor beta 1 in glioma and trigeminal neurinoma cells. Anticancer Res 1994, 14, 2419–2424. [Google Scholar]
- Schulz, R.; Vogel, T.; Mashima, T.; Tsuruo, T.; Krieglstein, K. Involvement of fractin in TGF-beta-induced apoptosis in oligodendroglial progenitor cells. Glia 2009, 57, 1619–1629. [Google Scholar]
- Xiao, B.G.; Bai, X.F.; Zhang, G.X.; Link, H. Transforming growth factor-beta1 induces apoptosis of rat microglia without relation to bcl-2 oncoprotein expression. Neurosci. Lett 1997, 226, 71–74. [Google Scholar]
- Jung, B.; Kim, M.O.; Yun, S.J.; Lee, E.H. Down-regulation of the expression of rat inhibitor-of-apoptosis protein-1 and −3 during transforming growth factor-beta1-mediated apoptosis in rat brain microglia. Neuroreport 2003, 14, 857–860. [Google Scholar]
- Dunker, N.; Schmitt, K.; Schuster, N.; Krieglstein, K. The role of transforming growth factor beta-2, beta-3 in mediating apoptosis in the murine intestinal mucosa. Gastroenterology 2002, 122, 1364–1375. [Google Scholar]
- Prehn, J.H.; Backhauss, C.; Krieglstein, J. Transforming growth factor-beta 1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemic injury in vivo. J. Cereb. Blood Flow Metab 1993, 13, 521–525. [Google Scholar]
