Density Functional Theory (DFT) Study of Edaravone Derivatives as Antioxidants

Abstract
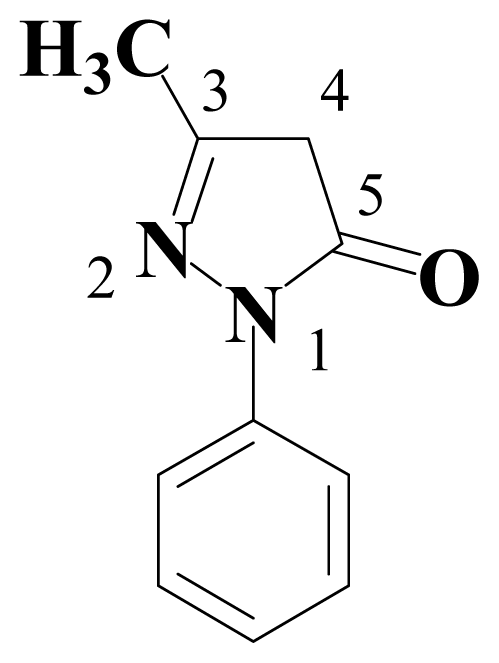
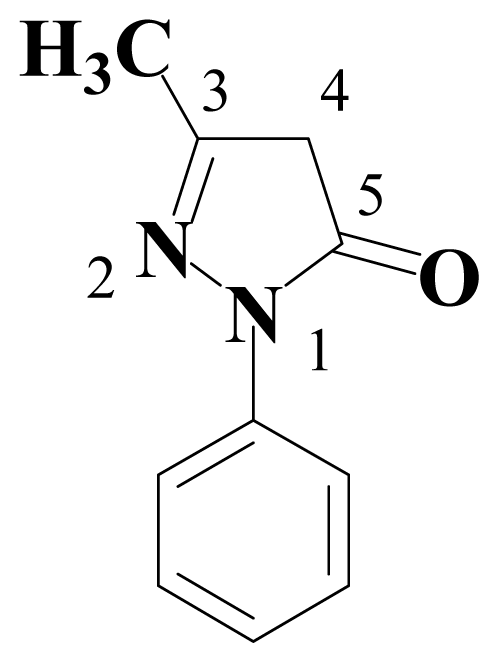
:1. Introduction
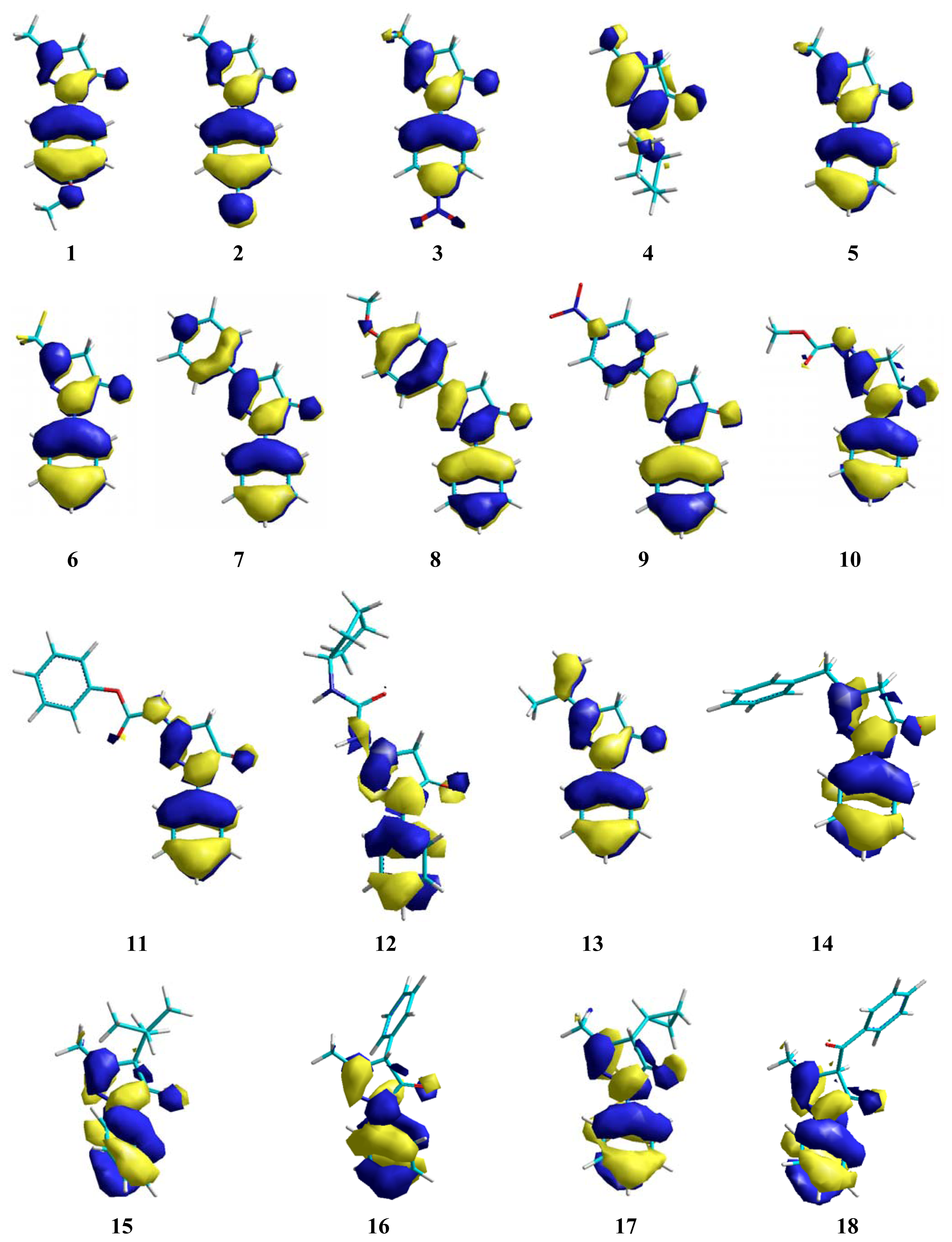
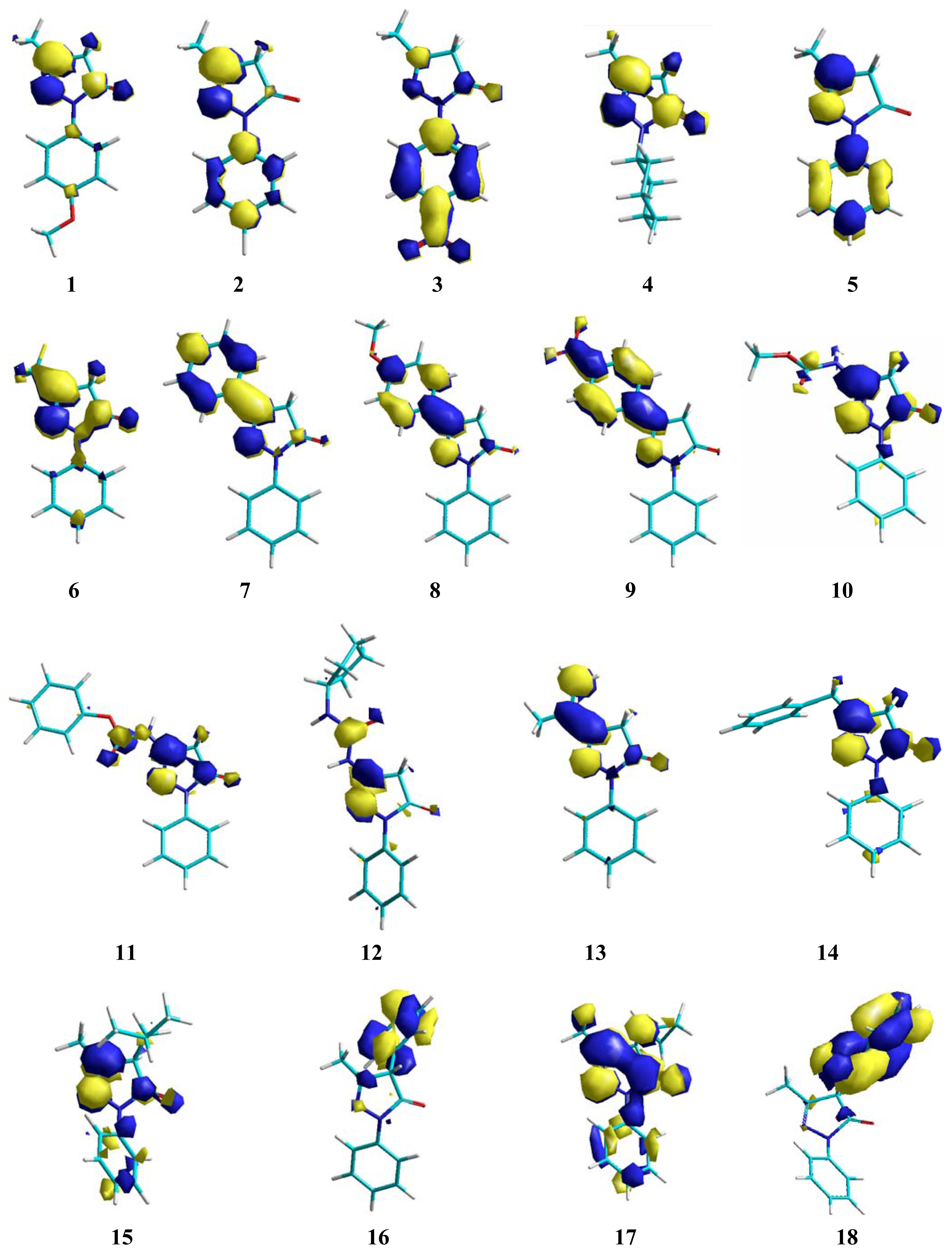
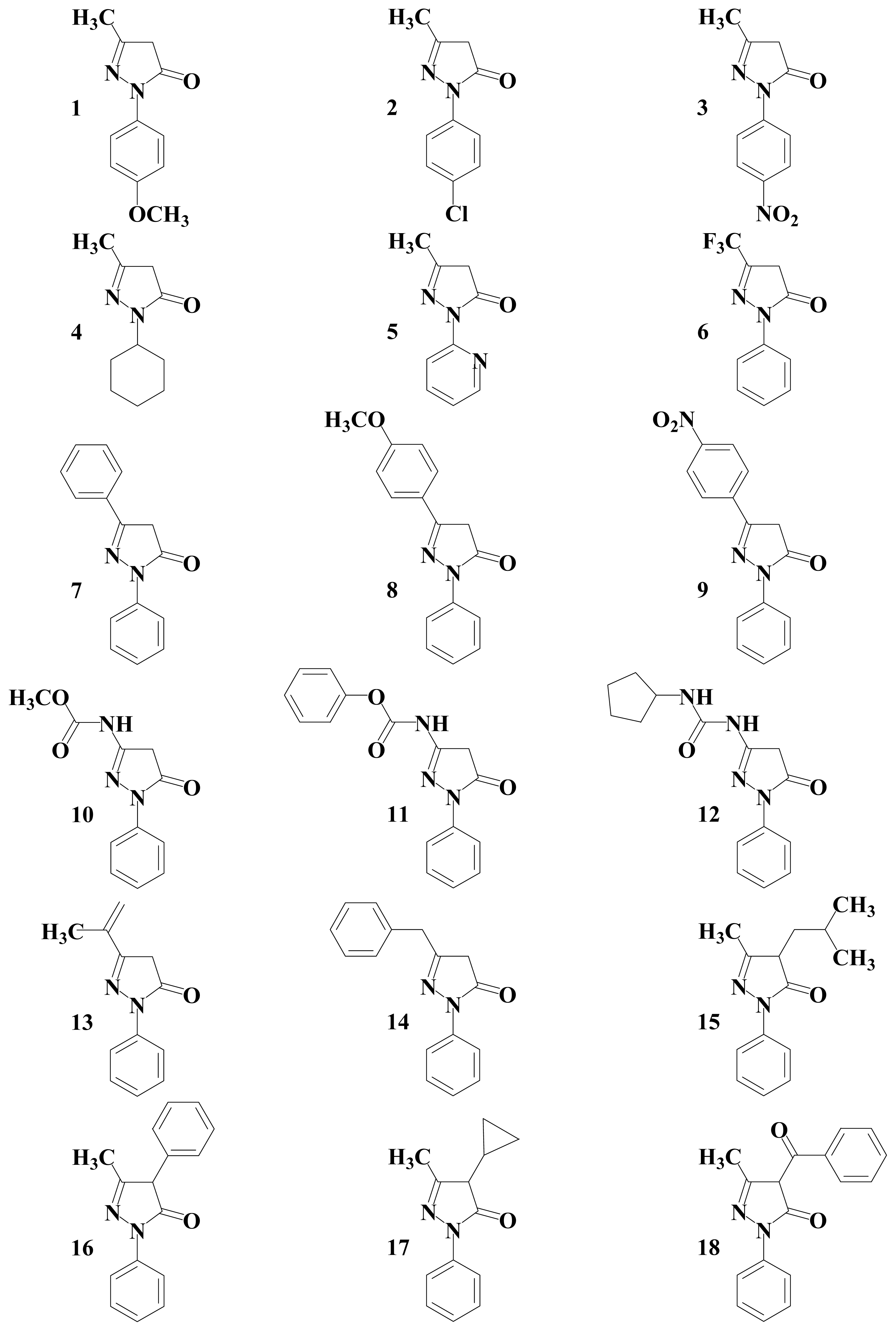
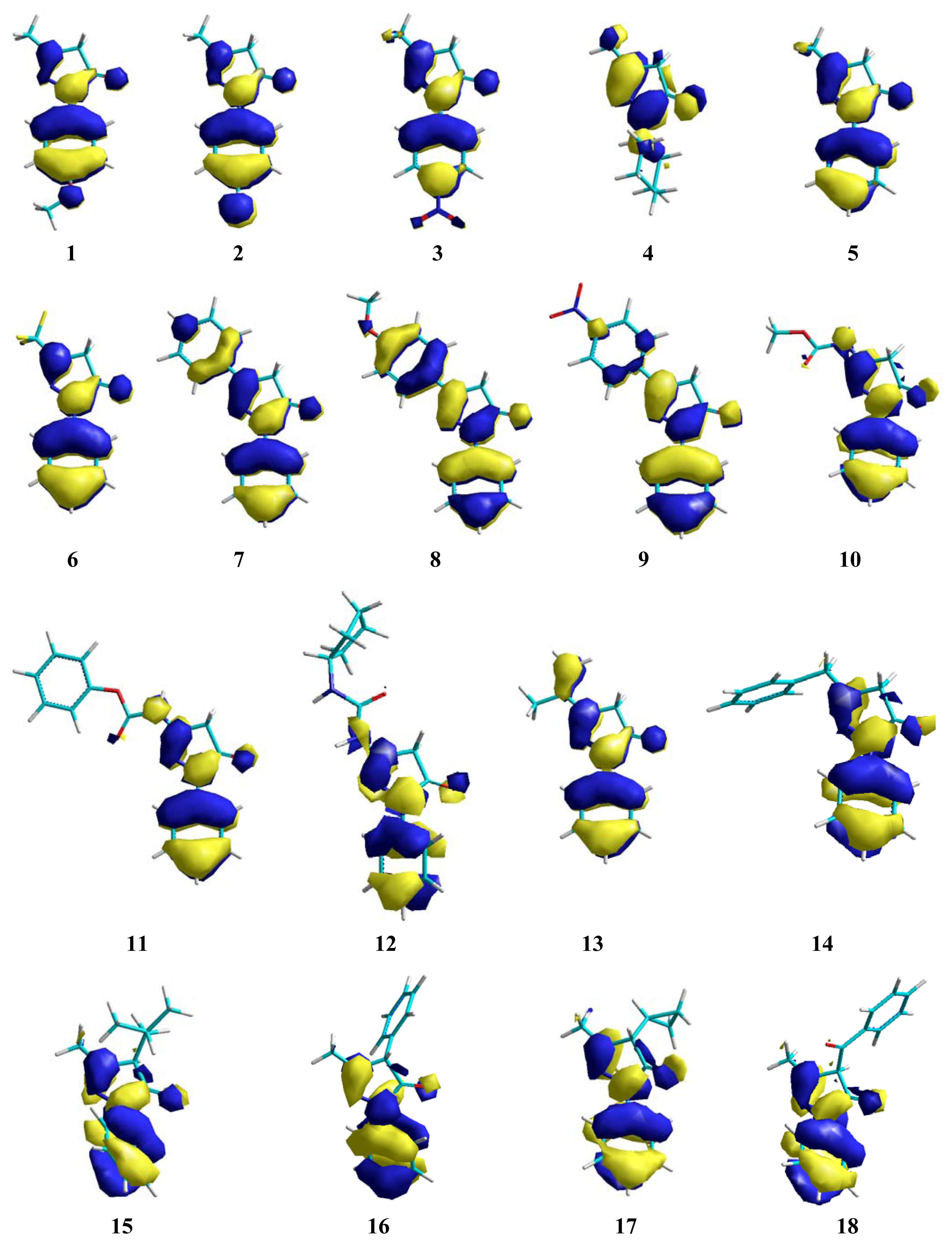
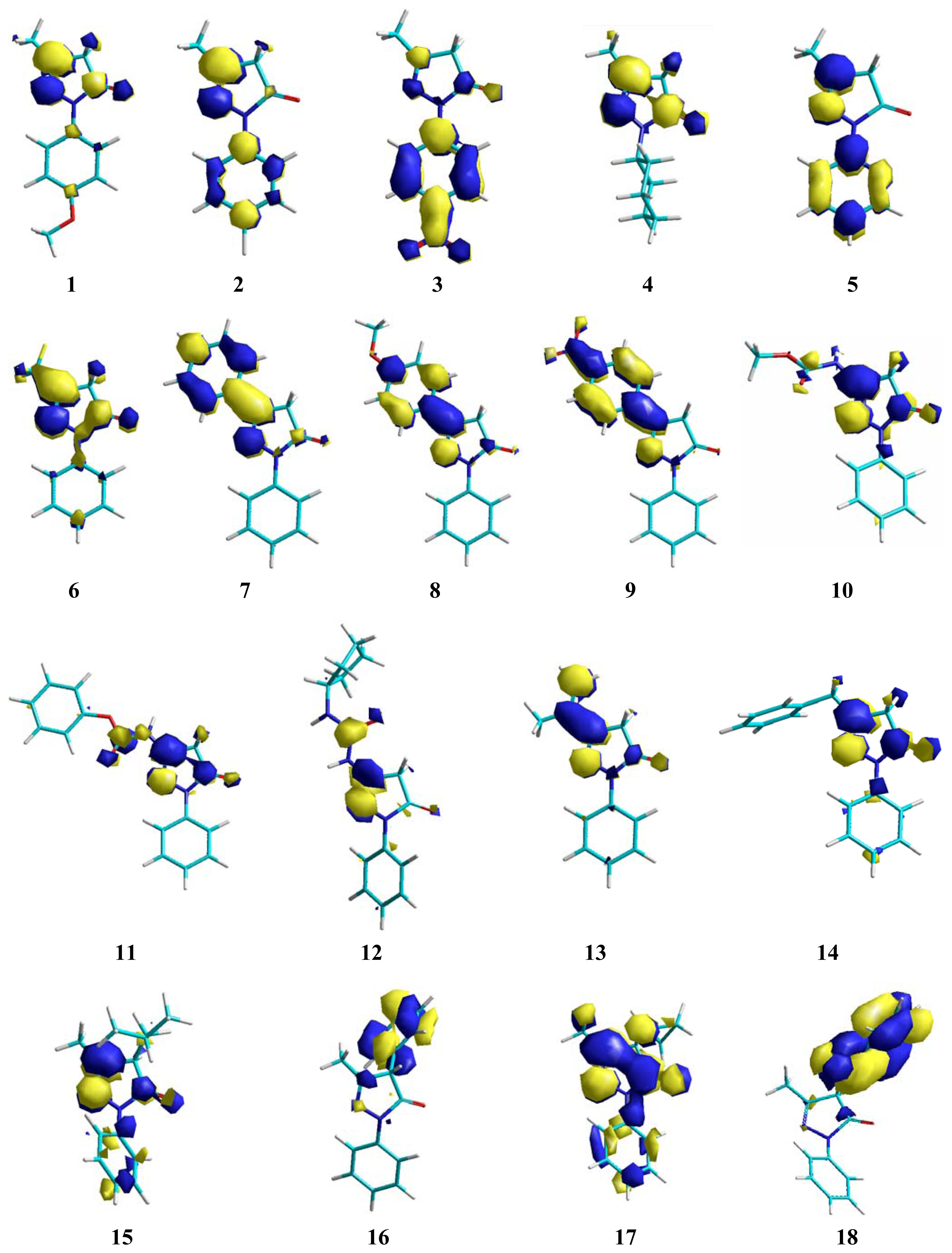
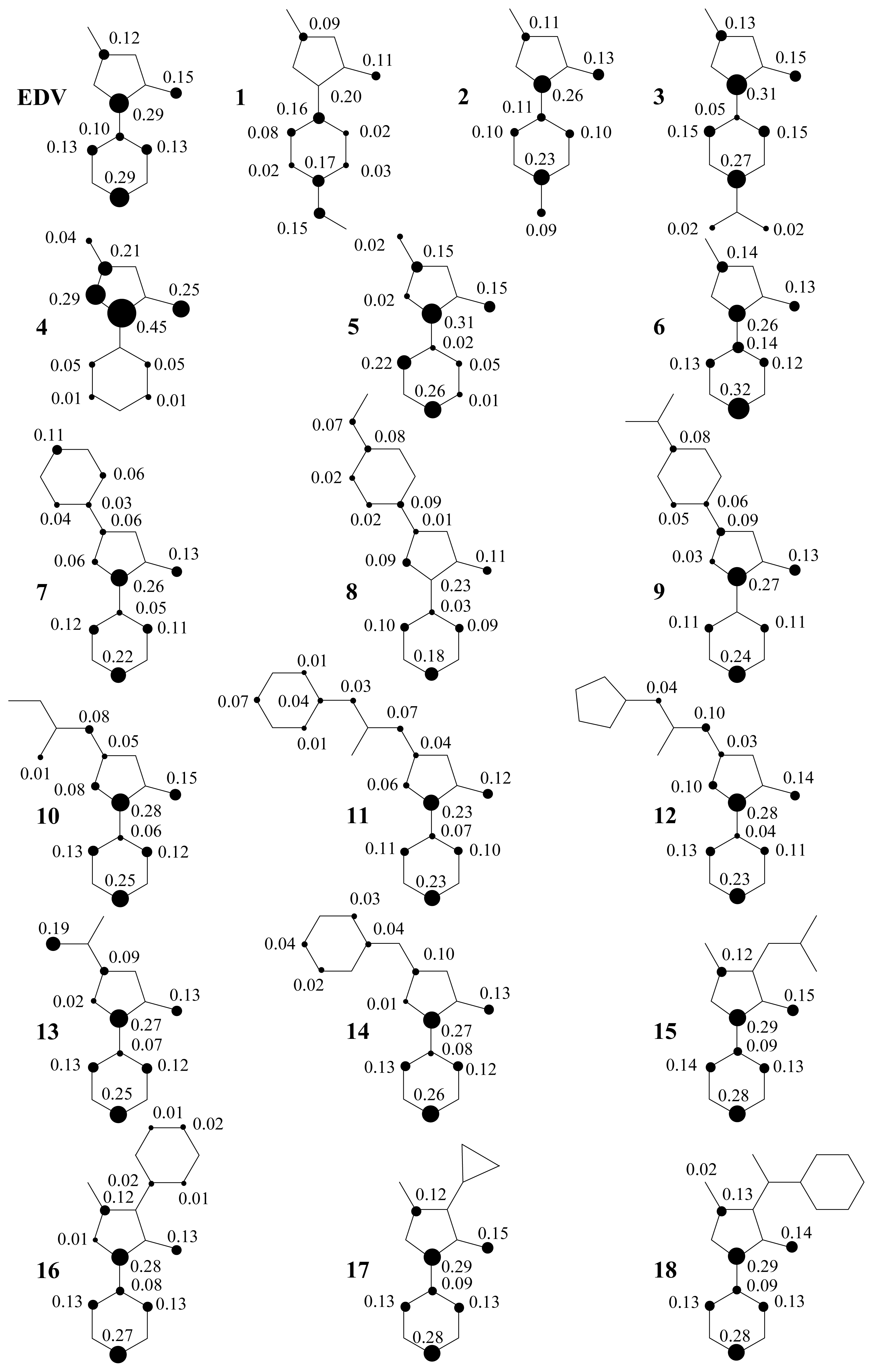
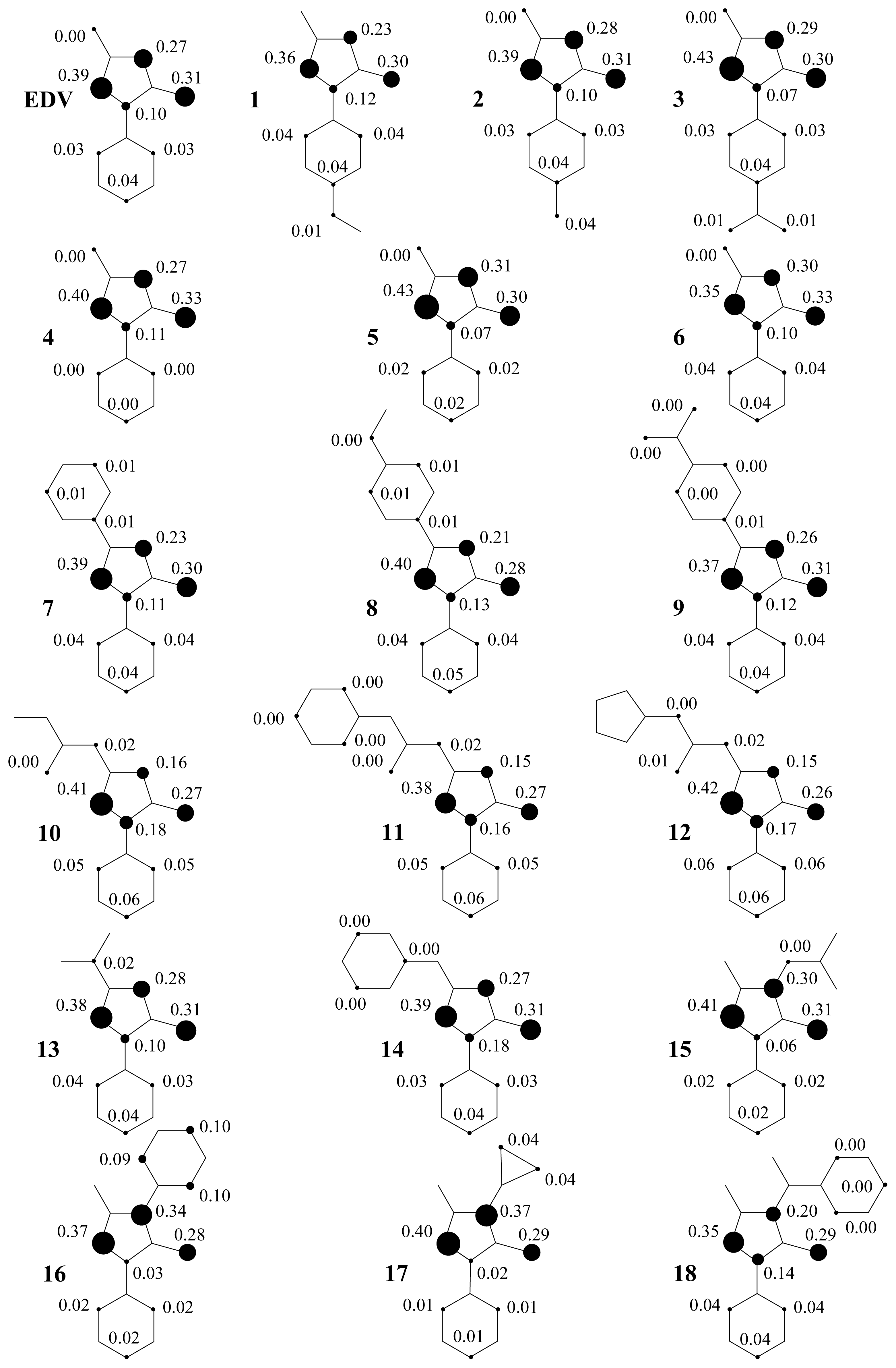
2. Results and Discussion
3. Computational Methods
4. Conclusions
Acknowledgments
References
- Tabrizchi, R. Edaravone Mitsubishi-Tokyo. Curr. Opin. Investig. Drugs 2000, 1, 347–354. [Google Scholar]
- Mizuno, A.; Umemura, K.; Nakashima, M. Inhibitory effect of MCI-186, a free radical scavenger, on cerebral ischemia following rat middle cerebral artery occlusion. Gen. Pharm 1998, 30, 575–578. [Google Scholar]
