A Computational Study on Thiourea Analogs as Potent MK-2 Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
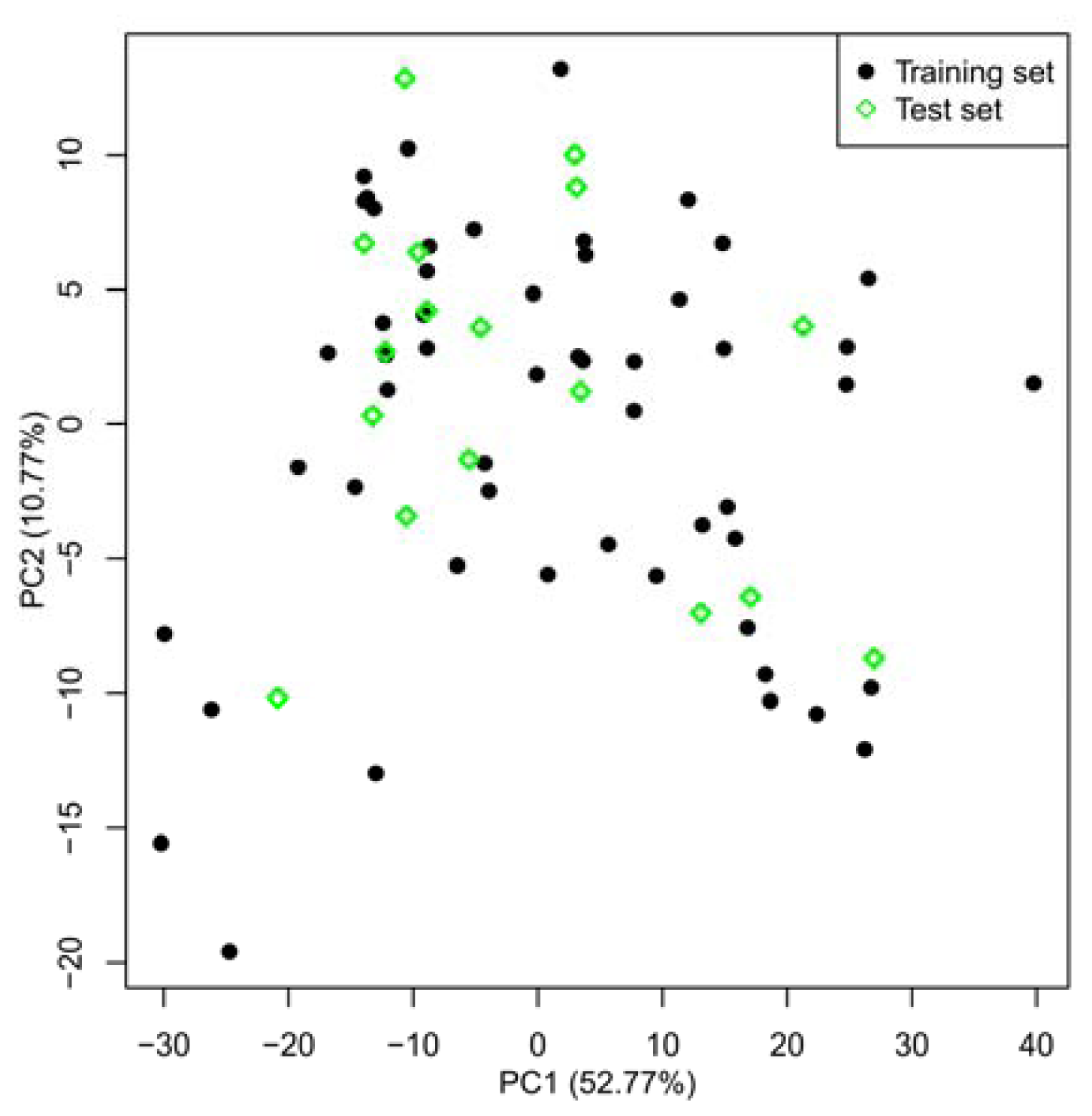
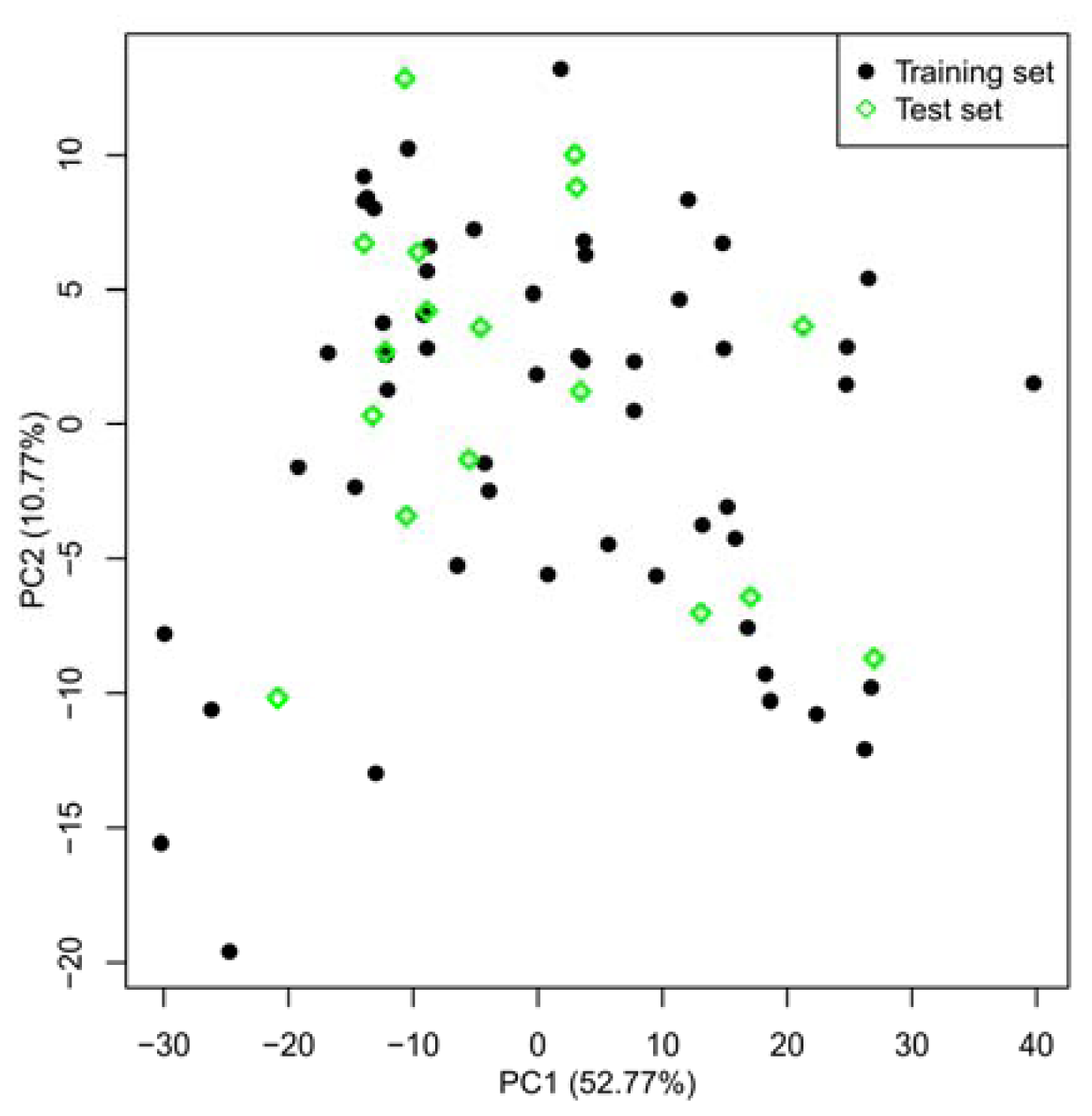
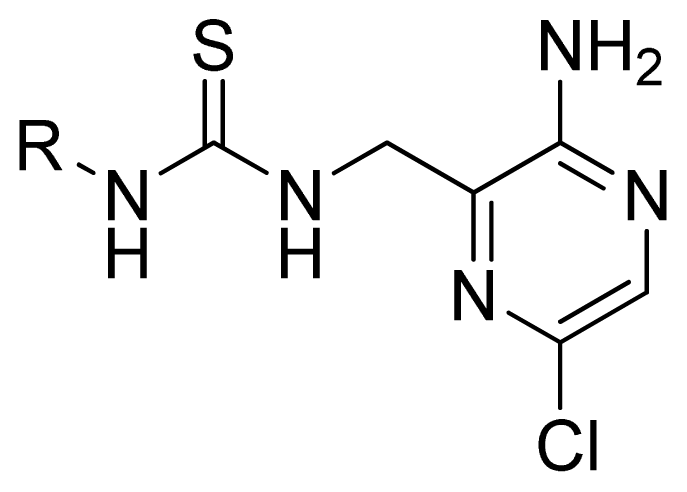
2.1. Selection of Training and Test Sets
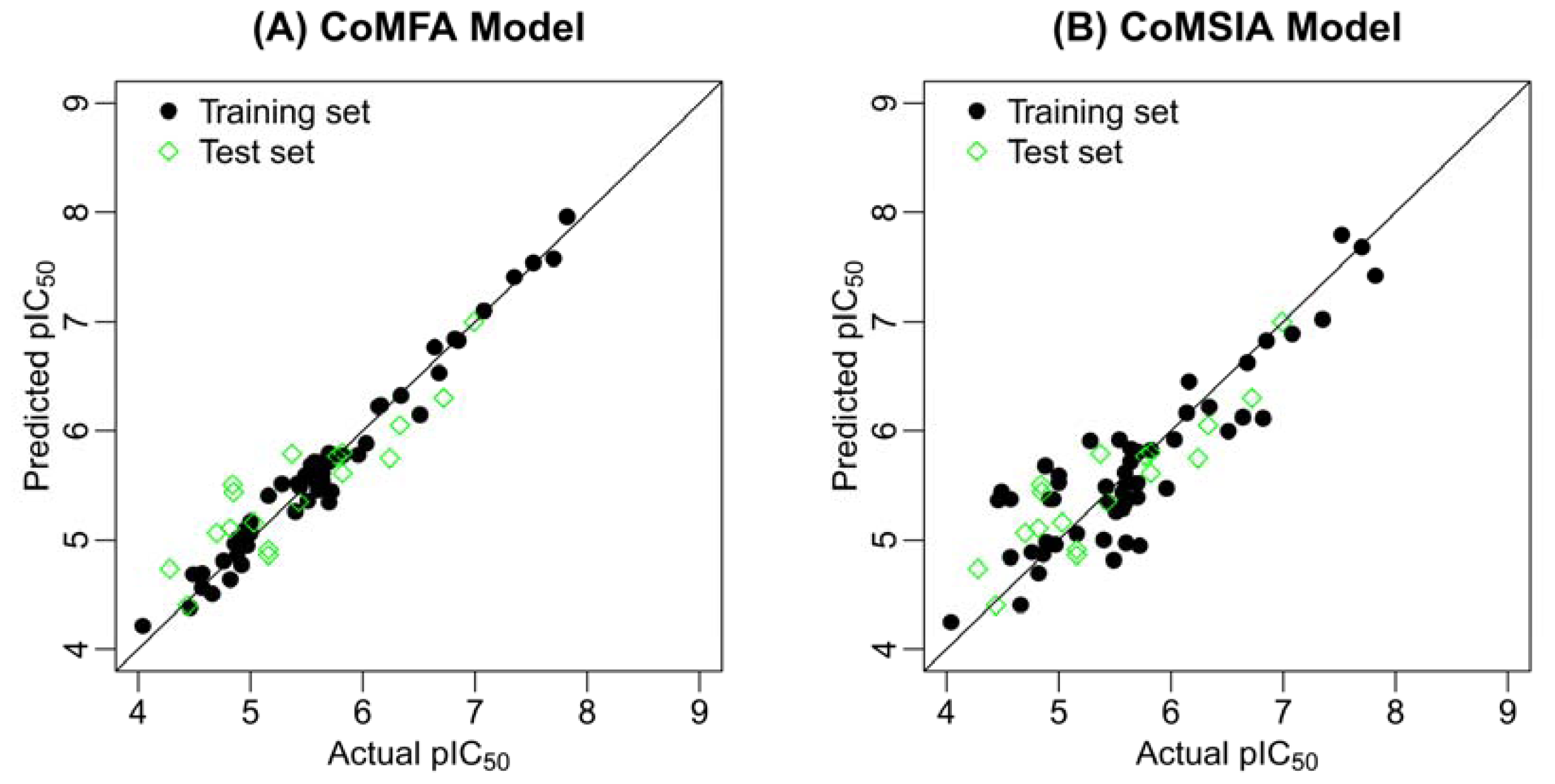
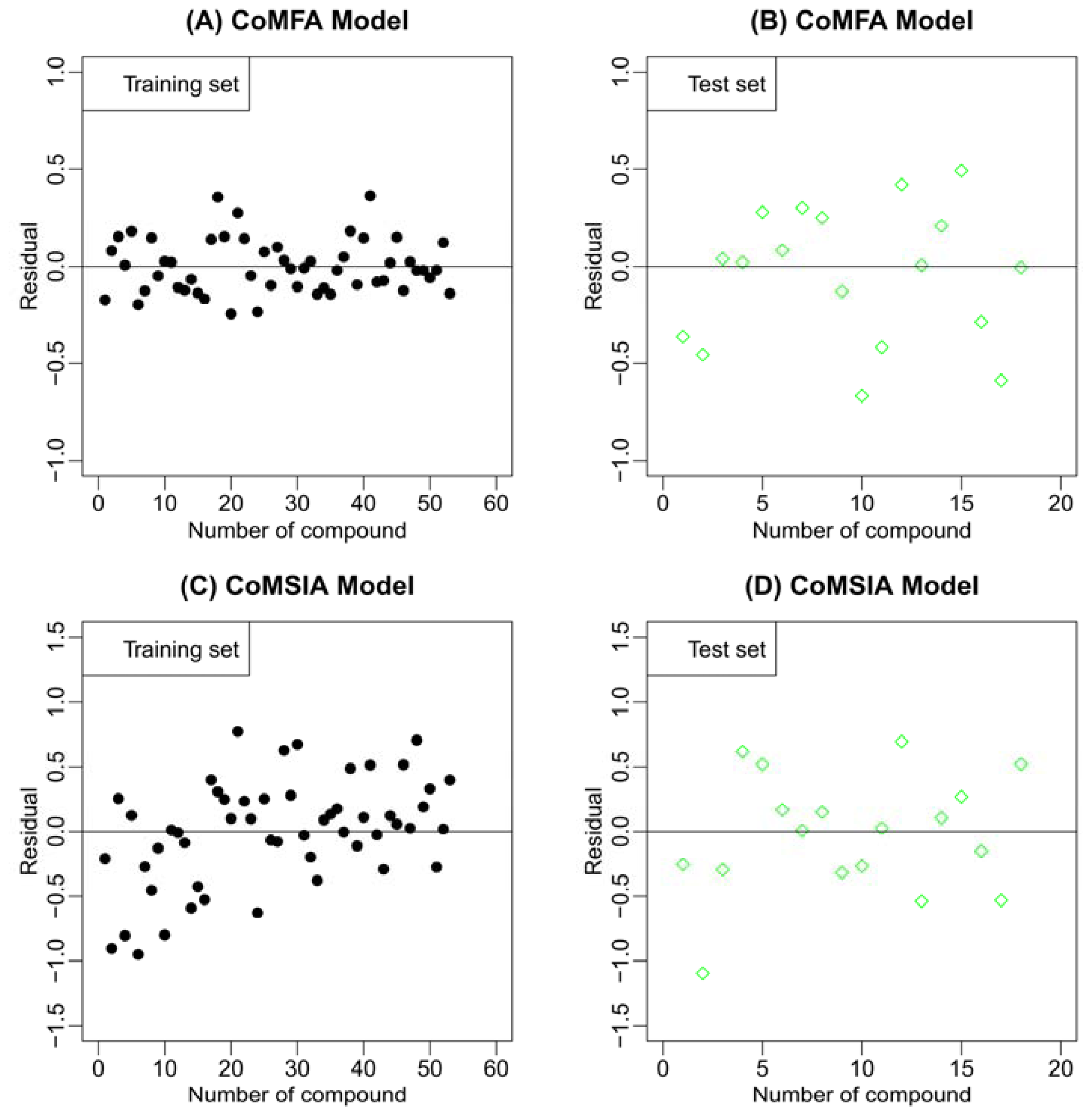
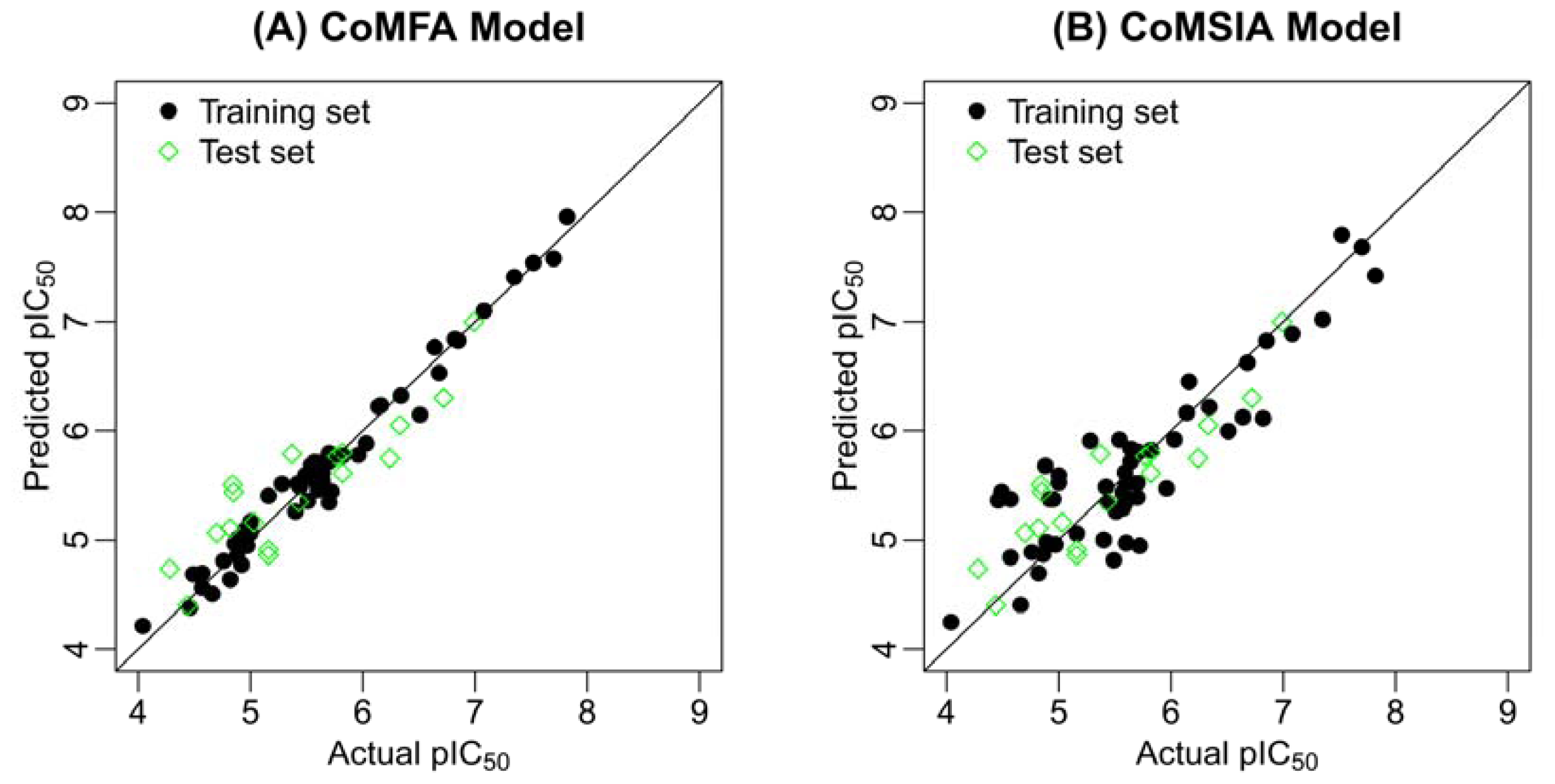
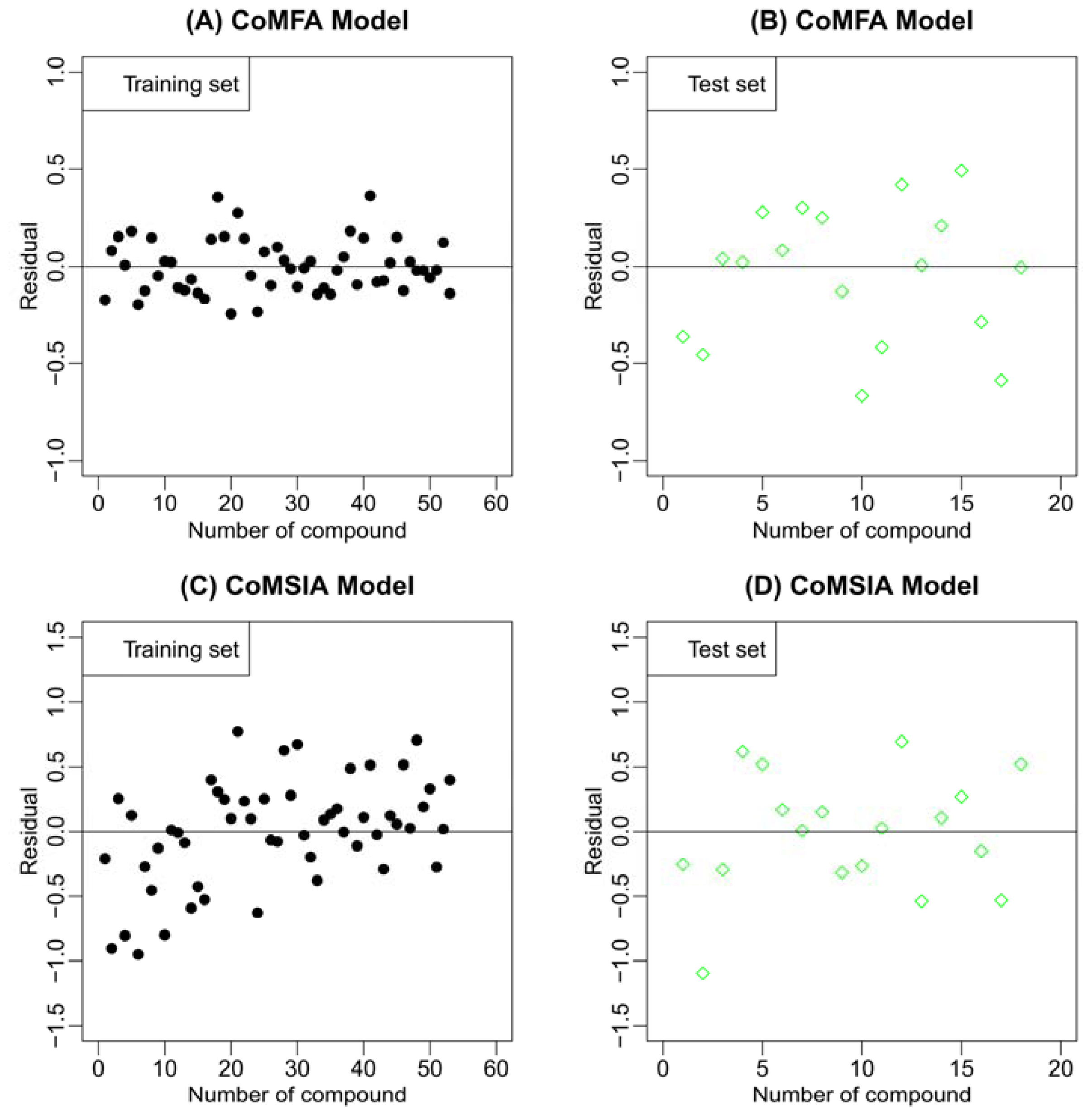
2.2. Results of CoMFA and CoMSIA
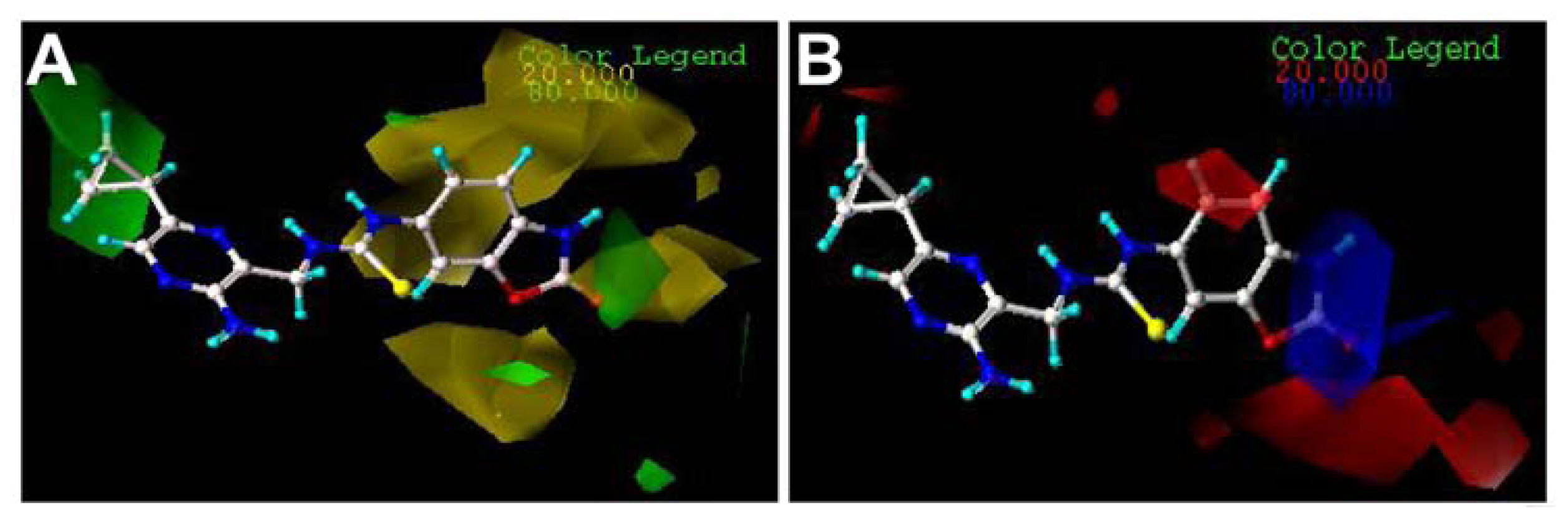
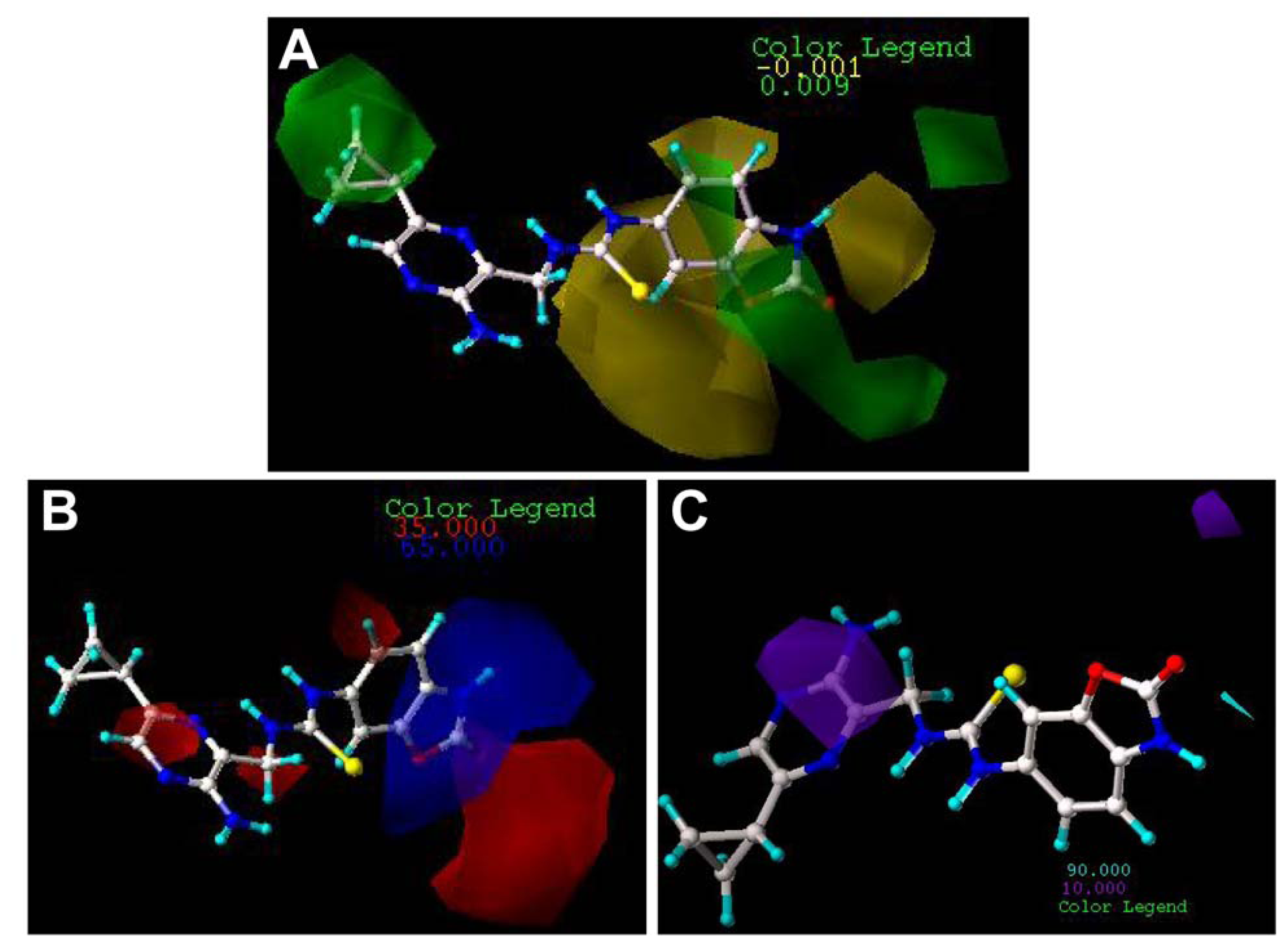
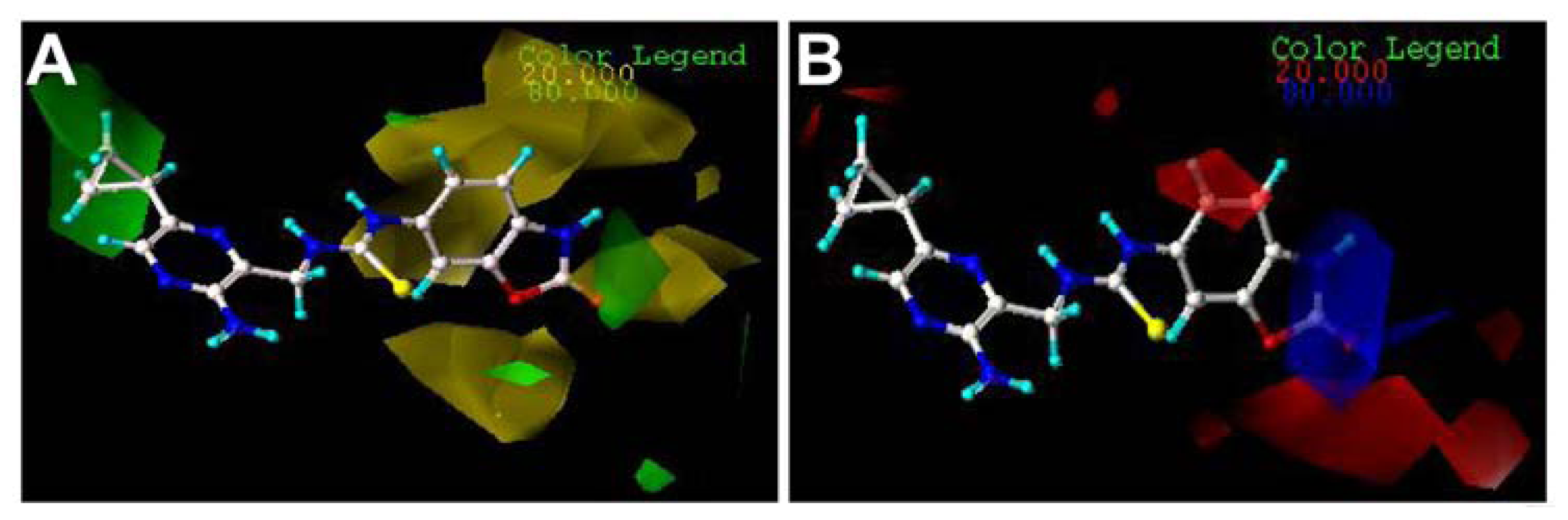
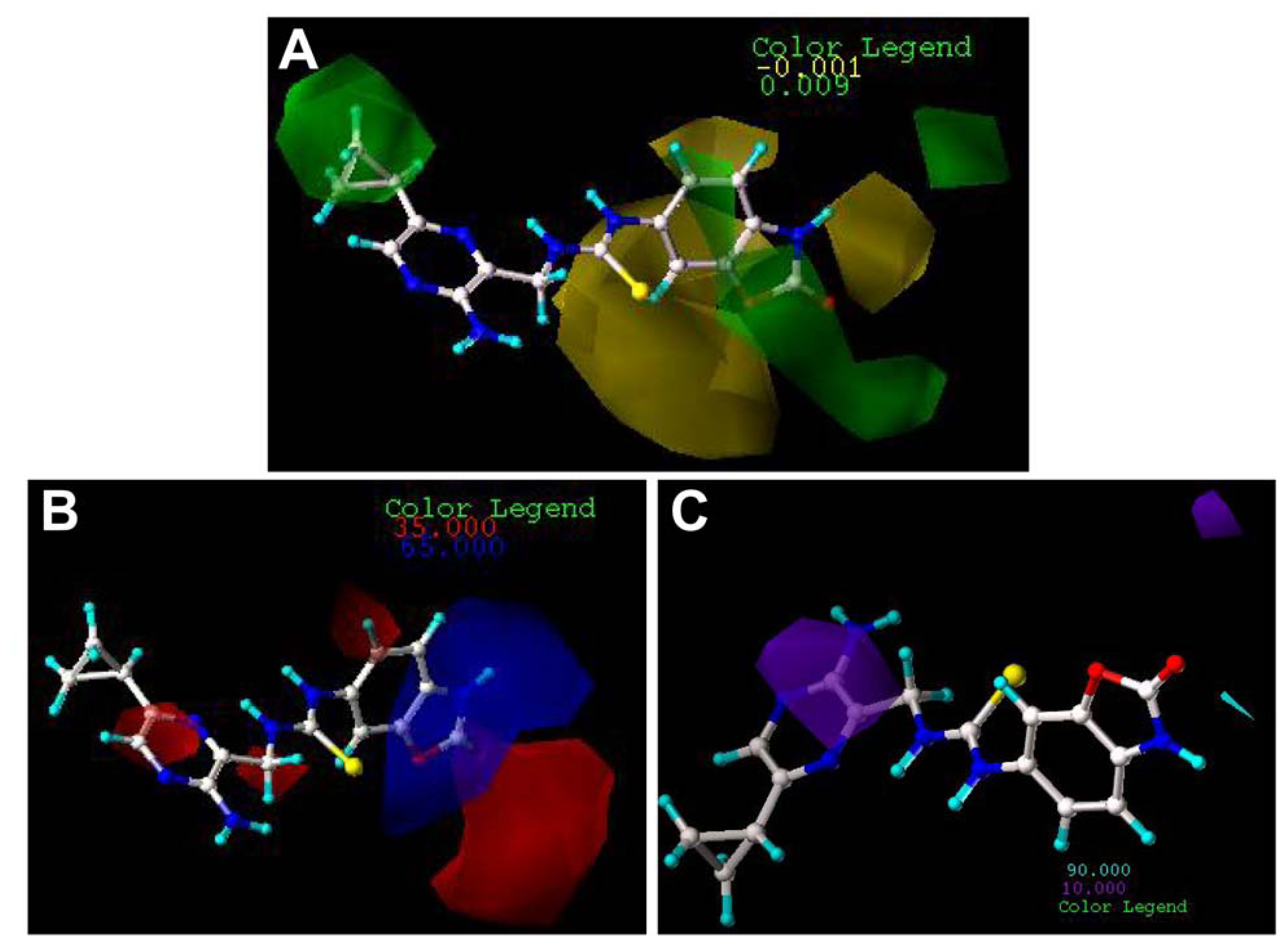
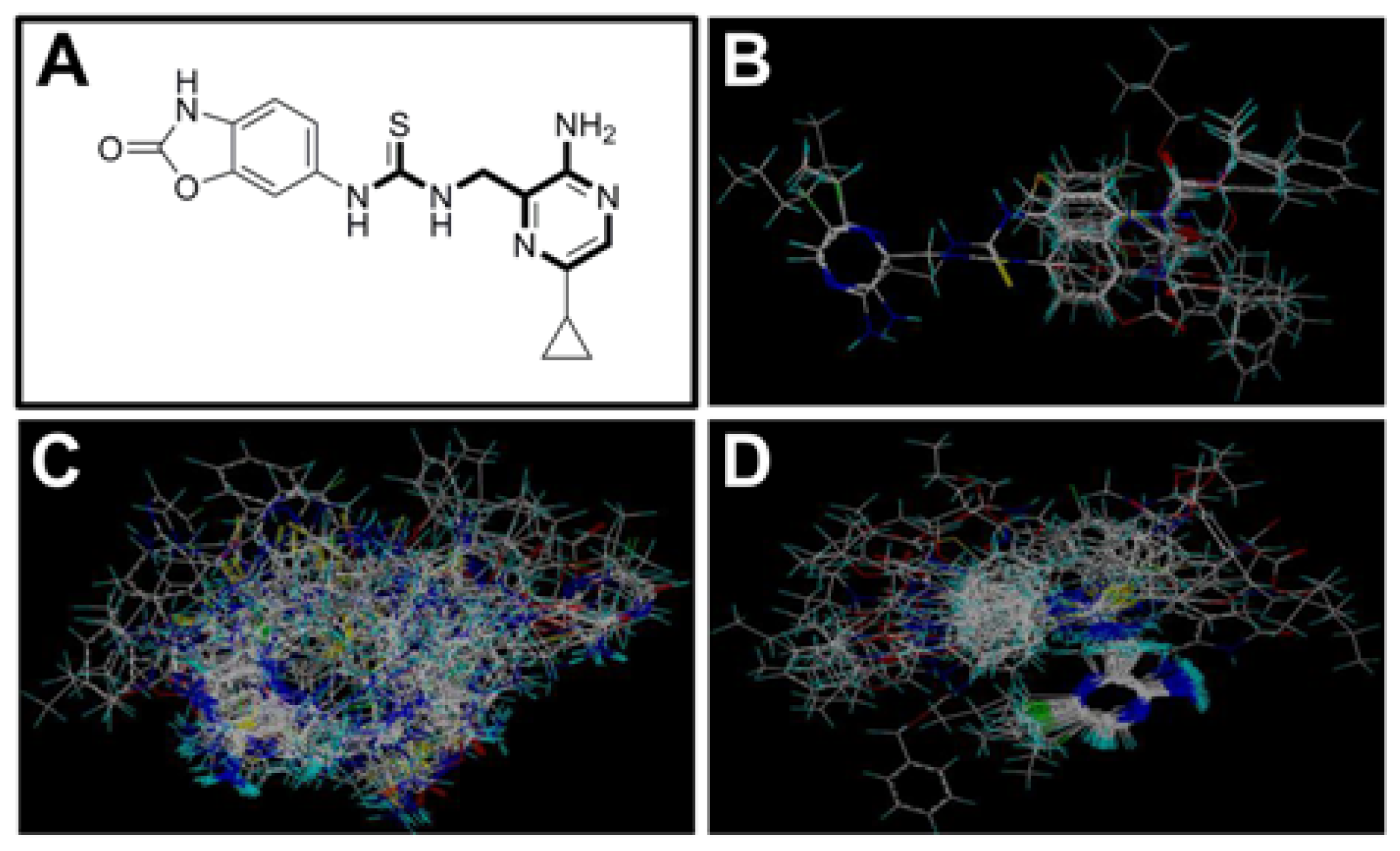
2.3. Contour Maps
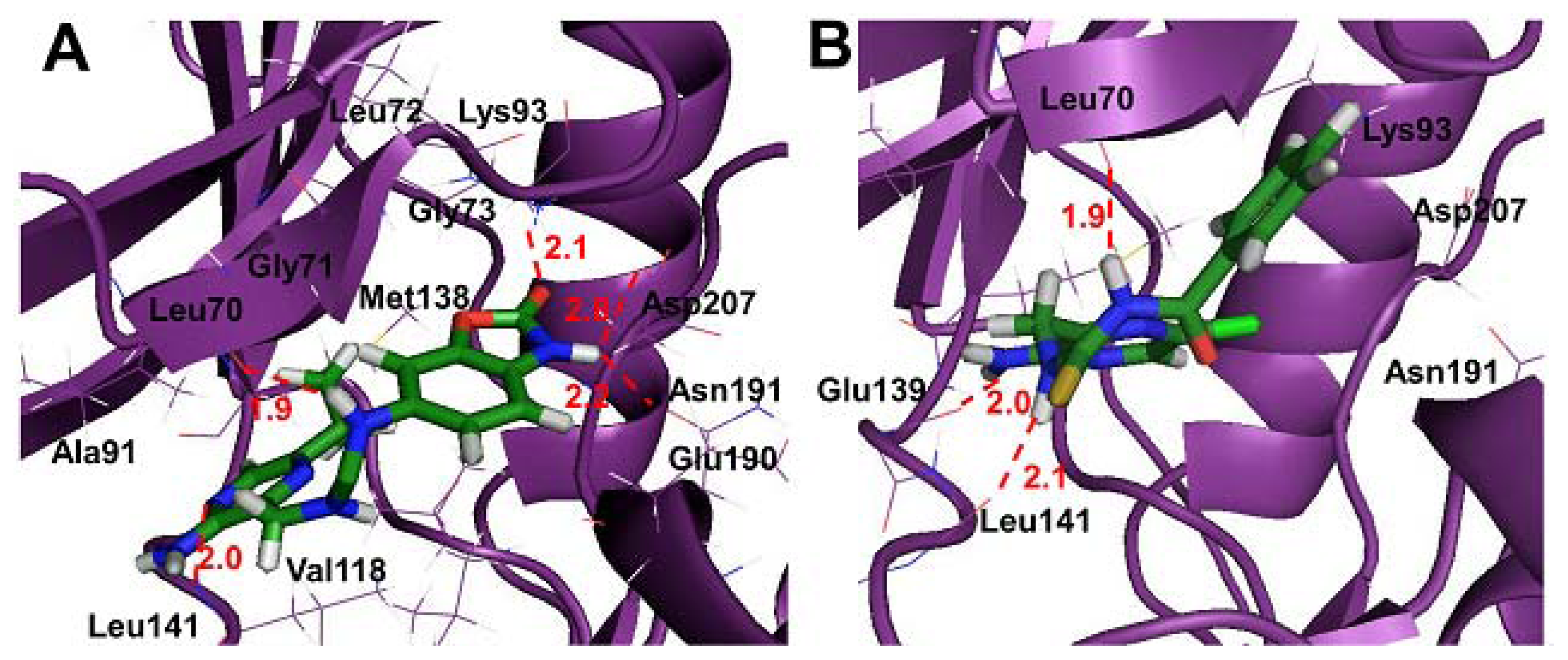
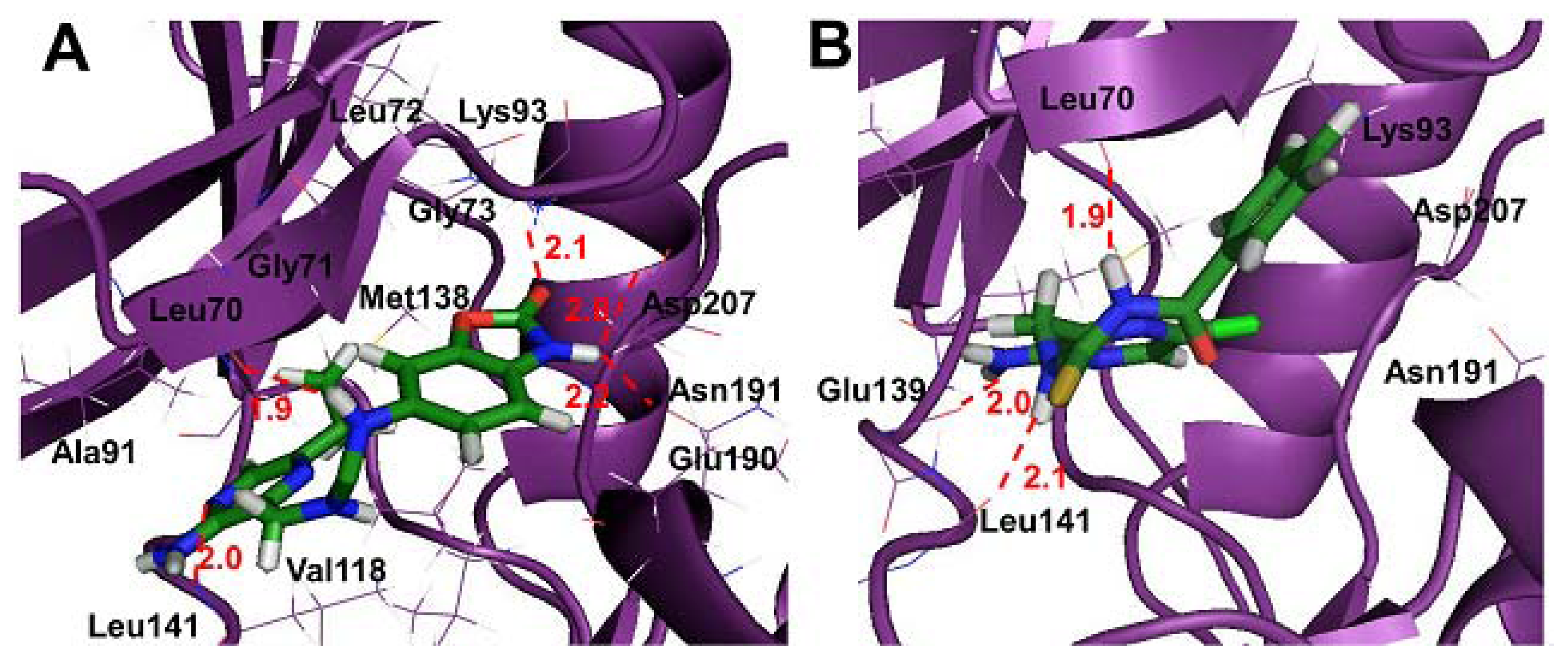
2.4. Molecular Docking
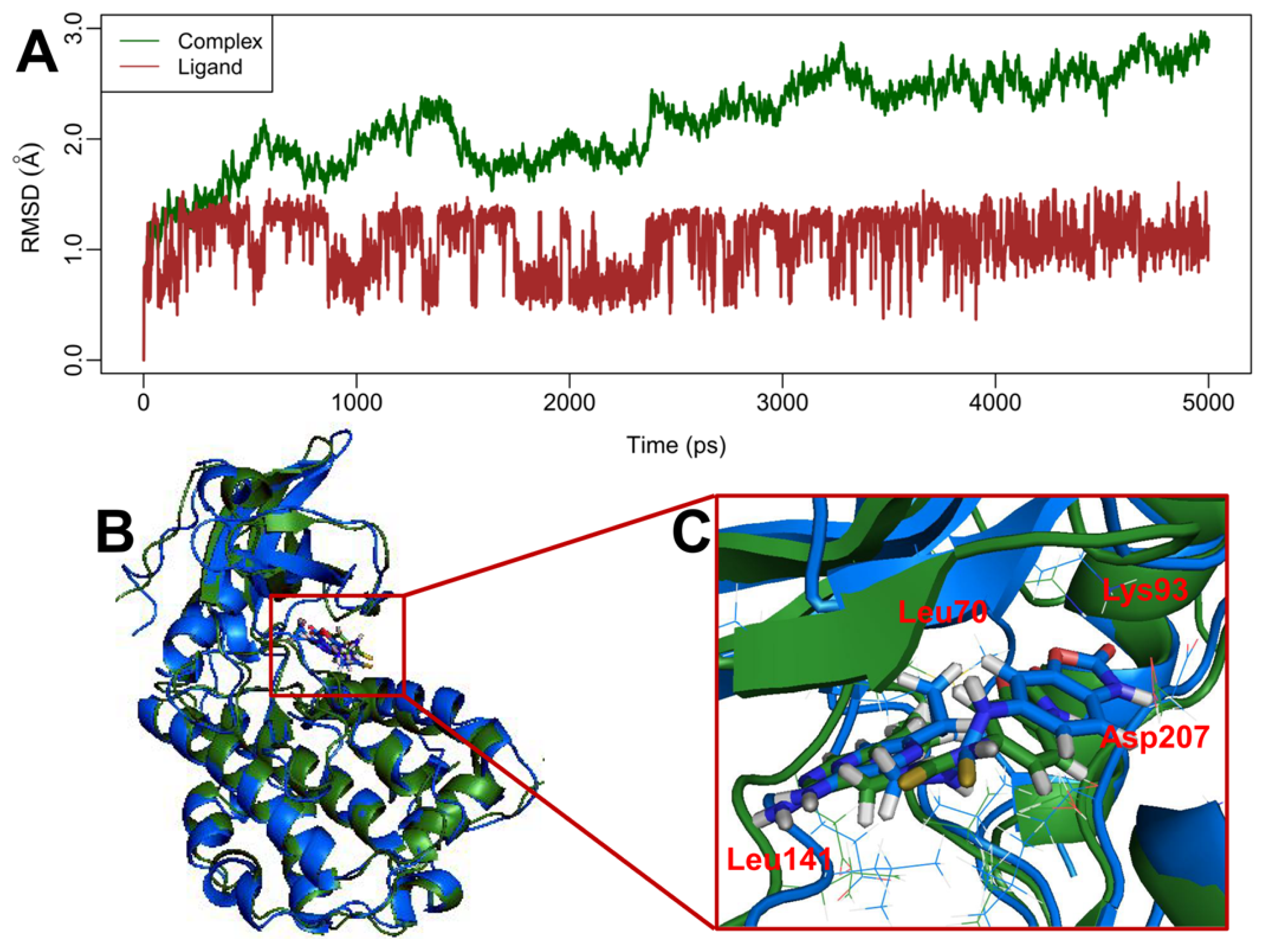
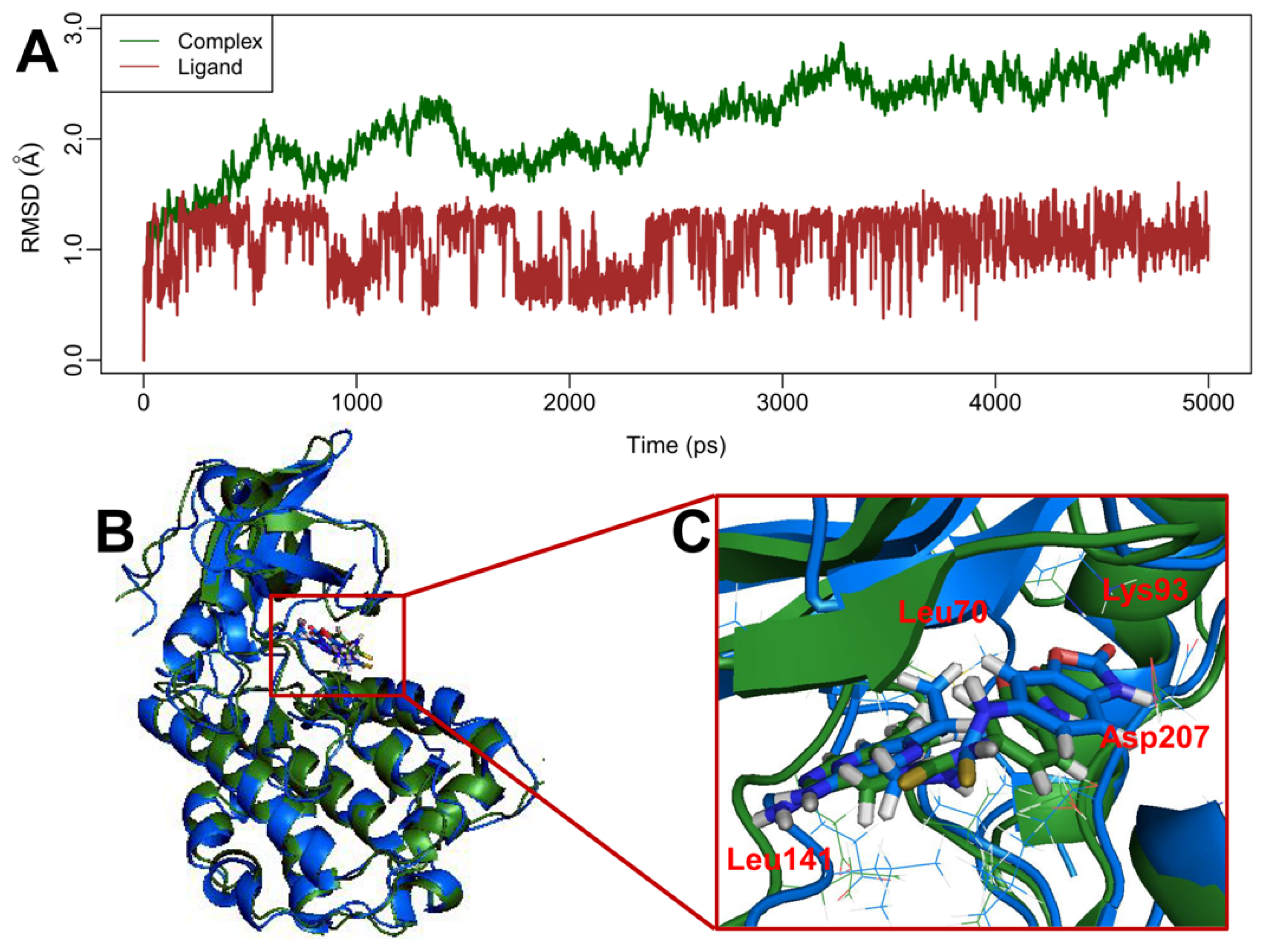
2.5. MD Simulations
3. Materials and Experimental Methods
3.1. Dataset
3.2. Molecular Modeling and Alignment
3.3. Docking Simulation
3.4. CoMFA and CoMSIA Calculations
3.5. Statistical Validation
3.6. Molecular Dynamics Simulations
4. Conclusions
Acknowledgement
References
- Camussi, G.; Lupia, E. The future role of anti-tumour necrosis factor (TNF) products in the treatment of rheumatoid arthritis. Drugs 1998, 55, 613–620. [Google Scholar]
- Barf, T.; Kaptein, A.; de Wilde, S.; van der Heijden, R.; van Someren, R.; Demont, D.; Schultz-Fademrecht, C.; Versteegh, J.; van Zeeland, M.; Seegers, N.; et al. Structure-based lead identification of ATP-competitive MK2 inhibitors. Bioorg. Med. Chem. Lett 2011, 21, 3818–3822. [Google Scholar]
- Lin, S.; Lombardo, M.; Malkani, S.; Hale, J.J.; Mills, S.G.; Chapman, K.; Thompson, J.E.; Zhang, W.X.; Wang, R.; Cubbon, R.M.; et al. Novel 1-(2-aminopyrazin-3-yl)methyl-2-thioureas as potent inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett 2009, 19, 3238–3242. [Google Scholar]
- Dominguez, C.; Powers, D.A.; Tamayo, N. p38 MAP kinase inhibitors: Many are made, but few are chosen. Curr. Opin. Drug Discov. Devel 2005, 8, 421–430. [Google Scholar]