- Ho, T.W.; Bristol, L.A.; Coccia, C.; Li, Y.; Milbrandt, J.; Johnson, E.; Jin, L.; Bar-Peled, O.; Griffin, J.W.; Rothstein, J.D. TGFbeta trophic factors differentially modulate motor axon outgrowth and protection from excitotoxicity. Exp. Neurol 2000, 161, 664–675. [Google Scholar]
- Docagne, F.; Nicole, O.; Gabriel, C.; Fernandez-Monreal, M.; Lesne, S.; Ali, C.; Plawinski, L.; Carmeliet, P.; MacKenzie, E.T.; Buisson, A.; Vivien, D. Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol. Cell. Neurosci 2002, 21, 634–644. [Google Scholar]
- Mesples, B.; Fontaine, R.H.; Lelievre, V.; Launay, J.M.; Gressens, P. Neuronal TGF-beta1 mediates IL-9/mast cell interaction and exacerbates excitotoxicity in newborn mice. Neurobiol. Dis 2005, 18, 193–205. [Google Scholar]
- Prehn, J.H.; Peruche, B.; Unsicker, K.; Krieglstein, J. Isoform-specific effects of transforming growth factors-beta on degeneration of primary neuronal cultures induced by cytotoxic hypoxia or glutamate. J. Neurochem 1993, 60, 1665–1672. [Google Scholar]
- Kane, C.J.; Brown, G.J.; Phelan, K.D. Transforming growth factor-beta2 increases NMDA receptor-mediated excitotoxicity in rat cerebral cortical neurons independently of glia. Neurosci. Lett 1996, 204, 93–96. [Google Scholar]
