Development of 101 Gene-based Single Nucleotide Polymorphism Markers in Sea Cucumber, Apostichopus japonicus

Abstract
:1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. Sampling and DNA Extraction
3.2. SNP Discovery and Genotyping
3.3. Data Analysis
4. Conclusions
Acknowledgments
References
- Sloan, N.A. References. In Echinodermata; Keegan, B.F., O’Connor, B.D.S., Eds.; A.A. Balkema: Rotterdam, The Netherlands, 1985; pp. 109–124. [Google Scholar]
- Chen, J.X. Present Status and Prospects of Sea Cucumber Industry in China. In Advances in Sea Cucumber Aquaculture and Management; Lovatelli, A., Conand, C., Purcell, S., Uthicke, S., Hamel, J.F., Mercier, A., Eds.; FAO: Rome, Italy, 2004; pp. 25–38. [Google Scholar]
- Collard, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar]
- Cho, R.J.; Mindrinos, M.; Richards, D.R.; Sapolsky, R.J.; Anderson, M.; Drenkard, E.; Dewdney, J.; Reuber, T.L.; Stammers, M.; Federspiel, N.; et al. Genome-wide mapping with biallelic markers in Arabidopsis thaliana. Nat. Genet 1999, 23, 203–207. [Google Scholar]
- Sun, W.J.; Li, Q.; Kong, L.F. Characterization of thirteen single nucleotide polymorphism markers in the sea cucumber (Apostichopus japonicus). Conserv. Genet. Resour 2010, 2, 141–144. [Google Scholar]
- Yang, A.F.; Sun, D.F.; Liu, S.K.; Dong, Y.; Chen, Z.; Zhou, Z.C. Characterization of fifteen SNP markers by mining EST in sea cucumber, Apostichopus japonicus. J. Genet. 2012, 91, e49–e53. [Google Scholar]
- Du, H.X.; Bao, Z.M.; Hou, R.; Wang, S.; Su, H.L.; Yan, J.J.; Tian, M.L.; Li, Y.; Wei, W.; Hu, X.L.; et al. Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka, 1867). PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Tsumura, Y.; Kado, T.; Takahashi, T.; Tani, N.; Ujino-Ihara, T.; Iwata, H. Genome scan to detect genetic structure and adaptive genes of natural populations of Cryptomeria japonica. Genetics 2007, 176, 2393–2403. [Google Scholar]
- Namroud, M.C.; Beaulieu, J.; Juge, N.; Laroche, J.; Bousquet, J. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol. Ecol 2008, 17, 3599–3613. [Google Scholar]
- NCBI nr database. Available online: ftp://ftp.ncbi.nlm.nih.gov/blast/db/ accessed on 25 January 2012.
- Zhan, A.B.; Bao, Z.M.; Lu, W.; Hu, X.L.; Peng, W.; Wang, M.L.; Hu, J.J. Development and characterization of 45 novel microsatellite markers for sea cucumber (Apostichopus japonicus). Mol. Ecol. Notes 2007, 7, 1345–1348. [Google Scholar]
- Wang, S.; Zhang, L.L.; Meyer, E.; Matz, M.V. Construction of a high-resolution genetic linkage map and comparative genome analysis for the reef-building coral Acropora millepora. Genome Biol 2009, 10. [Google Scholar] [CrossRef]