- Schlapbach, A.; Feifel, R.; Hawtin, S.; Heng, R.; Koch, G.; Moebitz, H.; Revesz, L.; Scheufler, C.; Velcicky, J.; Waelchli, R. Pyrrolo-pyrimidones: A novel class of MK2 inhibitors with potent cellular activity. Bioorg. Med. Chem. Lett 2008, 18, 6142–6146. [Google Scholar]
- Cheung, P.C.F.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J 2003, 22, 5793–5805. [Google Scholar]
- Winzen, R.; Kracht, M.; Ritter, B.; Wilhelm, A.; Chen, C.Y.A.; Shyu, A.B.; Müller, M.; Gaestel, M.; Resch, K.; Holtmann, H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J 1999, 18, 4969–4980. [Google Scholar]
- Goldberg, D.; Choi, Y.; Cogan, D.; Corson, M.; DeLeon, R.; Gao, A.; Gruenbaum, L.; Hao, M.; Joseph, D.; Kashem, M. Pyrazinoindolone inhibitors of MAPKAP-K2. Bioorg. Med. Chem. Lett 2008, 18, 938–941. [Google Scholar]
- Hegen, M.; Gaestel, M.; Nickerson-Nutter, C.L.; Lin, L.L.; Telliez, J.B. MAPKAP kinase 2-deficient mice are resistant to collagen-induced arthritis. J. Immunol 2006, 177, 1913–1917. [Google Scholar]
- Trujillo, J.; Meyers, M.; Anderson, D.; Hegde, S.; Mahoney, M.; Vernier, W.; Buchler, I.; Wu, K.; Yang, S.; Hartmann, S. Novel tetrahydro-β-carboline-1-carboxylic acids as inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett 2007, 17, 4657–4663. [Google Scholar]
- Anderson, D.; Meyers, M.; Vernier, W.; Mahoney, M.; Kurumbail, R.; Caspers, N.; Poda, G.; Schindler, J.; Reitz, D.; Mourey, R. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J. Med. Chem 2007, 50, 2647–2654. [Google Scholar]
- Anderson, D.R.; Hegde, S.; Reinhard, E.; Gomez, L.; Vernier, W.F.; Lee, L.; Liu, S.; Sambandam, A.; Snider, P.A.; Masih, L. Aminocyanopyridine inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett 2005, 15, 1587–1590. [Google Scholar]
- Wu, J.; Wang, J.; Abeywardane, A.; Andersen, D.; Emmanuel, M.; Gautschi, E.; Goldberg, D.; Kashem, M.; Lukas, S.; Mao, W. The discovery of carboline analogs as potent MAPKAP-K2 inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 4664–4669. [Google Scholar]