- Bae, J.J.; Xiang, Y.Y.; Martinez-Canabal, A.; Frankland, P.W.; Yang, B.B.; Lu, W.Y. Increased transforming growth factor-beta1 modulates glutamate receptor expression in the hippocampus. Int. J. Physiol. Pathophysiol. Pharmacol 2011, 3, 9–20. [Google Scholar]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H.; et al. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar]
- Pepper, M.S.; Vassalli, J.D.; Orci, L.; Montesano, R. Biphasic effect of transforming growth factor-beta 1 on in vitro angiogenesis. Exp. Cell. Res 1993, 204, 356–363. [Google Scholar]
- Gajdusek, C.M.; Luo, Z.; Mayberg, M.R. Basic fibroblast growth factor and transforming growth factor beta-1: Synergistic mediators of angiogenesis in vitro. J. Cell. Physiol 1993, 157, 133–144. [Google Scholar]
- Fajardo, L.F.; Prionas, S.D.; Kwan, H.H.; Kowalski, J.; Allison, A.C. Transforming growth factor beta1 induces angiogenesis in vivo with a threshold pattern. Lab. Invest 1996, 74, 600–608. [Google Scholar]
- Bertolino, P.; Deckers, M.; Lebrin, F.; ten Dijke, P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest 2005, 128, 585S–590S. [Google Scholar]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol 2009, 219, 449–458. [Google Scholar]
- Ueki, N.; Nakazato, M.; Ohkawa, T.; Ikeda, T.; Amuro, Y.; Hada, T.; Higashino, K. Excessive production of transforming growth-factor beta 1 can play an important role in the development of tumorigenesis by its action for angiogenesis: Validity of neutralizing antibodies to block tumor growth. Biochim. Biophys. Acta 1992, 1137, 189–196. [Google Scholar]