- Rozen, S.; Skaletsky, H. References. In Bioinformatics Methods and Protocols: Methods in Molecular Biology; Krawetz, S., Misener, S., Eds.; Humana: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res 2007, 35, 43–46. [Google Scholar]
- Yeh, F.C.; Boyle, T.J.B. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg. J. Bot 1997, 129, 157. [Google Scholar]
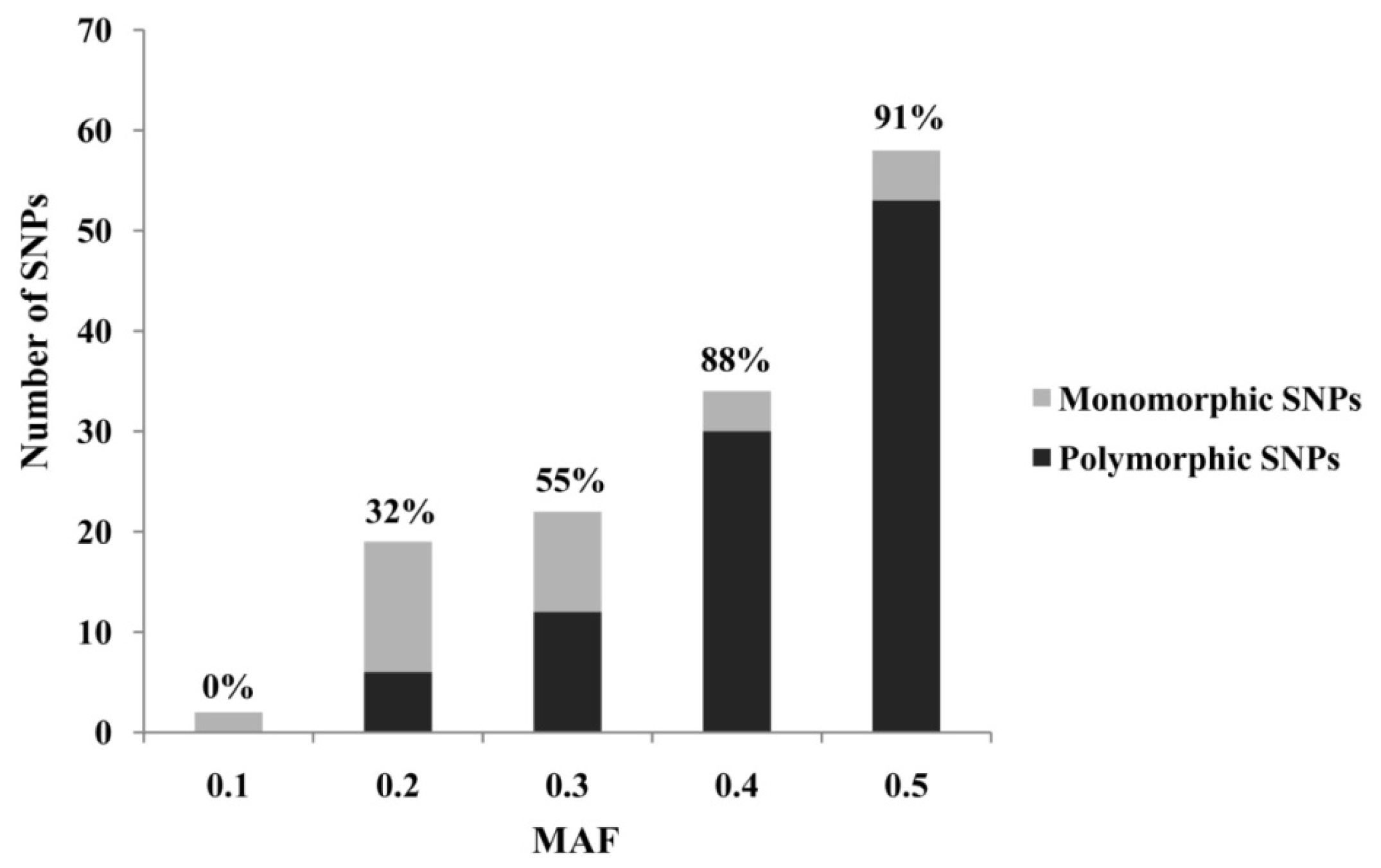

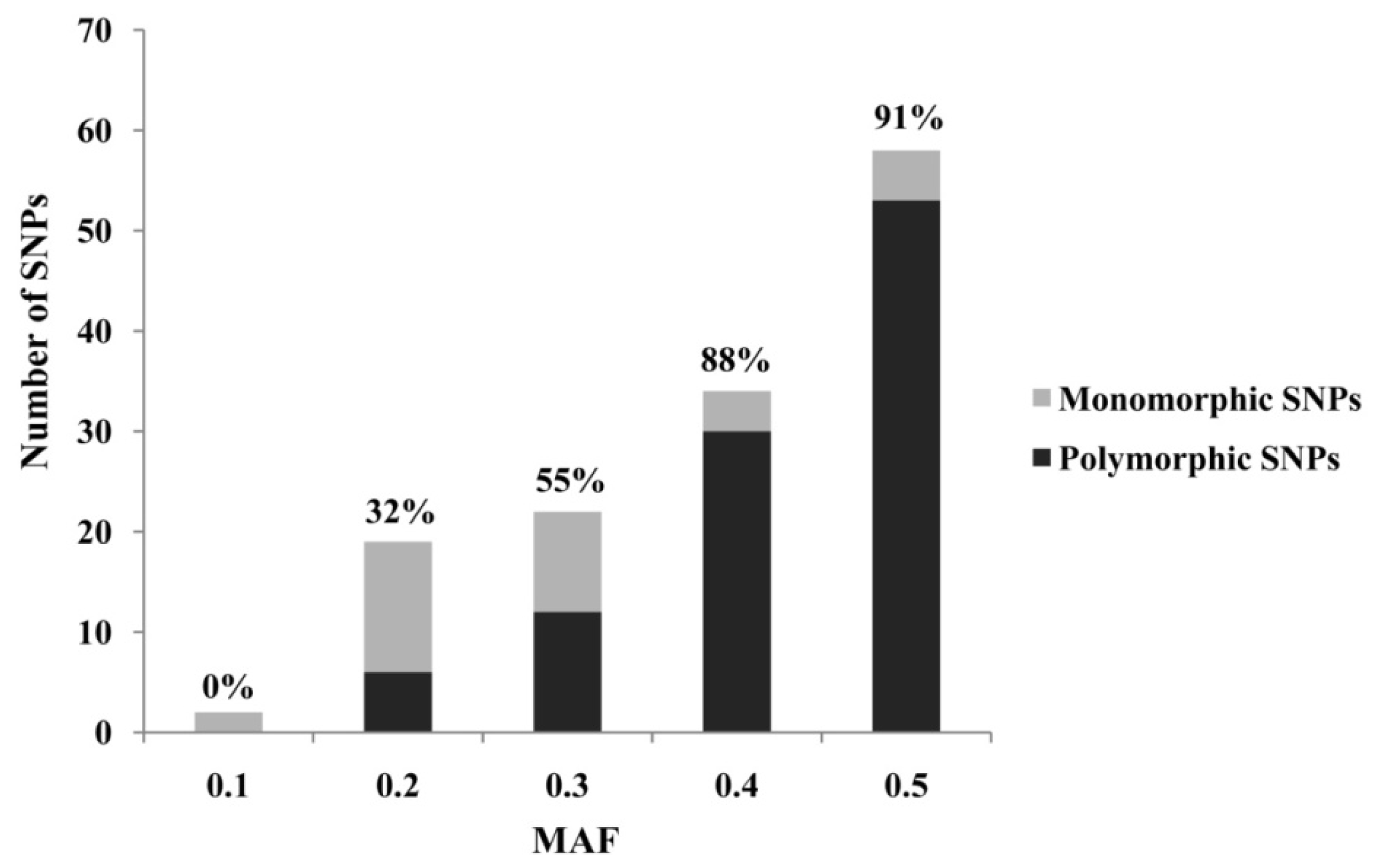{kind=link}
| Categories | Number of SNPs |
|---|---|
| Total number of tested SNPs | 200 |
| Successful PCR | 159 |
| Successful genotype calling | 135 |
| Polymorphic SNPs | 101 |
| Monomorphic SNPs | 34 |
| Failed SNPs | 65 |
| Locus ID | Gene Name | Primers and Probes (5′–3′) | Size (bp) | Ho | He | MA | MAF | p-Value |
|---|---|---|---|---|---|---|---|---|
| ApjSNP001_CT | similar to Mech2 protein | F:CCTCAGTCCCAATCACCACT R:ACACTGGCATACACCAGCAA P:CAATGACTTCTCCTTCTCCTACAGCTTCC | 98 | 0.250 | 0.431 | T | 0.128 | 0.239 |
| ApjSNP002_CT | Iron-sulfur cluster assembly 2 homolog | F:TGAATCAGGCAGTTGTGATGA R:GGTCCAGCTAGTCATGCTTTT P:TCAAAGAAATACACTATTTCATCCGATAAAGCAAG | 102 | 0.231 | 0.485 | C | 0.397 | 0.080 |
| ApjSNP003_AC | Protein strawberry notch homolog 2 | F:AGCGATTATATCCGATGCAG R:GCTGACCAGGGTAGTTCGAC P:TACAAGAGTAGCCAATCAGAGGCGAGC | 108 | 0.431 | 0.583 | A | 0.489 | 0.482 |
| ApjSNP004_AG | Thiosulfate sulfurtransferase | F:CAGTTGTAACTGCACCTCAGC R:ATGCCTACTTGGATGCCAGA P:ACCACAGGGTGTAGCCAGGTTCGTCAG | 70 | 0.314 | 0.342 | A | 0.178 | 0.578 |
| ApjSNP005_AG | Thiosulfate sulfurtransferase | F:AGGCATCCCTACGGGTATTT R:ACAGGAATGAAGTGGCTTGG P:TCGCTCTAGTCATTCCGCCTCAACG | 70 | 0.374 | 0.312 | A | 0.240 | 0.857 |
| ApjSNP006_CT | Dynein heavy chain 6, axonemal | F:GGGAGGTCTTACGAAGTGGA R:CGAGGAGTTCGGAGTAGCTTT P:ACCCTACCGACTGTCGCAGAGAATG | 97 | 0.271 | 0.273 | C | 0.384 | 0.345 |
| ApjSNP007_AG | Sodium-dependent phosphate transport protein 2B | F:ACCTTGGTGGCAGATATGGA R:TTCAGTGTCCGCAGATTTCTT P:AGTTGAATTAACGGCTCTCGAACCAG | 75 | 0.252 | 0.432 | G | 0.287 | 0.418 |
| ApjSNP008_GT | Testis-specific serine/threonine-protein kinase 1 | F:CCACAATTAGCGATGGGTTT R:CAAAGCTCCAGGACTTCTGC P:GTTGTAGTGAACGCATTGGTTTAGGAAGGAAAC | 102 | 0.349 | 0.488 | G | 0.410 | 0.058 |
| ApjSNP009_AC | Disintegrin and metalloproteinase domain-containing | F:CTAAAGGGGATCACCACGAC R:ATAAGCAGGCTTCCCTTTCG P:ACCTTTCGCCGGCCACGCCCT | 94 | 0.208 | 0.289 | A | 0.104 | 0.365 |
| ApjSNP010_GT | Zinc finger protein 62 homolog | F:CCACCAGATGTCTTTGATTCG R:TCACGACCAATACTGCTTGG P:AGATCCGACCCATGCAAGACCAAGGT | 106 | 0.428 | 0.512 | G | 0.448 | 0.552 |