- Xiong, Z.; Gao, D.; Cogan, D.; Goldberg, D.; Hao, M.; Moss, N.; Pack, E.; Pargellis, C.; Skow, D.; Trieselmann, T. Synthesis and SAR studies of indole-based MK2 inhibitors. Bioorg. Med. Chem. Lett 2008, 18, 1994–1999. [Google Scholar]
- Anderson, D.; Meyers, M.; Kurumbail, R.; Caspers, N.; Poda, G.; Long, S.; Pierce, B.; Mahoney, M.; Mourey, R. Benzothiophene inhibitors of MK2. Part 1: Structure-activity relationships, assessments of selectivity and cellular potency. Bioorg. Med. Chem. Lett 2009, 19, 4878–4881. [Google Scholar]
- Anderson, D.; Meyers, M.; Kurumbail, R.; Caspers, N.; Poda, G.; Long, S.; Pierce, B.; Mahoney, M.; Mourey, R.; Parikh, M. Benzothiophene inhibitors of MK2. Part 2: Improvements in kinase selectivity and cell potency. Bioorg. Med. Chem. Lett 2009, 19, 4882–4884. [Google Scholar]
- Kaptein, A.; Oubrie, A.; de Zwart, E.; Hoogenboom, N.; de Wit, J.; van de Kar, B.; van Hoek, M.; Vogel, G.; de Kimpe, V.; Schultz-Fademrecht, C.; et al. Discovery of selective and orally available spiro-3-piperidyl ATP-competitive MK2 inhibitors. Bioorg. Med. Chem. Lett 2011, 21, 3823–3827. [Google Scholar]
- Oubrie, A.; Kaptein, A.; de Zwart, E.; Hoogenboom, N.; Goorden, R.; van de Kar, B.; van Hoek, M.; de Kimpe, V.; van der Heijden, R.; Borsboom, J.; et al. Novel ATP competitive MK2 inhibitors with potent biochemical and cell-based activity throughout the series. Bioorg. Med. Chem. Lett 2012, 22, 613–618. [Google Scholar]
- Afantitis, A.; Melagraki, G.; Sarimveis, H.; Koutentis, P.A.; Markopoulos, J.; Igglessi-Markopoulou, O. A novel QSAR model for evaluating and predicting the inhibition activity of dipeptidyl aspartyl fluoromethylketones. QSAR Comb. Sci 2006, 25, 928–935. [Google Scholar]
- Kumar, H.; Kumar, R.; Grewal, B.K.; Sobhia, M.E. Insights into the Structural Requirements of PKCβII Inhibitors Based on HQSAR and CoMSIA Analyses. Chem. Biol. Drug Des 2011, 78, 283–288. [Google Scholar]
- Mercader, A.G.; Pomilio, A.B. QSAR study of flavonoids and biflavonoids as influenza H1N1 virus neuraminidase inhibitors. Eur. J. Med. Chem 2010, 45, 1724–1730. [Google Scholar]