- Shichinohe, H.; Kuroda, S.; Yasuda, H.; Ishikawa, T.; Iwai, M.; Horiuchi, M.; Iwasaki, Y. Neuroprotective effects of the free radical scavenger Edaravone (MCI-186) in mice permanent focal brain ischemia. Brain Res 2004, 1029, 200–206. [Google Scholar]
- Wang, L.F.; Zhang, H.Y. A theoretical investigation on DPPH radical-Scavenging mechanism of edaravone. Bioorg. Med. Chem. Lett 2003, 13, 3789–3792. [Google Scholar]
- Nakagawa, H.; Ohyama, R.; Kimata, A.; Suzuki, T.; Miyata, N. Hydroxyl radical scavenging by edaravone derivatives: Efficient scavenging by 3-methyl-1-(pyridin-2-yl)-5-pyrazolone with an intramolecular base. Bioorg. Med. Chem. Lett 2006, 16, 5939–5942. [Google Scholar]
- Lin, M.; Katsumura, Y.; Hata, K.; Muroya, Y.; Nakagawa, K. Pulse radiolysis study on free radical scavenger edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one). J. Photochem. Photobiol. B 2007, 89, 36–43. [Google Scholar]
- Gomes, B.A.Q.; Queiroz, A.N.; Borges, R.B. Tautomerism and Radical-Scavenging Activity of Edaravone by DFT Methods. J. Comp. Theory. Nanosci 2009, 6, 1637–1639. [Google Scholar]
- Chegaev, K.; Cena, C.; Giorgis, M.; Rolando, B.; Tosco, P.; Bertinaria, M.; Fruttero, R.; Carrupt, P.-A.; Gasco, A. Edaravone derivatives containing NO-donor functions. J Med Chem 2009, 52, 574–578. [Google Scholar]
- Pérez-González, A.; Galano, A. Ionization energies, proton affinities, and pKa values of a large series of edaravone derivatives: Implication for their free radical scavenging activity. J. Phys. Chem. B 2011, 115, 10375–10384. [Google Scholar]