| ApjSNP011_AG | Kelch-like protein 9 | F:CAGTCAGCCTAGCCCTACCA R:TCGTTGACCTTTGGTACTGATG P:GTGCAAACCAATCGCAAGTCATTGTCGT | 93 | 0.500 | 0.498 | G | 0.458 | 0.574 |
| ApjSNP012_CT | TATA box-binding protein-associated factor RNA | F:CCTTCACTGGTATGGCATGTT R:TGATCCATGTAGGGAGGCTTT P:GTACATTAACTCTCCACAAGCTCCCTTGTA | 88 | 0.314 | 0.468 | T | 0.240 | 0.045 |
| ApjSNP013_AT | Protocadherin Fat 3 | F:TGTTAGCACCTCTATCAAGGATGA R:TTCCATACCTCCTGCCAATC P:GTTCAAGGACACTTGATGGAAAGTGTAATGATT | 102 | 0.500 | 0.454 | A | 0.454 | 1.000 |
| ApjSNP014_GT | Seryl-tRNA synthetase, mitochondrial | F:ATTCGTGTCCAGTTCGCAAT R:GAGATCGGGCGATATAACCA P:TCATATCAATTTGTGCCTCGAGGATCGAC | 96 | 0.271 | 0.276 | G | 0.386 | 0.346 |
| ApjSNP015_CT | Creatine kinase, flagellar | F:TCACAGGCCATCGATCATAC R:CCTTTTCACCAACCTCTCCA P:TCTAAGAGGTGCTGGTGCCCAGTAC | 92 | 0.436 | 0.502 | C | 0.446 | 0.556 |
| ApjSNP016_AG | Fibrinogen-like protein | A F:AATGGCCTCAAGAAAGTGGA R:TCCAGTACCTAGATTTGAAGGACA P:GAATTCATGTGGAGTGAGCATCTTGGAAT | 108 | 0.430 | 0.583 | A | 0.483 | 0.497 |
| ApjSNP017_GT | Abhydrolase domain-containing protein 14B | F:CGGGGTCTACCTCATACAACC R:CCTCCGCCATCTACAGTGTT P:CATATATGGAGCCATTTGCTGTATATTGTAACATG | 78 | 0.293 | 0.444 | T | 0.475 | 0.854 |
| ApjSNP018_AG | Apolipoprotein A–I-binding protein | F:CATAGGTGTCCAGAAATGTTCG R:TGTCCCATGTCTAAAGCATAACTG P:CACAGAGTTCCCATGGGCAGATAGAAG | 93 | 0.073 | 0.083 | G | 0.083 | 1.000 |
| ApjSNP019_AG | N-acyl-phosphatidylethanolamine-hydrolyzing | F:CGTGCTCGGTTTTAATGTTG R:CATGGTGAAACCTGGTAGACG P:CCAAGCACAACCAGAACCGAGAAATCCA | 91 | 0.688 | 0.505 | A | 0.354 | 0.498 |
| ApjSNP020_AT | Polypeptide N-acetylgalactosaminyltransferase 11 | F:AAAGAGGTATCGACCTTGTCCA R:TGCTCGGACTGTATGTTCATC P:TGGAGGAACTTCCAGAAATCAATGCTGAG | 109 | 0.250 | 0.256 | A | 0.328 | 0.857 |
| ApjSNP021_AG | Hyalin | F:TTCAAGTGGTATCACGAAAACG R:CGTGCTATTGCCTTTGGATT P:GCTGAGGCTTCCAAAAGATGACGATTC | 92 | 0.108 | 0.333 | A | 0.290 | 0.557 |
| ApjSNP022_CG | Transmembrane protein 129 | F:TGGAATGCCACTAACACCAA R:TTGACACCACACCACCAATC P:TTGATATGTCTGCTGGGCTATTCTGGTA | 80 | 0.442 | 0.364 | G | 0.483 | 0.381 |
| ApjSNP023_CT | Mediator of RNA polymerase II transcription subunit | F:GCTGATGAGCAATCTTCACACT R:CAAGTTTCAGACGGGACCTG P:GTCTTGATTATCCACGAATCTGTGACATACCA | 95 | 0.146 | 0.505 | T | 0.489 | 0.051 |
| ApjSNP024_AG | AF339450_1 hillari | F:TCCATTGAACGGAGGACTTC R:CAAACATTTCAGCCTTGTGG P:GTCTGGGATGGGATGTAGTCGACACTTA | 108 | 0.419 | 0.484 | A | 0.395 | 0.376 |
| ApjSNP025_AC | Proteasome subunit beta type-5 | F:TCCAGATCGCTACGGTCTTC R:ACGACCAGGTAGCTGCAGAG P:TGGTGTATCAAGGAAATTCAAACCCAGCTGT | 81 | 0.250 | 0.250 | A | 0.423 | 0.125 |