- Deeb, O.; Goodarzi, M. Exploring QSARs for inhibitory activity of non-peptide HIV-1 protease inhibitors by GA-PLS and GA-SVM. Chem. Biol. Drug Des 2010, 75, 506–514. [Google Scholar]
- Darnag, R.; Mazouz, E.L.M.; Schmitzer, A.; Villemin, D.; Jarid, A.; Cherqaoui, D. Support vector machines: Development of QSAR models for predicting anti-HIV-1 activity of TIBO derivatives. Eur. J. Med. Chem 2010, 45, 1590–1597. [Google Scholar]
- Yang, Z.Q.; Sun, P.H.; Chen, W.M. 3D-QSAR analysis on pyrrolopyridine analogs as mitogen-activated protein kinase-activated protein kinase 2 (MK-2) inhibitors. Lett. Drug Des. Discov 2007, 4, 557–561. [Google Scholar]
- Kaushik, U.; Sharma, V.; Kumar, V. Computation of pharmacophore models for the prediction of mitogen-activated protein kinase activated protein kinase-2 inhibitory activity of pyrrolopyridines. Med. Chem. Res 2011. [Google Scholar] [CrossRef]
- Nayana, R.S.; Bommisetty, S.K.; Singh, K.; Bairy, S.K.; Nunna, S.; Pramod, A.; Muttineni, R. Structural analysis of carboline derivatives as inhibitors of MAPKAP K2 using 3D QSAR and docking studies. J. Chem. Inf. Model 2009, 49, 53–67. [Google Scholar]
- Ren, Y.; Liu, H.; Yao, X.; Liu, M. Prediction of ozone tropospheric degradation rate constants by projection pursuit regression. Anal. Chim. Acta 2007, 589, 150–158. [Google Scholar]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model 2008, 48, 1337–1344. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graphics Modell 2002, 20, 269–276. [Google Scholar]
- Golbraikh, A.; Shen, M.; Xiao, Z.Y.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput.-Aided Mol. Des 2003, 17, 241–253. [Google Scholar]
- Roy, P.; Roy, K. On some aspects of variable selection for partial least squares regression models. QSAR Comb. Sci 2008, 27, 302–313. [Google Scholar]
- Velcicky, J.; Feifel, R.; Hawtin, S.; Heng, R.; Huppertz, C.; Koch, G.; Kroemer, M.; Moebitz, H.; Revesz, L.; Scheufler, C.; et al. Novel 3-aminopyrazole inhibitors of MK-2 discovered by scaffold hopping strategy. Bioorg. Med. Chem. Lett 2010, 20, 1293–1297. [Google Scholar]
