Solution NMR Structure of Hypothetical Protein CV_2116 Encoded by a Viral Prophage Element in Chromobacterium violaceum

Abstract
:1. Introduction
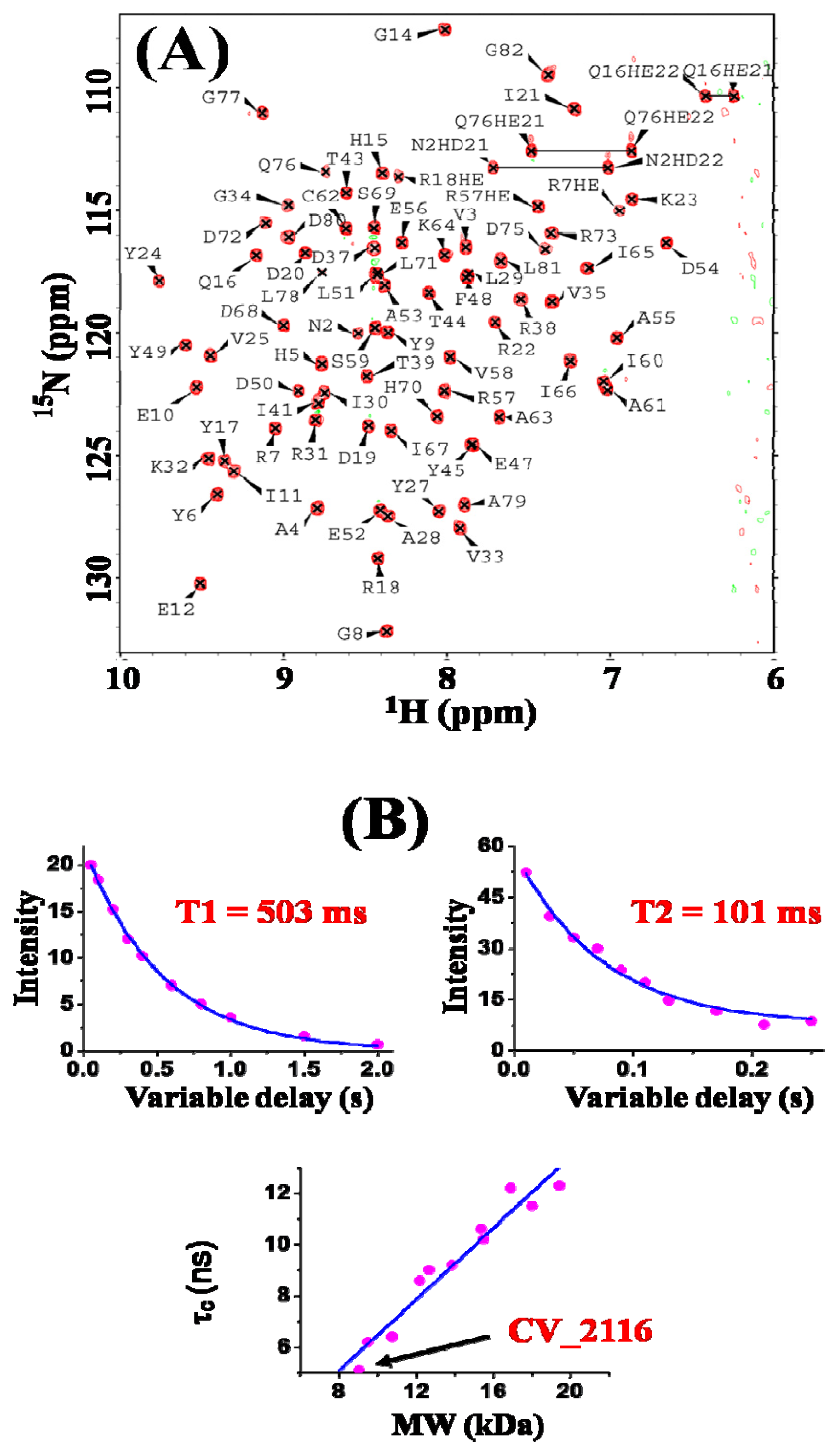
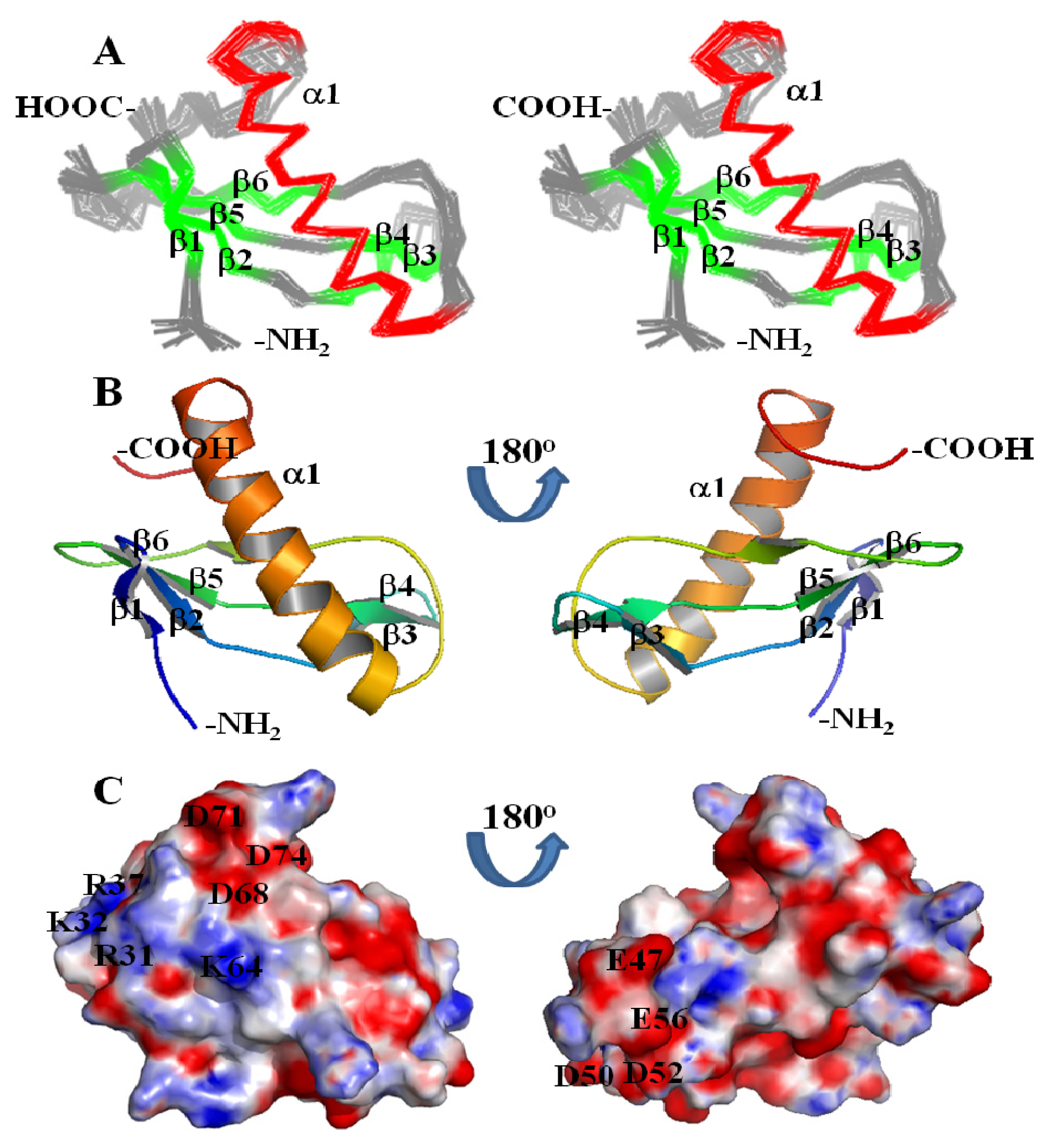
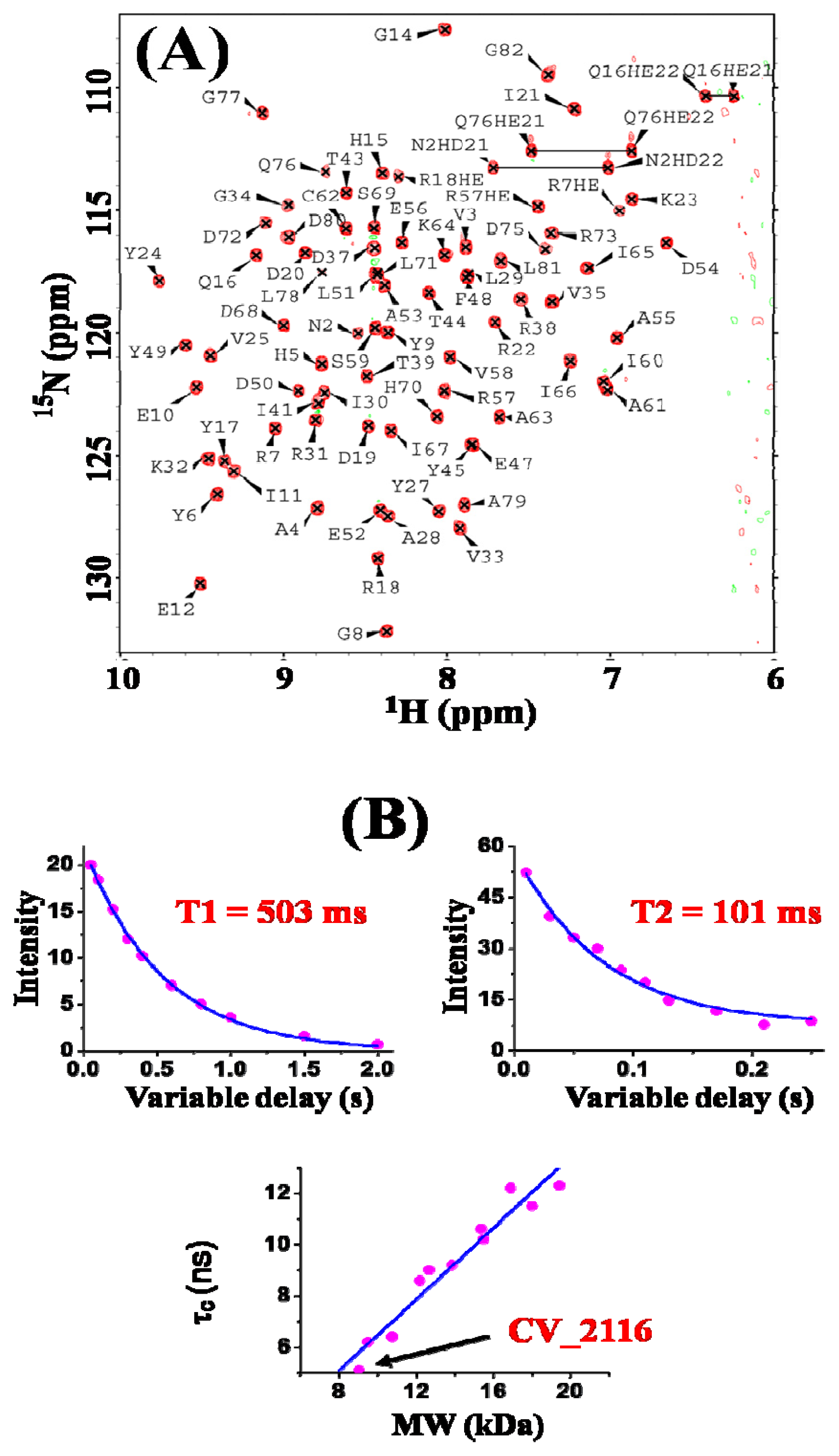
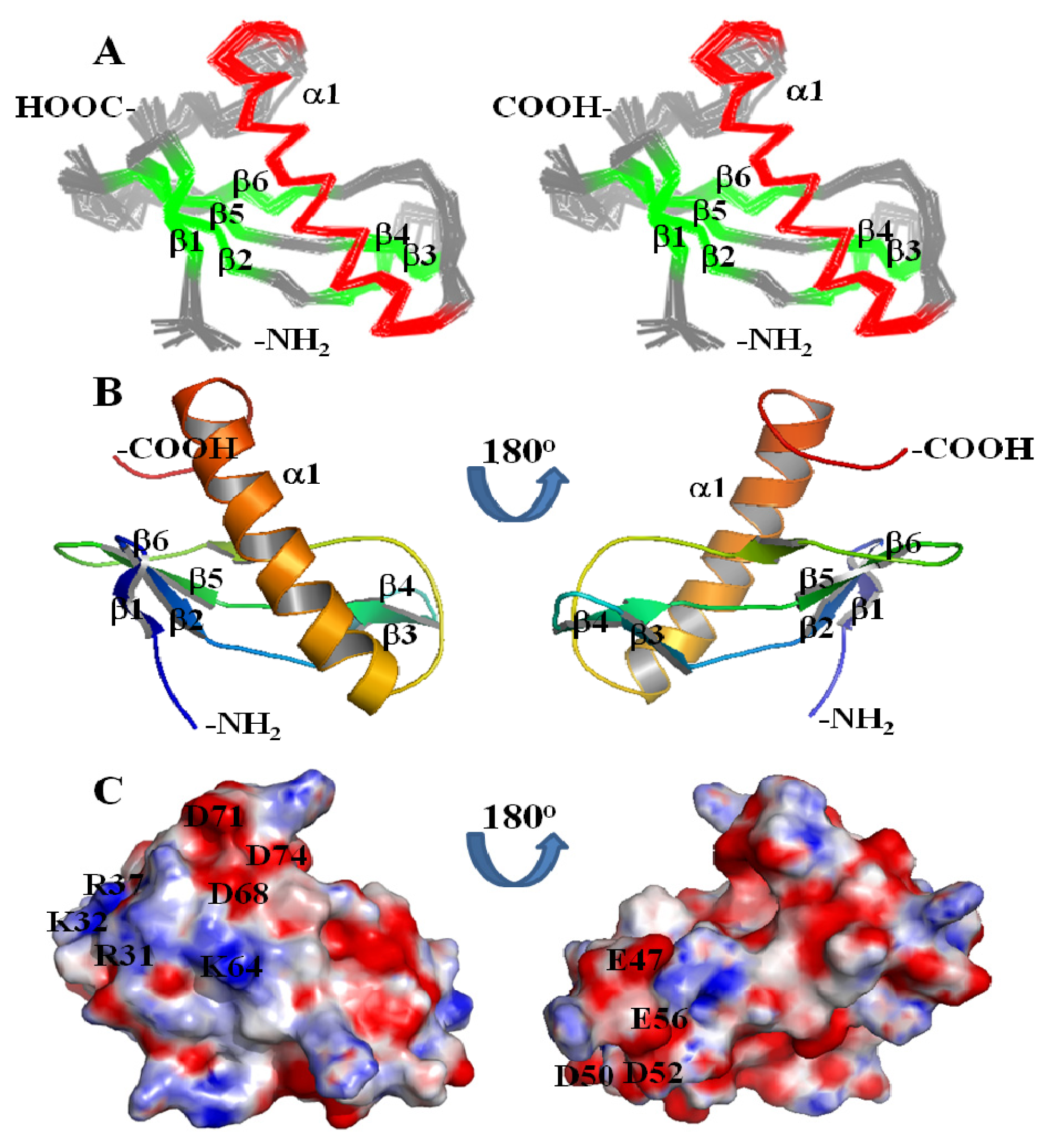
2. Results and Discussion
3. Experimental Section
3.1. Preparation of Protein Samples