| ApjSNP026_AG | Dynactin subunit 5 | F:GCCTGTTGCTGTTAACTTTCG R:CTGGCATGTAACTCTATGAAACTC P:GTTAAGTGAAAGTTGACTGCCTCAGTATTGTA | 110 | 0.316 | 0.365 | A | 0.461 | 0.724 |
| ApjSNP027_AG | Apoptosis-inducing factor 2 | F:CAGAGAAAGCTGGAGATGATGA R:ATGATTTCAACTGGGCCATC P:GATGATGAACCGCAGAAGGGTTCGAA | 88 | 0.516 | 0.467 | A | 0.361 | 0.324 |
| ApjSNP028_CT | Uncharacterized protein C6orf163 | F:ATAGTTGGGTGTGGCTTTGC R:CCGATGCAGTGATGGAAATA P:AAATGTCACCTAACTGTGATTGATCCTCGCC | 104 | 0.209 | 0.190 | C | 0.105 | 0.698 |
| ApjSNP029_AT | F-box/LRR-repeat protein 2 | F:CCGTGATCCTAAATGAGGCTA R:CGCTAAGAGTAAGAGAAAGAAGCA P:GCCTAACCATACTGGATTGGCTAGCAGT | 98 | 0.271 | 0.237 | A | 0.135 | 0.762 |
| ApjSNP030_CT | TBC1 domain family member 10B | F:CCGGAGACGTAAAAGCACTC R:TCGTCGTGTCTGGTATCCAC P:AAGTCTGGACAGCTGTTAGCTAAGGGC | 91 | 0.191 | 0.174 | T | 0.095 | 0.754 |
| ApjSNP031_CG | Stejaggregin-A subunit alpha | F:ATCGGTGCTAGACCCAAAGA R:TCCTTCTCTGGTGAATTGATTG P:CATCCCAACGACGGACCGATATGGTA | 81 | 0.150 | 0.245 | G | 0.264 | 0.358 |
| ApjSNP032_AC | Lysine-specific demethylase 6A | F:CGAAGGCAACCAAGTAGGAC R:TGCCACCTCGATCATTTTCT P:CGCTGGTGTTAATAACTTCATAGTCCGTTAC | 91 | 0.138 | 0.833 | C | 0.383 | 0.497 |
| ApjSNP033_AG | ATP synthase subunit beta, mitochondrial | F:GAGTAACAACGGCCCAGAAA R:TACAGTGCCTACACCGGTCA P:GGTCTGACCGCTATTGGGATCAATCTGC | 76 | 0.458 | 0.467 | A | 0.232 | 0.854 |
| ApjSNP034_GT | Ubiquitin carboxyl-terminal hydrolase 8 | F:GGCTTGAAGAAACATGGGTAA R:CCAGTAGATTGCATCTTTCCATC P:TCATGTTCACTTCTTTATACCACACGATGACAT | 110 | 0.292 | 0.314 | G | 0.035 | 1.000 |
| ApjSNP035_CT | Uncharacterized protein C7orf26 homolog | F:CGGTGGTGAGGTGTCTACATT R:GGAATAGGCAACTCGAGGAA P:GTCGGTGAAGTACGAAGCCTTCATGAA | 76 | 0.449 | 0.367 | T | 0.485 | 0.498 |
| ApjSNP036_AC | hypothetical protein | F:AAGATGCCAGACAGCAACAA R:CATGACTGCGTCTTCTGCTC P:CAGGAATCTCACAGACGAGAGGGAACT | 100 | 0.545 | 0.413 | C | 0.264 | 0.857 |
| ApjSNP037_AG | DNA replication licensing factor MCM8 | F:GGAACCGGAGAGATGACAGA R:CCAGCGTCGTCACCTTTTAC P:AGAGCAAGATCAACAGAATGAGGACAAAGTA | 95 | 0.492 | 0.502 | A | 0.458 | 0.557 |
| ApjSNP038_AG | LRP2-binding protein | F:GATGAAAGTACCTGGGAGGAA R:AGCTGATCATCGGTCCATCT P:GGAGATTGAAGATTGATCCCACTGACAAACTC | 83 | 0.750 | 0.625 | G | 0.147 | 0.381 |
| ApjSNP039_AG | Endoplasmin | F:ATAACGTCGGACGAGCATTC R:AGCAACCACCATCTCTCTGC P:AAGGGTTTGGAGTAAAACAGTCGGATGCCC | 76 | 0.409 | 0.479 | G | 0.387 | 0.051 |
| ApjSNP040_CT | heat shock protein 90 kDa beta | F:CTTTGAAGATATGATGCCCAAG R:TTGTGTTGCTGCAGGGTTT P:ACTCCGATGACCTGCCTCTCAATGTGA | 102 | 0.348 | 0.291 | C | 0.174 | 0.084 |
| ApjSNP041_CT | Titin | F:AGCCATCGAGAATGAGAAGC R:TGATGGTCTGTTCGATCCAC P:GGTCACCGACTACGACAAGATCTCCTGC | 82 | 0.382 | 0.314 | T | 0.192 | 0.091 |