- Lovering, F.; Kirincich, S.; Wang, W.; Combs, K.; Resnick, L.; Sabalski, J.E.; Butera, J.; Liu, J.; Parris, K.; Telliez, J.B. Identification and SAR of squarate inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem 2009, 17, 3342–3351. [Google Scholar]
- Hillig, R.C.; Eberspaecher, U.; Monteclaro, F.; Huber, M.; Nguyen, D.; Mengel, A.; Muller-Tiemann, B.; Egner, U. Structural basis for a high affinity inhibitor bound to protein kinase MK2. J. Mol. Biol 2007, 369, 735–745. [Google Scholar]
- Murumkar, P.R.; Gupta, S.D.; Zambre, V.P.; Giridhar, R.; Yadav, M.R. Development of predictive 3D-QSAR CoMFA and CoMSIA models for β-aminohydroxamic acid-derived tumor necrosis factor-α converting enzyme inhibitors. Chem. Biol. Drug Des 2009, 73, 97–107. [Google Scholar]
- Murumkar, P.R.; Giridhar, R.; Yadav, M.R. 3D-quantitative structure-activity relationship studies on benzothiadiazepine hydroxamates as inhibitors of tumor necrosis factor-α converting enzyme. Chem. Biol. Drug Des 2008, 71, 363–373. [Google Scholar]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 2003, 31, 3381–3385. [Google Scholar]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The universal protein resource (UniProt). Nucleic Acids Res 2005, 33, D154–D159. [Google Scholar]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Walker, R.; Zhang, W.; Merz, K.; et al. AMBER 11; University of California: San Francisco, CA, USA, 2010. [Google Scholar]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc 2001, 123, 5221–5230. [Google Scholar]
- Nettels, D.; Müller-Späth, S.; Küster, F.; Hofmann, H.; Haenni, D.; Rüegger, S.; Reymond, L.; Hoffmann, A.; Kubelka, J.; Heinz, B.; et al. Single-molecule spectroscopy of the temperature-induced collapse of unfolded proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 20740–20745. [Google Scholar]
- Agrawal, P.M.; Rice, B.M.; Zheng, L.; Thompson, D.L. Molecular dynamics simulations of hexahydro-1,3,5-trinitro-1,3,5-s-triazine (RDX) using a combined sorescu−rice−thompson AMBER force field. J. Phys. Chem. B 2006, 110, 26185–26188. [Google Scholar]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS analysis | Alignment methods | |||||