3.2. NMR Spectroscopy and Data Collection
3.3. NMR Structure Calculation
4. Conclusions
Acknowledgments
References
- Kodach, L.L.; Bos, C.L.; Durán, N.; Peppelenbosch, M.P.; Ferreira, C.V.; Hardwick, J.C.H. Violacein synergistically increases 5-fluorouracil cytotoxicity, induces apoptosis and inhibits Akt-mediated signal transduction in human colorectal cancer cells. Carcinogenesis 2006, 27, 508–516. [Google Scholar]
- De Almeida, D.F.; Hungria, M.; Guimarães, C.T.; Antônio, R.V.; Almeida, F.C.; de Almeida, L.G.P.; de Almeida, R.; Andrade, E.M.; Araripe, J.; de Araújo, M.F.F.; et al. The complete genome sequence of Chromobacterium violaceum reveals remarkable and exploitable bacterial adaptability. Proc. Nat. Acad. Sci. USA 2003, 100, 11660–11665. [Google Scholar]
- De Almeida, R.; Trevilato, P.B.; Bartoleti, L.A.; Proença-Módena, J.L.; Hanna, E.S.; Gregoracci, G.B.; Brocchi, M. Bacteriophages and insertion sequences of Chromobacterium violaceum ATCC 12472. Genet. Mol. Res 2004, 3, 76–84. [Google Scholar]
- Protein: Q7NW74_CHRVO (Q7NW74). Availbale online: http://pfam.sanger.ac.uk/protein/Q7NW74 accessed on 15 February 2012.