| ApjSNP042_AG | Midasin | F:CAGCCTGGAAGACCCTCAGT R:TTGGACTTCCACCATCAGAA P:AACCAGGCTACGATTTCATGGACCGGT | 88 | 0.800 | 0.691 | G | 0.291 | 0.635 |
| ApjSNP043_CT | Scavenger receptor cysteine-rich type 1 protein M130 | F:GGTTCACAACCTCAGGATGAC R:CTTCTGCACACCGCACTTT P:GAAATTACAACCTGCTTTAGTGTCCAGAGATAG | 95 | 0.317 | 0.505 | C | 0.476 | 0.200 |
| ApjSNP044_AC | FK506-binding protein 15 | F:TCATACACTCAGGGCATCCA R:GCGTAGGCATATGACGAGAGA P:CAGTTTTGTGAGTGTCTTGACAGTGATAGTGG | 90 | 0.583 | 0.473 | A | 0.332 | 0.149 |
| ApjSNP045_AC | Titin | F:CGTTGAGATCCAAGTCAATGAG R:TGTAGGTGAGTGGTGAACGTG P:TAGAAAGAATGGACAGCGTCCCTGGAGT | 105 | 0.512 | 0.502 | A | 0.456 | 0.897 |
| ApjSNP046_AG | Radial spoke head protein 4 homolog A | F:GGGGAAGATGAGGTAGAAACG R:GCTCATACCGATTCCTGCTT P:ACTCCCAAACCTACCGGAACTTATGTTTTAGA | 81 | 0.113 | 0.109 | G | 0.056 | 0.623 |
| ApjSNP047_CT | Phenylalanyl-tRNA synthetase beta chain | F:TGGCAAATCAATCGGATTCT R:AACGGTTCAATGGTTATCTCTAGG P:CTCAAAGTTTGAGCTTCCAAACCCATGTGGA | 102 | 0.326 | 0.300 | T | 0.178 | 0.653 |
| ApjSNP048_AG | Mitochondrial inner membrane protein | F:CCGATGAGAGGGGTATTCAA R:CCCCCATTCTCGTCTATCAG P:GGGAGAGGTGGGAGAATATCCAGAGATA | 98 | 0.222 | 0.468 | A | 0.361 | 0.002 * |
| ApjSNP049_CT | Sulfotransferase family cytosolic 1B member 1 | F:CCAGGGTAAAGTCAAAGGTCA R:ACTGTAGCCCAGAACGATGC P:TCCTTTCATTTTCCCCTCGTACAAGTCATGT | 82 | 0.524 | 0.479 | T | 0.278 | 0.401 |
| ApjSNP050_CT | RalA-binding protein 1 | F:GGTTGAGGAGTTCTTGGGAGT R:CATCAGCATGATCCAACACA P:CTGAATGATTTGCCAACTTGTAACTACACCTTAGA | 105 | 0.250 | 0.408 | C | 0.275 | 0.018 |
| ApjSNP051_GT | Alpha-amylase B | F:TTCGATTCATCTGGTGCTTG R:CTTGACCTTCGCAGGTGTTT P:TGGAGAGAGATCCGTAACATGGTCGAATTGT | 107 | 0.096 | 0.481 | T | 0.390 | 0.005 * |
| ApjSNP052_GT | Putative vitellogenin receptor | F:CAGTCTGAAAGAACCACTGAAGA R:CGAGTATAGGAGGCTGAAAACG P:GCCCAGAAGATATCGCCTCTCTTCAAATAGG | 98 | 0.411 | 0.485 | G | 0.400 | 0.758 |
| ApjSNP053_CT | UDP-N-acetylglucosamine--peptide | F:TCGAAGCTAGATTACTGTGAGCA R:TCTGAAGGAGATGCAGGACA P:TGATTTGGATGGCTCTGGTATAGCACTCA | 101 | 0.071 | 0.503 | T | 0.404 | 0.000 * |
| ApjSNP054_CT | Kanadaptin | F:CAAGCCGTACATGAAAGCAA R:TGTCCAGGTACGAGTCATCG P:AGAAGAAGAAGAATTGGGCGGACGATCT | 88 | 0.585 | 0.506 | C | 0.489 | 0.307 |
| ApjSNP055_GT | Epidermal growth factor receptor | F:TCACGTTCCACCAGATTTTG R:ATGATGGGGGTAATGGCATA P:TGACCAATAGCATATTCGATGTGATGTCACCA | 104 | 0.253 | 0.435 | G | 0.424 | 0.518 |
| ApjSNP056_CT | hypothetical protein | F:ATGCCACCCTCTTAATCTGG R:CTTGCCTGGGTTTTCCATAC P:TCAGACCGGTGCTTCTGACAGTACATT | 107 | 0.125 | 0.117 | T | 0.318 | 0.442 |