- Abe, K.; Chu, P.J.; Ishihara, A.; Saito, H. Transforming growth factor-beta 1 promotes re-elongation of injured axons of cultured rat hippocampal neurons. Brain Res 1996, 723, 206–209. [Google Scholar]
- Yu, G.; Fahnestock, M. Differential expression of nerve growth factor transcripts in glia and neurons and their regulation by transforming growth factor-beta1. Brain Res. Mol. Brain Res 2002, 105, 115–125. [Google Scholar]
- Ferrer, I.; Lopez, E.; Pozas, E.; Ballabriga, J.; Marti, E. Multiple neurotrophic signals converge in surviving CA1 neurons of the gerbil hippocampus following transient forebrain ischemia. J. Comp. Neurol 1998, 394, 416–430. [Google Scholar]
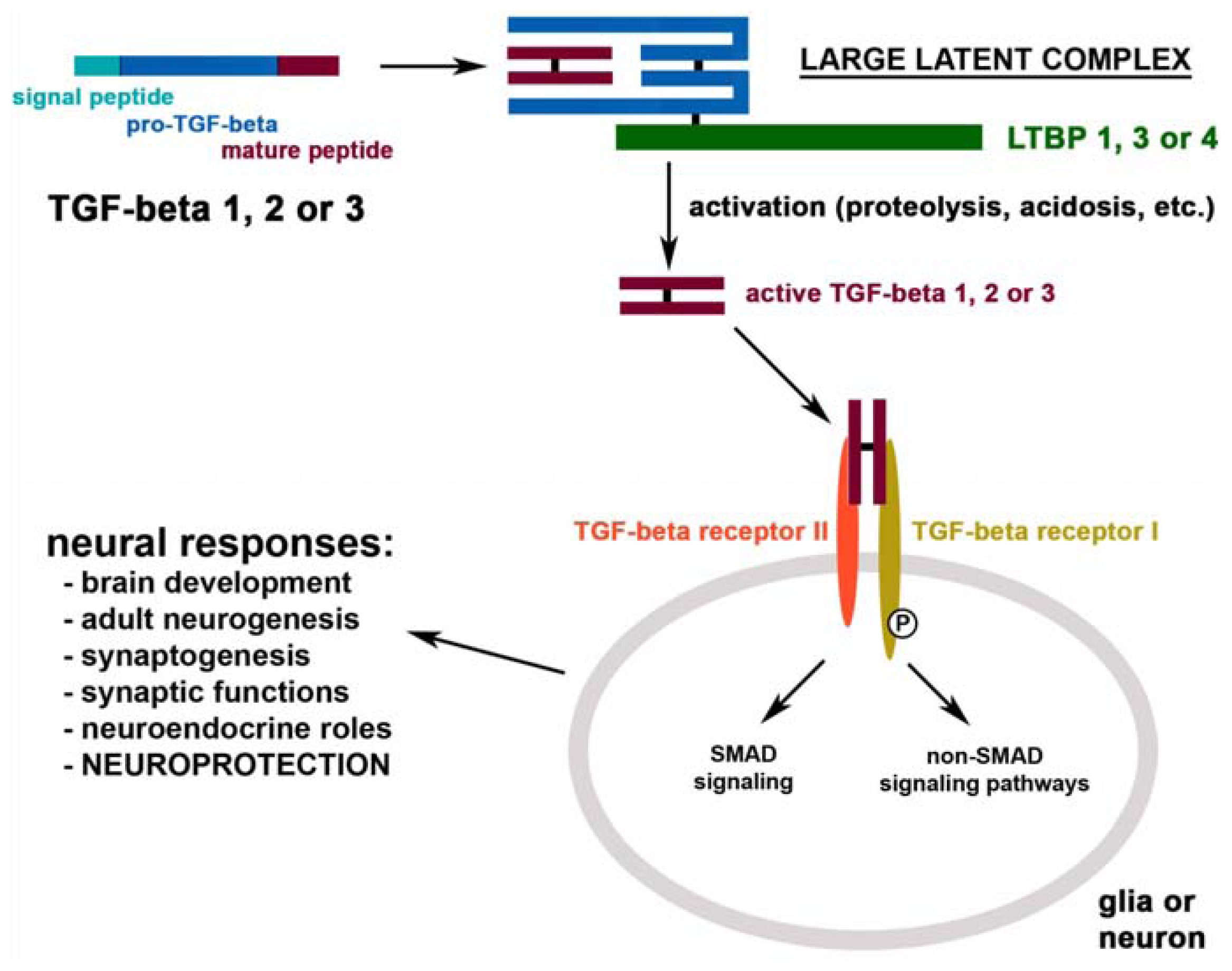


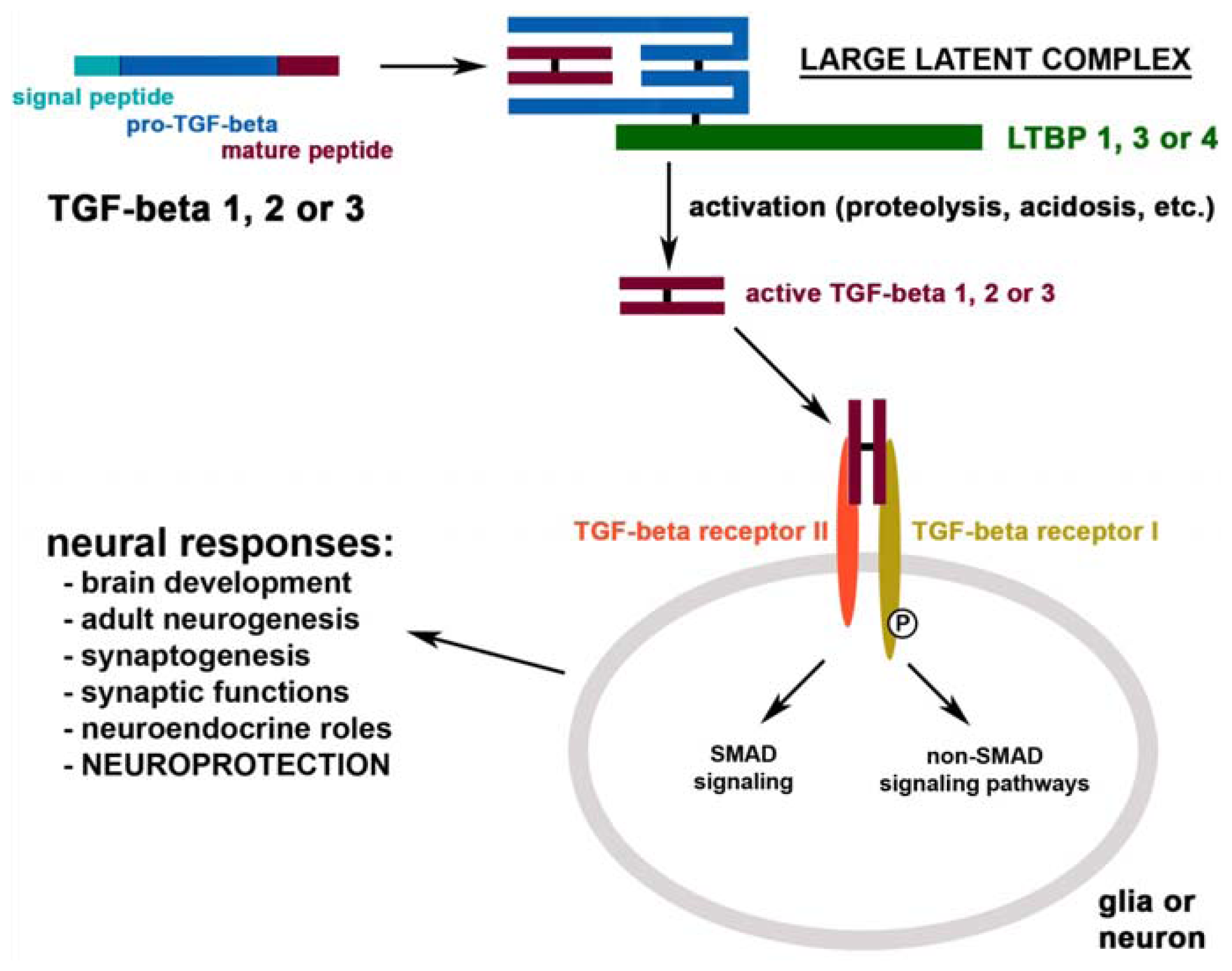{kind=link}
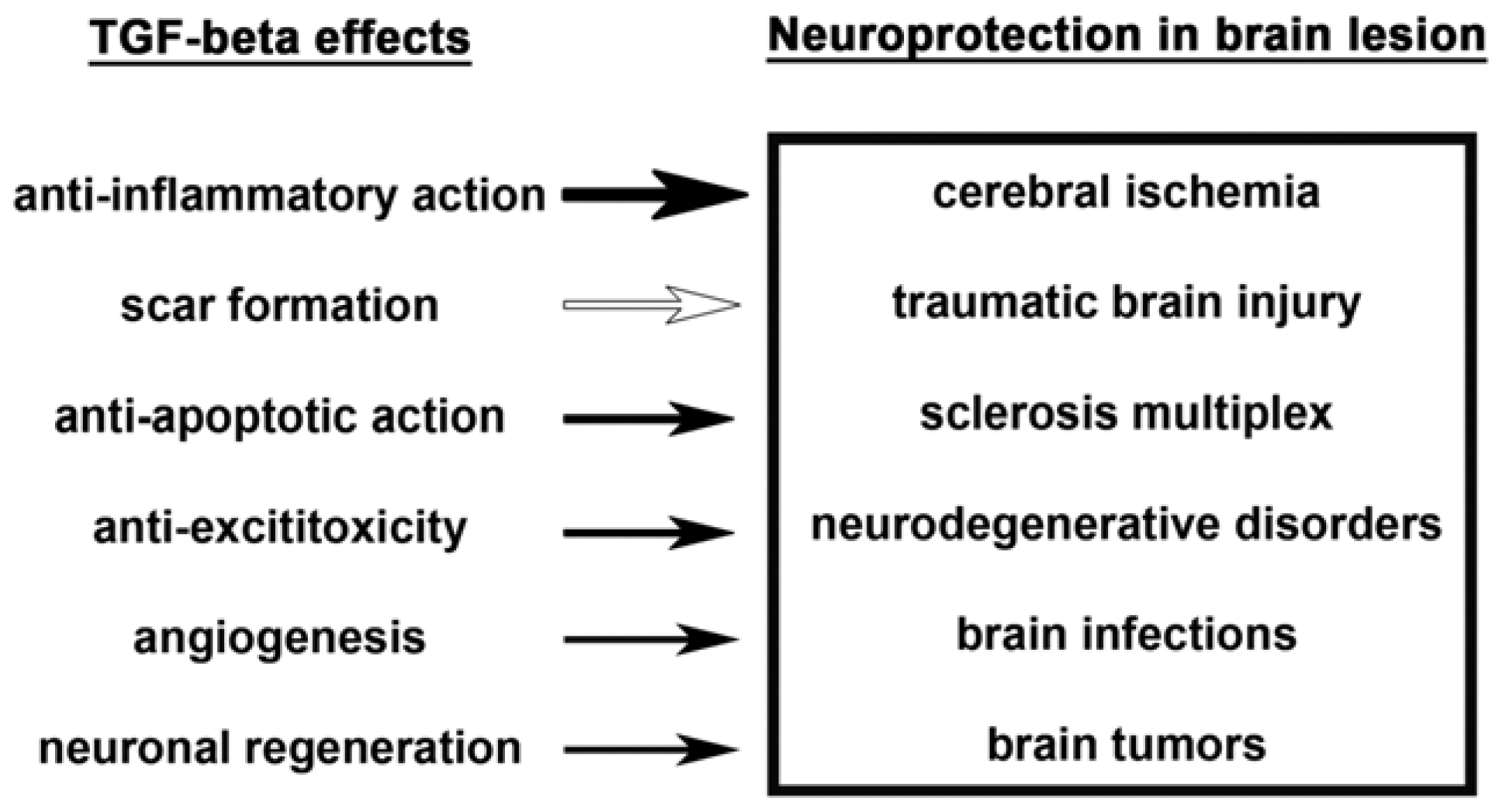{kind=link}
| Type of TGF-β | Brain Regions |
|---|---|
| TGF-β1 | hippocampus; amygdala, central nucleus; hypothalamus, preoptic and paraventricular nuclei; midbrain; pons and medulla oblongata, reticular formation, motor nuclei, superior olive, area postrema; choroid plexus |