- Bauer, M.; Kube, M.; Teeling, H.; Richter, M.; Lombardot, T.; Allers, E.; Würdemann, C.A.; Quast, C.; Kuhl, H.; Knaust, F.; et al. Whole genome analysis of the marine Bacteroidetes “Gramella forsetii” reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol 2006, 8, 2201–2213. [Google Scholar]
- Rhee, M.S.; Moritz, B.E.; Xie, G.; del Rio, T.G.; Dalin, E.; Tice, H.; Bruce, D.; Goodwin, L.; Chertkov, O.; Brettin, T.; et al. Complete Genome Sequence of a thermotolerant sporogenic lactic acid bacterium, Bacillus coagulans strain 36D1. Stand. Genomic Sci 2011, 5. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Yang, Y.; Ramelot, T.A.; Cort, J.R.; Wang, H.; Ciccosanti, C.; Jiang, M.; Janjua, H.; Acton, T.B.; Xiao, R.; Everett, J.K.; et al. Solution NMR structure of Dsy0195 homodimer from Desulfitobacterium hafniense: First structure representative of the YabP domain family of proteins involved in spore coat assembly. J. Struct. Funct. Genomics 2011, 12, 175–179. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Cryst 1993, 26, 283–291. [Google Scholar]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar]
- CATH: Protein Structure Classification Database. Available online: http://www.cathdb.info/ accessed on 15 February 2012.
- SCOP: Structural Classification of Proteins. Available online: http://scop.mrc-lmb.cam.ac.uk/scop/ accessed on 15 February 2012.
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic. Acids. Res 2010, 38, W545–W549. [Google Scholar]
- Kumazaki, T.; Ishii, S. Comparative study on fibers isolated from four R-type pyocins, phage-tail-like bacteriocins of Pseudomonas aeruginosa. J. Biochem 1982, 92, 1559–1566. [Google Scholar]
- Haggard-Ljungquist, E.; Jacobsen, E.; Rishovd, S.; Six, E.W.; Sunshine, M.G.; Lindqvist, B.H.; Kim, K.-J.; Barreiro, V.; Koonin, E.V.; Calendar, R.; et al. Bacteriophage P2: Genes involved in baseplate assembly. Virology 1995, 213, 109–121. [Google Scholar]
- Rucinsky, T.E.; Gregory, J.P.; Cota-Robles, E.H. Organization of bacteriophage tail-like particles in cells of Chromobacterium violaceum. J. Bacteriol 1972, 110, 754–757. [Google Scholar]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brüssow, H. Prophage genomics. Microbiol. Mol. Biol. Rev 2003, 67, 238–276. [Google Scholar]
- Parkhill, J.; Dougan, G.; James, K.D.; Thomson, N.R.; Pickard, D.; Wain, J.; Churcher, C.; Mungall, K.L.; Bentley, S.D.; Holden, M.T.G.; et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 2001, 413, 848–852. [Google Scholar]
- Neri, D.; Szyperski, T.; Otting, G.; Senn, H.; Wuethrich, K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry 1989, 28, 7510–7516. [Google Scholar]
- Goddard, T.; Kneller, D. SPARKY, verison 3; University of California: San Fransisco, CA, USA, 2008. Available online: http://www.cgl.ucsf.edu/home/sparky/ accessed on 15 February 2012.