- Pérez-González, A.; Galano, A. OH radical scavenging activity of edaravone: Mechanism and kinetics. J. Phys. Chem. B 2011, 115, 1306–1314. [Google Scholar]
- Mendes, A.P.S.; Borges, R.S.; Chaves Neto, A.M.J.; de Macedo, L.G.M.; da Silva, A.B.F. The basic antioxidant structure for flavonoid derivatives. J. Mol. Model 2012. [Google Scholar] [CrossRef]
- Queiroz, A.N.; Gomes, B.A.Q.; Moraes, W.M., Jr; Borges, R.S. A theoretical antioxidant pharmacophore for resveratrol. Eur. J. Med. Chem 2009, 44, 1644–1649. [Google Scholar]
- Diniz, J.E.M.; Borges, R.S.; Alves, C.N. A DFT study for paracetamol and 3,5-disubstituted analogues. J. Mol. Struct. (TheoChem) 2004, 673, 93–97. [Google Scholar]
- Alves, C.N.; Borges, R.S.; da Silva, A.B.F. Density functional theory study of metabolic derivatives of the oxidation of paracetamol. Int. J. Quantum Chem 2006, 106, 2617–2623. [Google Scholar]
- Reis, M.; Lobato, B.; Lameira, J.; Santos, A.S.; Alves, C.N. A theoretical study of phenolic compounds with antioxidant properties. Eur. J. Med. Chem 2007, 42, 440–446. [Google Scholar]
- Freire, A.D.T.; Landivar, L.M.C.; Queiroz, A.N.; Borges, R.S. A theoretical study for oxidative metabolism of salicylates. J. Comp. Theor. Nanosci 2009, 6, 1140–1142. [Google Scholar]
- Bentes, A.L.A.; Borges, R.S.; Monteiro, W.R.; de Macedo, L.G.M.; Alves, C.N. Structure of dihydrochalcones and related derivatives and their scavenging and antioxidant activity against oxygen and nitrogen radical species. Molecules 2011, 16, 1749–1760. [Google Scholar]