| ApjSNP057_CG | RuvB-like 2 | F:CCATAACACCGATGACACCA R:GAAGCTGATAAGATGGAAGTAGCC P:CATTGTCAAGGCAGTCATCTTGTCAGGA | 108 | 0.295 | 0.388 | C | 0.258 | 0.159 |
| ApjSNP058_AG | Eyes absent homolog 1 | F:CGTATCCCGTACCACAACCT R:AACCCGTAGGGAACCTGACT P:GGTGTGCAACCAAACGCTGGGTACGG | 79 | 0.400 | 0.501 | G | 0.247 | 0.485 |
| ApjSNP059_CT | WD repeat and FYVE domain-containing protein 3 | F:TTCCAGGGATTTGACAGAGG R:TGGCATCTAAAGCTGCTAGTCT P:TCCAGGAGAGATCCTAGGGTGTACTGGG | 110 | 0.530 | 0.500 | C | 0.446 | 0.984 |
| ApjSNP060_AT | similar to LOC398543 protein | F:CCACTACACATCGGTGACCA R:CATCTCCTTCCGATAACACAGTT P:AGATGAAGAATGTATTATTAACGCTGCACACT | 110 | 0.095 | 0.433 | A | 0.309 | 0.008 * |
| ApjSNP061_CT | Coiled-coil domain-containing protein C6orf97 | F:GCTGTTGCCGATGAAACAAT R:CAAATTGAACGAGATGGAGACA P:AGAATATCCTGCCTTGGGATAACGTAAACC | 110 | 0.479 | 0.447 | T | 0.329 | 0.489 |
| ApjSNP062_CT | Uncharacterized gene 48 protein | F:CAGAAGGATAAAGTCCAAGAGACC R:TTCTCCTTTCTGTCCATCCTG P:ACAGGCCTATAGCTACGATCAGGAATCG | 86 | 0.182 | 0.220 | T | 0.198 | 0.809 |
| ApjSNP063_AT | Uncharacterized protein C2orf73 homolog | F:CACATGTGTCACCTCTGGCTA R:ACTGGAACAGCGCCTTTAGA P:CAGCTCAAACCCTCACAACTATGCAAG | 73 | 0.479 | 0.586 | A | 0.311 | 0.252 |
| ApjSNP064_AT | Methionine synthase | F:TCGATACCCTTCACCAAAGAAT R:CGAGGGTCTTGGGAAAGGA P:CCAGGCTTCATCATCAACAGCTTTCTAA | 103 | 0.486 | 0.495 | T | 0.432 | 0.654 |
| ApjSNP065_CT | Tubulin alpha chain | F:CATAGCTTCGGTGGTGGAAC R:GCTTCGATTTCTTGCCGTAG P:GGATTTGCAGCTCTACTTCTTGAACGCG | 85 | 0.061 | 0.091 | T | 0.091 | 1.000 |
| ApjSNP066_AG | TATA element modulatory factor | F:TGGTGCTCAGCTGAATCTGT R:TGGTCTCTTCGTGAGCCTCT P:GAAACAACAAGACAACCTCGAGAGGCTT | 86 | 0.415 | 0.100 | G | 0.321 | 0.007 * |
| ApjSNP067_AG | TATA element modulatory factor | F:GCAACTGGAGGCAGAGAGAG R:GGCCTGCTCGAGTTTACCT P:AGAGACCAAGGAAGAGCTGGAAGAGAA | 79 | 0.315 | 0.400 | A | 0.206 | 0.486 |
| ApjSNP068_CT | Uncharacterized protein KIAA1704 homolog | F:TGACACCTATGGACCGTCTCT R:GGAGGTAATGGTGGACCAAA P:GGATTCAAAGGTGTCGACAAAGAGTCTGAAC | 90 | 0.412 | 0.504 | C | 0.477 | 0.135 |
| ApjSNP069_CT | WD repeat-containing protein KIAA1875 | F:GGGTCTTCCAGCCAATGATA R:ACCACGGCTACGTTTGAGTC P:TACTGGTTGATCGCTCTGGAAGAAACAGGA | 103 | 0.326 | 0.225 | C | 0.471 | 0.390 |
| ApjSNP070_AG | Glycoprotein 3-alpha-l-fucosyltransferase A | F:CCAGGAAGGGGTAGACTTGC R:ATCTCGCCGTTCAAGTTGTT P:CTCAGGAAGTTCTAGAGAGGAAGGATGTC | 102 | 0.528 | 0.469 | G | 0.334 | 0.051 |
| ApjSNP071_AG | N/A | F:CGAAACTATAGTGACCTCTTGGTTA R:CAAGCCCTAGTCTCTTCATTCG P:CAGAATTTCTCTCGAAGTCCTTTGCCAG | 104 | 0.364 | 0.470 | A | 0.364 | 0.189 |