|---|---|---|---|---|---|---|
| I | II | III | ||||
| CoMFA | CoMSIA | CoMFA | CoMSIA | CoMFA | CoMSIA | |
| q2 | 0.536 | 0.556 | −0.035 | 0.263 | 0.311 | 0.400 |
| SEP | 0.642 | 0.595 | 0.909 | 0.759 | 0.741 | 0.699 |
| PCs | 8 | 3 | 3 | 2 | 3 | 4 |
| r2ncv | 0.974 | 0.779 | 0.873 | 0.810 | 0.897 | 0.843 |
| SEE | 0.151 | 0.420 | 0.318 | 0.385 | 0.287 | 0.357 |
| F value | 207.641 | 57.509 | 112.475 | 106.868 | 141.747 | 64.605 |
| r2cv(mean) | 0.630 | 0.606 | 0.169 | 0.273 | 0.401 | 0.390 |
| r2bs | 0.989 | 0.820 | - | - | - | - |
| SEEbs | 0.099 | 0.374 | - | - | - | - |
| r2pred | 0.810 | 0.669 | 0.416 | 0.480 | 0.444 | 0.156 |
| Relative Contribution (%) | ||||||
| S | 0.496 | 0.250 | 0.348 | - | 0.475 | 1.000 |
| E | 0.504 | 0.503 | 0.652 | 0.483 | 0.525 | - |
| H | - | - | - | 0.517 | - | - |
| D | - | 0.247 | - | - | - | - |
| A | - | - | - | - | - | |
| Model | r2pred | r2test | (r2test − r2o)/r2test | k | r2m |
|---|---|---|---|---|---|
| CoMFA | 0.810 | 0.807 | 0.013 | 0.993 | 0.723 |
| CoMSIA | 0.669 | 0.677 | 0.054 | 0.998 | 0.548 |
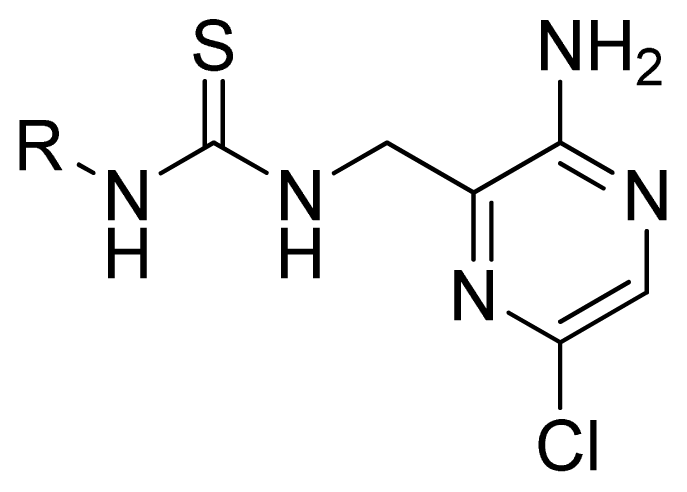 | |||||
|---|---|---|---|---|---|
| Compd. | R | Experimental pIC50 | Predicted pIC50 | ||
| CoMFA | CoMSIA | ||||
| 1 | Ph | 5.699 | 5.344 | 5.391 | |
| 2* | Et | 4.699 | 5.063 | 4.954 | |
| 3 | i-Pr | 4.886 | 5.013 | 4.978 | |
| 4 | t-Bu | 5.602 | 5.569 | 4.973 | |
| 5 | c-Pr | 4.921 | 4.773 | 5.374 | |
| 6* | c-Pentyl | 4.276 | 4.735 | 5.375 | |
| 7 | c-Heptanyl | 4.456 | 4.380 | 5.364 | |
| 8* | Bn | 4.444 | 4.401 | 4.736 | |
| 9 | MeOCH2CH2 | 4.658 | 4.507 | 4.405 | |
| 10 | Benzoyl | 4.041 | 4.213 | 4.249 | |
| 11 | 1-Naphthyl | 5.495 | 5.595 | 4.816 | |
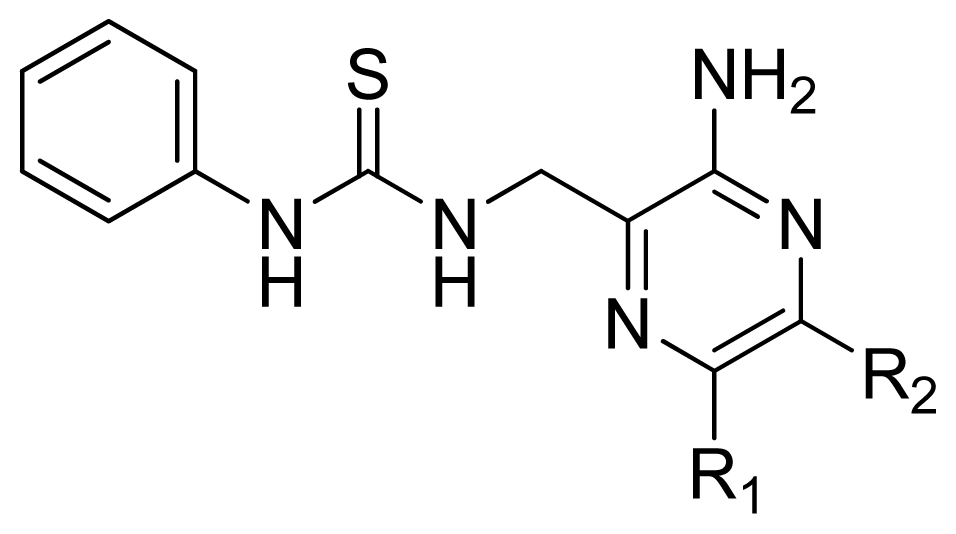 | |||||
| Compd. | R1 | R2 | Exprimental pIC50 | Predicted pIC50 | |
| CoMFA | CoMSIA | ||||
| 12 | H | H | 4.495 | 4.687 | 5.440 |
| 13 | Me | H | 5.398 | 5.261 | 5.001 |
| 14 * | Et | H | 5.824 | 5.799 | 5.203 |
| 15 | n-Pr | H | 5.638 | 5.614 | 5.837 |
| 16 * | c-Pr | H | 6.328 | 6.051 | 5.811 |
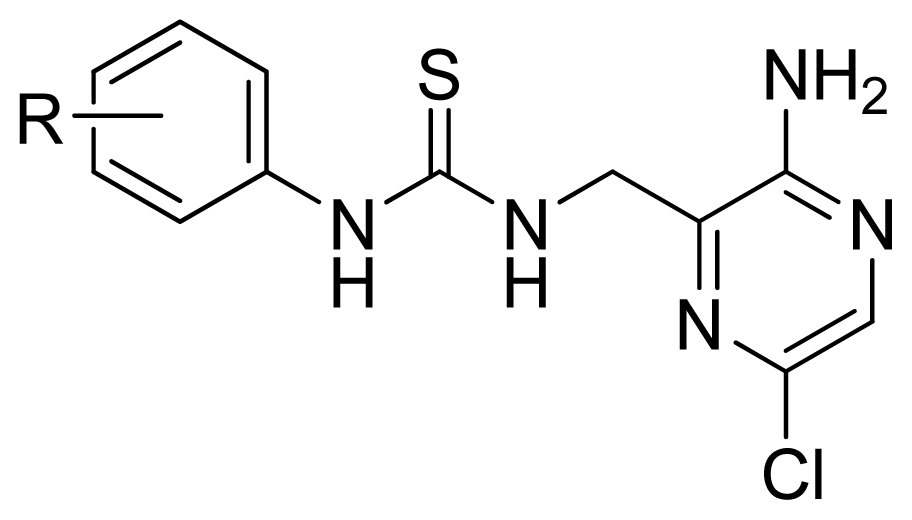 | |||||