| TGF-β2 | cerebral cortex, layer V; dentate gyrus; thalamus, parafascicular and midline nuclei; hypothalamus, posterior part and mamillary body; midbrain, raphe nuclei; pons and medulla oblongata, reticular formation, motor nuclei, superior olive, area postrema; cerebellar cortex, Purkinje cell layer; choroid plexus |
| TGF-β3 | cerebral cortex, layers IV,VI; dentate gyrus; amygdala, basal nucleus; thalamus, anterior, ventral, midline and reticular nuclei; hypothalamus, arcuate and supramamillary nuclei; midbrain, superior colliculus; pons and medulla oblongata, reticular formation, motor nuclei, superior and inferior olive, area postrema |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dobolyi, A.; Vincze, C.; Pál, G.; Lovas, G. The Neuroprotective Functions of Transforming Growth Factor Beta Proteins. Int. J. Mol. Sci. 2012, 13, 8219-8258. https://doi.org/10.3390/ijms13078219
Dobolyi A, Vincze C, Pál G, Lovas G. The Neuroprotective Functions of Transforming Growth Factor Beta Proteins. International Journal of Molecular Sciences. 2012; 13(7):8219-8258. https://doi.org/10.3390/ijms13078219
Chicago/Turabian StyleDobolyi, Arpád, Csilla Vincze, Gabriella Pál, and Gábor Lovas. 2012. "The Neuroprotective Functions of Transforming Growth Factor Beta Proteins" International Journal of Molecular Sciences 13, no. 7: 8219-8258. https://doi.org/10.3390/ijms13078219