| ApjSNP072_AG | N/A | F:GAGTTAGACCCTCGGCTAGGTA R:GCAAAGAGCCTAGCCTTTAGGT P:TGCATCAGTACTAGCAGCATGGAAAACT | 87 | 0.388 | 0.412 | G | 0.333 | 0.247 |
| ApjSNP073_AG | N/A | F:AAATGTACAGACCCGCATGA R:CTGGAAAAACAGTGTGAACCAA P:TGTAAAATTAATGAGCCGTTCGAACCAAGAG | 107 | 0.225 | 0.309 | A | 0.188 | 0.104 |
| ApjSNP074_AT | N/A | F:GATGGTGAAAATCACGGAGAA R:TTCTATGTCTTGTTGATGCAGAGAC P:CACAATAACCTGGAAATATCAACCTTAGAAGAATTCA | 103 | 0.300 | 0.404 | A | 0.275 | 0.108 |
| ApjSNP075_AT | N/A | F:GACCACGATGACAGCCAGTA R:CTCGCCAAGTCAGGAAAAAG P:AGGATCGTCATTCGGGCACTCTTGG | 95 | 0.630 | 0.879 | T | 0.450 | 0.328 |
| ApjSNP076_CT | N/A | F:AACTCTCGATGGAATGCAAAG R:AACAGACTCGGTCGCATCTC P:GATAGTTCTGACAGCGATTTAGGAGACTAA | 108 | 0.175 | 0.392 | C | 0.263 | 0.001 * |
| ApjSNP077_CT | N/A | F:AACCATCCTGTAGCGAAACC R:CGGGGACGAGGATATTGTTA P:GTGTTGAATGAAGTCGTTCGCGTAAATGC | 103 | 0.175 | 0.339 | T | 0.213 | 0.004 * |
| ApjSNP078_GT | N/A | F:GCCAAGCAACATACAGAAGGA R:TAGTTGGGCTGTCTTGCTGA P:TTGCTGCATTAATGTTTAGATGATGATGTGTCT | 87 | 0.563 | 0.907 | T | 0.487 | 0.637 |
| ApjSNP079_AG | N/A | F:TGGGCAGAAGAAAATTTGGA R:GAGTGGCACATGACTTGGTG P:CTGCAATTGGACAACCCCATGCTCAT | 99 | 0.475 | 0.469 | G | 0.375 | 0.084 |
| ApjSNP080_CT | N/A | F:GGGCGCTATCAGACTTTGAC R:GCACCCTCTATTTTAGCTGTTCA P:TCTTGCTAGCTAATGGGAAAGAACGTTAT | 110 | 0.200 | 0.292 | C | 0.175 | 0.062 |
| ApjSNP081_CT | N/A | F:CTGGTTGCAATAGGTTATTTGG R:TGAATACATGCCGTTTCTGA P:GTTGGATTCAGAACACAGACTGCCATTCC | 103 | 0.075 | 0.073 | C | 0.038 | 0.780 |
| ApjSNP082_CT | N/A | F:CAGAAACGGCATGTATTCAAAC R:CCCGACCACAAGGAAAGATA P:AGGGGAGTTTGTGATGACAAATTGTTGCAG | 94 | 0.500 | 0.404 | C | 0.275 | 0.098 |
| ApjSNP083_AC | N/A | F:CACGATGCCCTGTGTGTAAT R:GTCGGCCTCCTGACTAACAG P:GCGCAGCAGAAACGGCGTGGA | 108 | 0.325 | 0.453 | C | 0.338 | 0.073 |
| ApjSNP084_CG | N/A | F:GGGTGGTGCATTTTCTTCAT R:TGGCTTCAGTTACACCATCCT P:ATCCTTGTGGTCGCCTGATCTTGTGTT | 75 | 0.150 | 0.444 | G | 0.325 | 0.000 * |
| ApjSNP085_AG | N/A | F:CGTCATTCGCTCCAAATACC R:GTCGTAGAGAGACATAACGATAACTGA P:CCATAATGCATAGTGGCTGCAGCATAA | 110 | 0.833 | 0.896 | A | 0.487 | 0.093 |
| ApjSNP086_AG | N/A | F:CGACAATATACTACAAATGCCCTGT R:GATGATGAATGGGTTGTTTGTG P:CAAGGCGAGTTCGTCACACGAAAAGT | 83 | 0.050 | 0.461 | G | 0.350 | 0.000 * |
| ApjSNP087_AC | N/A | F:CACTCTGGCCTTGCACTCTT R:TGTGAGAACAATAGGTTCACAGGT P:GGGCAAACTGATGTCATGTTCACAGGTATGT | 109 | 0.450 | 0.353 | C | 0.225 | 0.252 |
| ApjSNP088_AG | N/A | F:ATGAAGCATGCGTGAATGAG R:CGATTTCACTGCTGTCATCAA P:AACTGTGGAGATGGTAACATATTCTATGAAGAGAA | 83 | 0.250 | 0.222 | G | 0.125 | 0.256 |
| ApjSNP089_AG | N/A | F:TGGTGAGAAGCATCCACAGA R:GTTGTTTTGAAGGCACTGATGA P:AAGTTCTTAAATGCAGAACTGGGTCAGAACA | 93 | 0.325 | 0.468 | A | 0.363 | 0.051 |