- Huang, Y.J.; Tejero, R.; Powers, R.; Montelione, G.T. A topology-constrained distance network algorithm for protein structure determination from NOESY data. Proteins 2006, 62, 587–603. [Google Scholar]
- Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol 2004, 278, 353–378. [Google Scholar]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor-NIH for NMR molecular structure determination. Progr. Nucl. Magn. Reson. Spectrosc 2006, 48, 47–62. [Google Scholar]
- Cai, M.; Huang, Y.; Suh, J.-Y.; Louis, J.M.; Ghirlando, R.; Craigie, R.; Clore, J.M. Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J. Biol. Chem 2007, 282, 14525–14535. [Google Scholar]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins 2007, 66, 778–795. [Google Scholar]
- Huang, Y.J.; Powers, R.; Montelione, G.T. Protein NMR recall, precision, and F-measure scores (RPF scores): Structure quality assessment measures based on information retrieval statistics. J. Am. Chem. Soc 2005, 127, 1665–1674. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Conformationally-Restricting Constraints b | |
| Distance Constraints | |
| Intra-residue (I = j) | 306 |
| Sequential (|i–j| = 1) | 357 |
| Medium-range (1 < |i–j| < 5) | 193 |
| Long-range (|i–j| ≥ 5) | 419 |
| Total | 1275 |
| Distance constraints per residue | 15.5 |
| Hydrogen bond constraints | |
| Long-range (|i–j| ≥ 5)/total | 20/56 |
| Dihedral angle constraints | 92 |
| Total number of restricting constraints | 1423 |
| Number of constraints per residue | 5.4 |
| Residue Constraint Violations b | |
| Average Number of Distance Violations Per Structure | |
| 0.1–0.2 Å | 4.85 |
| 0.2–0.5 Å | 1.15 |
| >0.5 Å | 0 |
| Average RMS distance violation/constraint (Å) | 0.01 |
| Maximum distance violation (Å) | 0.29 |
| Average Number of Dihedral Angle Violations Per Structure | |
| 1–10° | 0.7 |
| >10° | 0 |
| Average RMS dihedral angle violation/constraint (degree) | 0.15 |
| Maximum dihedral angle violation (degree) | 2.60 |
| RMSD from Average Coordinates (Å) c | |
| Backbone/Heavy atoms | 0.5/0.9 |
| Ramachandran Plot Statistics c | |
| Most favored regions (%) | 96.3 |
| Additional allowed regions (%) | 3.6 |
| Generously allowed (%) | 0.1 |
| Disallowed regions (%) | 0 |
| Global Quality Scores (raw/Z-score) b | |
| Verify3D | 0.24/−3.53 |
| Prosall | 0.30/−1.45 |
| Procheck(phi-psi) c | 0.21/1.14 |
| Procheck(all) c | 0.23/1.36 |
| Molprobity clash | 16.49/−1.30 |
| RPF Scores d | |
| Recall | 0.98 |
| Precision | 0.91 |
| F-measure | 0.94 |
| DP-score | 0.80 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, Y.; Ramelot, T.A.; Cort, J.R.; Garcia, M.; Yee, A.; Arrowsmith, C.H.; Kennedy, M.A. Solution NMR Structure of Hypothetical Protein CV_2116 Encoded by a Viral Prophage Element in Chromobacterium violaceum. Int. J. Mol. Sci. 2012, 13, 7354-7364. https://doi.org/10.3390/ijms13067354
Yang Y, Ramelot TA, Cort JR, Garcia M, Yee A, Arrowsmith CH, Kennedy MA. Solution NMR Structure of Hypothetical Protein CV_2116 Encoded by a Viral Prophage Element in Chromobacterium violaceum. International Journal of Molecular Sciences. 2012; 13(6):7354-7364. https://doi.org/10.3390/ijms13067354
Chicago/Turabian StyleYang, Yunhuang, Theresa A. Ramelot, John R. Cort, Maite Garcia, Adelinda Yee, Cheryl H. Arrowsmith, and Michael A. Kennedy. 2012. "Solution NMR Structure of Hypothetical Protein CV_2116 Encoded by a Viral Prophage Element in Chromobacterium violaceum" International Journal of Molecular Sciences 13, no. 6: 7354-7364. https://doi.org/10.3390/ijms13067354