| Compd. | R | Experimental pIC50 | Predicted pIC50 | ||
| CoMFA | CoMSIA | ||||
| 17 | 4-Me | 5.569 | 5.714 | 5.436 | |
| 18 * | 4-Cl | 5.432 | 5.348 | 5.263 | |
| 19 | 4-MeO | 5.721 | 5.444 | 4.948 | |
| 20 | 4-i-Pr | 5.585 | 5.516 | 5.337 | |
| 21 | 4-t-Bu | 4.863 | 4.968 | 4.868 | |
| 22 | 4-Br | 5.509 | 5.357 | 5.261 | |
| 23 | 4-NO2 | 5.004 | 5.168 | 5.527 | |
| 24 * | 4-CN | 5.161 | 4.859 | 5.153 | |
| 25 | 4-BnO | 5.569 | 5.582 | 5.290 | |
| 26 * | 4-Ac | 5.161 | 4.910 | 5.010 | |
| 27 | 4-EtOC(=O)- | 4.971 | 4.949 | 4.958 | |
| 28 | 4-NMe2 | 5.155 | 5.405 | 5.060 | |
| 29 | 4-(Morphorin-1-yl) | 4.762 | 4.809 | 4.889 | |
| 30 | 4-AcNH- | 5.284 | 5.515 | 5.908 | |
| 31 | 4-NH2 | 5.699 | 5.795 | 5.811 | |
| 32 | 4-BnOC(=O)NH- | 6.337 | 6.322 | 6.218 | |
| 33 | 2-Me | 5.444 | 5.488 | 5.342 | |
| 34 | 2-Cl | 5.959 | 5.778 | 5.473 | |
| 35 | 2-MeO | 5.585 | 5.599 | 5.619 | |
| 36 | 2-F | 5.699 | 5.720 | 5.524 | |
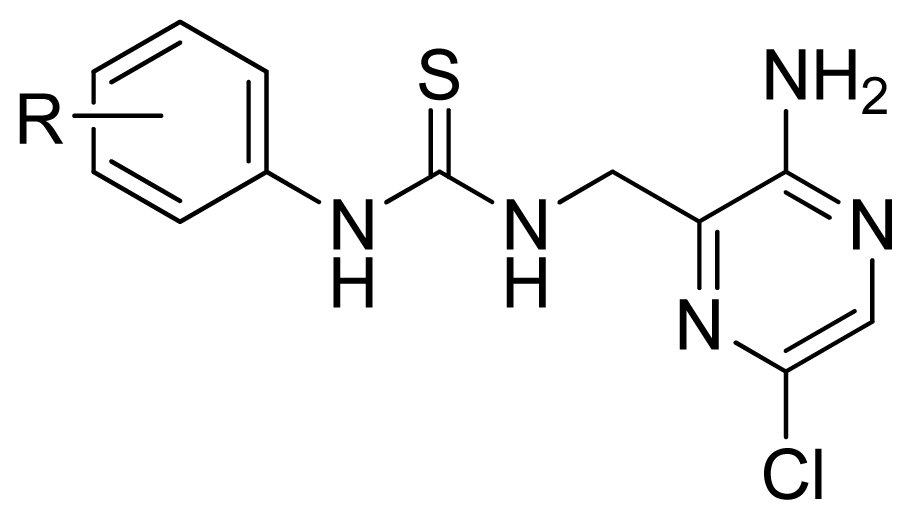 | |||||
| Compd. | R | Experimental pIC50 | Predicted pIC50 | ||
| CoMFA | CoMSIA | ||||
| 37 | 2-MeS | 5.602 | 5.711 | 5.511 | |
| 38 * | 3-Me | 5.032 | 5.160 | 5.348 | |
| 39 | 3-Cl | 4.947 | 5.088 | 5.375 | |
| 40 * | 3-MeO | 4.839 | 5.507 | 5.107 | |
| 41 | 2,4-Di-MeO | 5.602 | 5.457 | 5.365 | |
| 42 * | 2,4-Di-Cl | 5.367 | 5.787 | 5.345 | |
| 43 | 3,5-Di-Cl | 4.569 | 4.564 | 5.374 | |
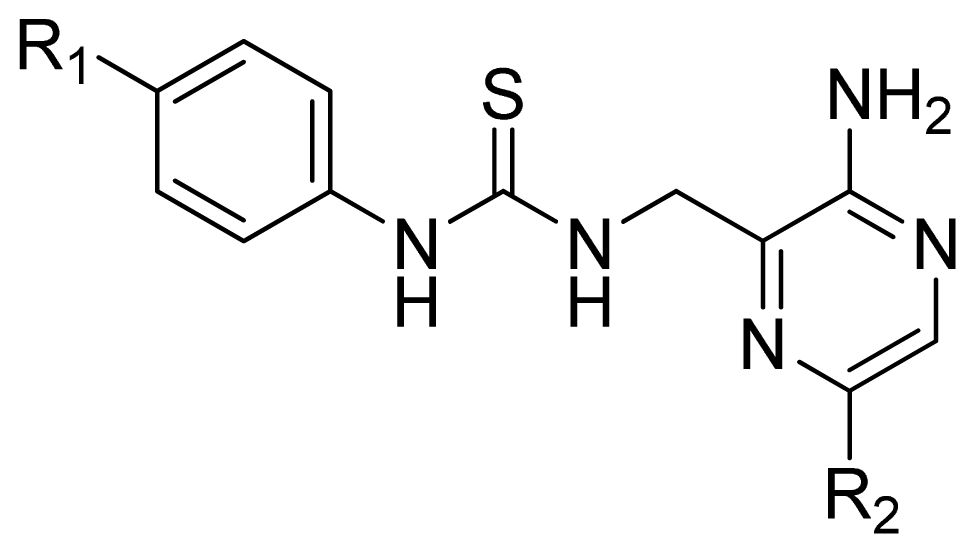 | |||||
| Compd. | R1 | R2 | Experimental pIC50 | Predicted pIC50 | |
| CoMFA | CoMSIA | ||||
| 44 | MeOC(=O)NH- | Cl | 6.027 | 5.884 | 5.920 |
| 45 | EtOC(=O)NH- | Cl | 6.509 | 6.147 | 5.997 |
| 46 | n-BuOC(=O)NH- | Cl | 6.638 | 6.765 | 6.124 |
| 47 | i-BuOC(=O)NH- | Cl | 6.824 | 6.841 | 6.114 |
| 48 * | t-BuOC(=O)NH- | Cl | 6.721 | 6.299 | 6.025 |
| 49 | i-BuOC(=O)N(Ph)- | Cl | 5.824 | 5.772 | 5.824 |
| 50 * | MeNHC(=O)NH- | Cl | 5.770 | 5.765 | 6.309 |
| 51 | t-BuNHC(=O)NH- | Cl | 6.155 | 6.234 | 6.451 |
| 52 | t-BuOC(=O)NH- | Me | 6.678 | 6.530 | 6.624 |
| 53 | PhOC(=O)NH- | Me | 6.137 | 6.221 | 6.165 |
| 54 | EtOC(=O)N(Me)- | Me | 5.000 | 5.067 | 5.592 |