| ApjSNP090_CT | N/A | F:TTGTACCGAGAAAGGGATGTTT R:CCTGAACAACATCTGCCTGA P:AGAGTATATTTCAAACGAAAACGGGAGTAGGGT | 110 | 0.161 | 0.373 | T | 0.242 | 0.002 * |
| ApjSNP091_CT | N/A | F:TGCGTCATTCTAACCAACCA R:AACACTTATGTAGGCGAGTCTTGA P:CAAAGCGCTTCATTTTCACAGCAACTA | 102 | 0.200 | 0.380 | C | 0.250 | 0.004 * |
| ApjSNP092_CT | N/A | F:TGACTGGACGTCAGATGTGG R:GTGGGCTTCCAGACACAGAT P:GGTTGCATCAAGGTCCCTGGGTACATACA | 81 | 0.075 | 0.073 | C | 0.038 | 0.780 |
| ApjSNP093_AG | N/A | F:TGAAATGTGGTGTGACTTGC R:TGTGTGACTTCAGCATCTCTGT P:GAATTGTATAATTGGATGCTGTGTGTCACTTAT | 80 | 0.222 | 0.282 | G | 0.167 | 0.227 |
| ApjSNP094_GT | N/A | F:TCTGCTAAGTTGTTGAGAGGATG R:CGAACGGTTGGTATTTGTGA P:TTCTGGTCACTTGCCCCAGGTTCCAC | 108 | 0.171 | 0.358 | T | 0.229 | 0.003 * |
| ApjSNP095_AG | N/A | F:ATTTGCGGCTCTTCTGTTCA R:TGAAGTGAACTCACCCACGA P:AAACTTGGCAACGAAGACGTCAGCAT | 110 | 0.225 | 0.367 | A | 0.238 | 0.018 |
| ApjSNP096_CT | N/A | F:TCATTCCTGTATTGCTACTACTCTGTG R:TGTGGTATGCCCATCGATTT P:TAAACAATAGTACTTAATGGCATTGAAGACAACAAAC | 109 | 0.333 | 0.491 | C | 0.409 | 0.060 |
| ApjSNP097_CG | N/A | F:CACAGTGATGTGTATGTACGTTCG R:GACCTTCGCTTTGTGCCTAC P:ACACACCGTATATACCGAATCTGGAAATTATCTT | 94 | 0.316 | 0.337 | C | 0.211 | 0.698 |
| ApjSNP098_CT | N/A | F:CTGTGTCAGAGAGGAAGAGTGC R:CGAAAGCTATTTCAAACCCAGT P:GGGTACTATCAAAATTGACTCACAAAGCGAC | 107 | 0.158 | 0.147 | C | 0.079 | 0.512 |
| ApjSNP099_AG | N/A | F:GACCTTCTGCTCTGCCTGAC R:CGGATATCAACAAACCAGAGC P:TCCTCATCTTCGGTGTCTTGCGAAC | 97 | 0.075 | 0.162 | G | 0.088 | 0.080 |
| ApjSNP100_GT | N/A | F:TCCACTGAGCCATCCTGATT R:GAAGAAAAACATGTCCCGATG P:AGTGGCTCCCCCTGGAATGTAATCCTG | 103 | 0.505 | 0.547 | T | 0.458 | 0.279 |
| ApjSNP101_GT | N/A | F:CTGCTGAAGTATGACAACATTAGAGAC R:CTAGTACTTTCTTCTTCAGTAGTTGG P:CTATTGAAAGCTCGATAGGCACATCCTG | 109 | 0.075 | 0.240 | T | 0.138 | 0.000 * |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Du, H.; Bao, Z.; Yan, J.; Tian, M.; Mu, X.; Wang, S.; Lu, W. Development of 101 Gene-based Single Nucleotide Polymorphism Markers in Sea Cucumber, Apostichopus japonicus. Int. J. Mol. Sci. 2012, 13, 7080-7097. https://doi.org/10.3390/ijms13067080
Du H, Bao Z, Yan J, Tian M, Mu X, Wang S, Lu W. Development of 101 Gene-based Single Nucleotide Polymorphism Markers in Sea Cucumber, Apostichopus japonicus. International Journal of Molecular Sciences. 2012; 13(6):7080-7097. https://doi.org/10.3390/ijms13067080
Chicago/Turabian StyleDu, Huixia, Zhenmin Bao, Jingjing Yan, Meilin Tian, Xiaoyu Mu, Shi Wang, and Wei Lu. 2012. "Development of 101 Gene-based Single Nucleotide Polymorphism Markers in Sea Cucumber, Apostichopus japonicus" International Journal of Molecular Sciences 13, no. 6: 7080-7097. https://doi.org/10.3390/ijms13067080