- Silva, E.R.; Queiroz, A.N.; Almeida, E.D.; Borges, R.S. A DFT study of aminophenol stability. J. Comp. Theor. Nanosci 2009, 6, 1694–1696. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem 1989, 10, 209–220. [Google Scholar]
- Barone, V. Recent Advances in Density Functional Methods; Chong, D.P., Ed.; World Scientific: Singapore, 1995; Volume 1, pp. 287–334. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem 1996, 100, 12974–12980. [Google Scholar]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc 1983, 105, 7512–7516. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc 1999, 121, 1922–1924. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HOMO (eV) | IP (kcal mol−1) | BDECH (kcal mol−1) | ΔEiso (kcal mol−1) |
|---|---|---|---|---|
| EDV | −5.73 | 173.0 | 82.1 | 0 |
| 1 | −5.27 | 161.0 | 81.3 | −0.8 |
| 2 | −5.86 | 174.2 | 82.4 | 0.3 |
| 3 | −6.44 | 187.5 | 83.3 | 1.2 |
| 4 | −6.00 | 183.0 | 80.6 | −1.4 |
| 5 | −6.01 | 179.8 | 82.8 | 0.7 |
| 6 | −6.22 | 184.7 | 81.0 | −1.0 |
| 7 | −5.65 | 165.7 | 82.0 | −0.1 |
| 8 | −5.44 | 158.9 | 82.1 | 0 |
| 9 | −6.06 | 174.8 | 81.8 | −0.3 |
| 10 | −5.54 | 166.6 | 81.9 | −0.2 |
| 11 | −5.59 | 165.8 | 81.8 | −0.3 |
| 12 | −5.42 | 162.4 | 82.5 | 0.4 |
| 13 | −5.71 | 169.6 | 83.9 | 1.8 |
| 14 | −5.73 | 170.1 | 82.6 | 0.5 |
| 15 | −5.69 | 170.4 | 75.9 | −6.1 |
| 16 | −5.71 | 169.8 | 73.3 | −8.8 |
| 17 | −5.73 | 171.5 | 74.5 | −7.6 |
| 18 | −5.73 | 173.0 | 83.8 | 1.7 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Borges, R.S.; Queiroz, A.N.; Mendes, A.P.S.; Araújo, S.C.; França, L.C.S.; Franco, E.C.S.; Leal, W.G.; Da Silva, A.B.F. Density Functional Theory (DFT) Study of Edaravone Derivatives as Antioxidants. Int. J. Mol. Sci. 2012, 13, 7594-7606. https://doi.org/10.3390/ijms13067594
Borges RS, Queiroz AN, Mendes APS, Araújo SC, França LCS, Franco ECS, Leal WG, Da Silva ABF. Density Functional Theory (DFT) Study of Edaravone Derivatives as Antioxidants. International Journal of Molecular Sciences. 2012; 13(6):7594-7606. https://doi.org/10.3390/ijms13067594
Chicago/Turabian StyleBorges, Rosivaldo S., Auriekson N. Queiroz, Anna P. S. Mendes, Sanderson C. Araújo, Luiz C. S. França, Edna C. S. Franco, Walace G. Leal, and Albérico B. F. Da Silva. 2012. "Density Functional Theory (DFT) Study of Edaravone Derivatives as Antioxidants" International Journal of Molecular Sciences 13, no. 6: 7594-7606. https://doi.org/10.3390/ijms13067594