| 55 | t-BuOC(=O)O- | Me | 5.538 | 5.684 | 5.918 |
| 56 | EtC(=O)NH- | Me | 4.876 | 4.854 | 5.679 |
| 57 | n-PrC(=O)NH- | Me | 5.638 | 5.541 | 5.719 |
| 58 * | n-BuC(=O)NH- | Me | 5.824 | 5.612 | 5.715 |
| 59 | BnC(=O)NH- | Me | 5.420 | 5.518 | 5.487 |
| 60 * | PhC(=O)NH- | Me | 6.237 | 5.748 | 5.971 |
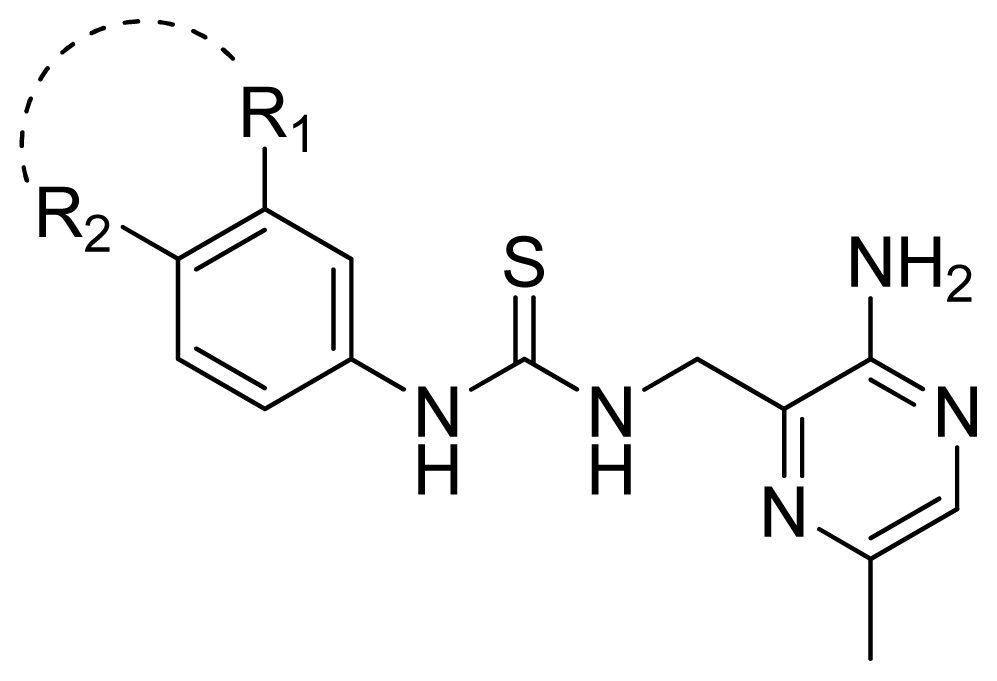 | |||||
| Compd. | R1-R2 | Experimental pIC50 | Predicted pIC50 | ||
| CoMFA | CoMSIA | ||||
| 61 | 3-NHC(=O)NH-4 | 4.824 | 4.639 | 4.696 | |
| 62 | 3-CH2C(=O)NH-4 | 4.569 | 4.696 | 4.842 | |
| 63 * | 3-SC(=O)NH-4 | 4.824 | 5.106 | 4.972 | |
| 64 * | 3-NHC(=O)O-4 | 4.854 | 5.437 | 5.382 | |
 | |||||
| Compd. | R1 | R2 | Experimental pIC50 | Predicted pIC50 | |
| CoMFA | CoMSIA | ||||
| 65 | 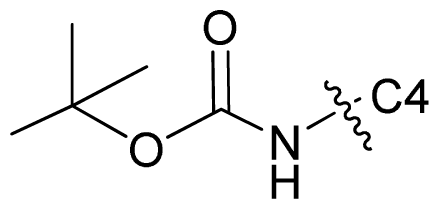 | Et | 6.854 | 6.827 | 6.824 |
| 66 | 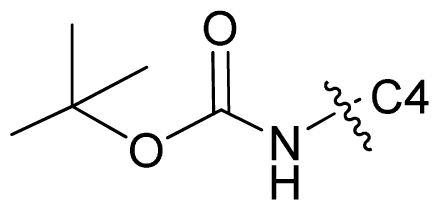 | n-Pr | 7.347 | 7.409 | 7.020 |
| 67* | 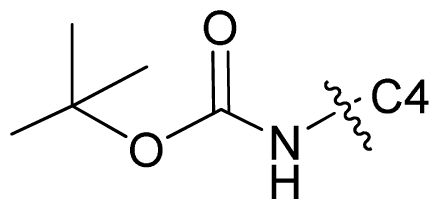 | c-Pr | 6.987 | 6.996 | 6.470 |
| 68 | 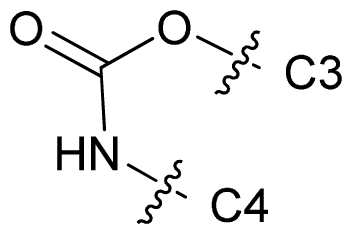 | n-Pr | 7.699 | 7.577 | 7.681 |
| 69 | 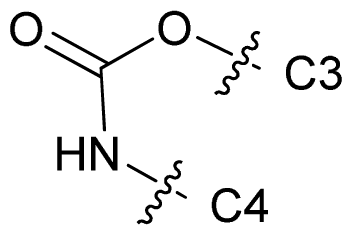 | n-Bu | 7.523 | 7.539 | 7.795 |
| 70 | 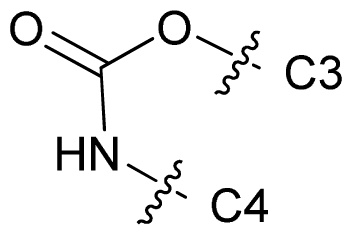 | c-Pr | 7.824 | 7.960 | 7.422 |
| 71 | 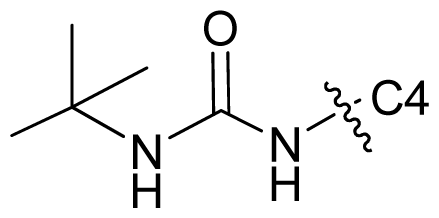 | c-Pr | 7.081 | 7.100 | 6.889 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hao, M.; Ren, H.; Luo, F.; Zhang, S.; Qiu, J.; Ji, M.; Si, H.; Li, G. A Computational Study on Thiourea Analogs as Potent MK-2 Inhibitors. Int. J. Mol. Sci. 2012, 13, 7057-7079. https://doi.org/10.3390/ijms13067057
Hao M, Ren H, Luo F, Zhang S, Qiu J, Ji M, Si H, Li G. A Computational Study on Thiourea Analogs as Potent MK-2 Inhibitors. International Journal of Molecular Sciences. 2012; 13(6):7057-7079. https://doi.org/10.3390/ijms13067057
Chicago/Turabian StyleHao, Ming, Hong Ren, Fang Luo, Shuwei Zhang, Jieshan Qiu, Mingjuan Ji, Hongzong Si, and Guohui Li. 2012. "A Computational Study on Thiourea Analogs as Potent MK-2 Inhibitors" International Journal of Molecular Sciences 13, no. 6: 7057-7079. https://doi.org/10.3390/ijms13067057